1. Introduction
Neuroblastoma, one of the most common cancers in children, is highly complex in terms of its genetic, physiological and clinical heterogeneity. It is this complexity that makes it extremely challenging to diagnose when and how neuroblastoma develops and further design the precise intervention [
1,
2]. In practice, several clinical parameters, such as age at diagnosis, tumor stage, genomic amplification of
MYCN oncogene, and ploidy, are widely used as the markers of neuroblastoma risk [
3,
4,
5,
6]. Increasing studies have attempted to stratify neuroblastoma risk based on the pattern of differential gene expression [
1,
4,
5,
7,
8]. Sets of genes of prognostic importance have been identified to link with neuroblastoma; for example, Oberthuer et al. [
4] established a 144-gene predictor for classifying the stratification of neuroblastoma patients, Formicola et al. [
5] used 18 genes to predict the outcome of stage 4 patients, and Utnes et al. [
6] identified 20 mRNAs and six lncRNAs of clinical relevance to the prediction of tumor recurrence and response to neuroblastoma therapy.
Existing approaches for gene-based stratification are mostly based on the differential expression of single genes. As a widely used approach for general cancer research, such reductionist thinking can simplify the identification of key major genes for neuroblastoma risk [
7]. It is becoming increasingly clear that a deeper understanding of cancer, including neuroblastoma, requires not only a detailed characterization of individual genes, but also of their interactions, which are encapsulated into an intricate but highly coordinated network [
9,
10]. As a complex disease, it is likely that neuroblastoma involves numerous genes that interact and work together to form a complex cellular network. As such, by regarding neuroblastoma as a network disease, we can develop and apply a holistic, systems-oriented approach to better reveal and interpret its genomic causes [
11,
12].
Computational tools play a critical role in reconstructing gene regulatory networks. Many approaches can only infer a single context-agnostic interaction network from a large number of samples, failing to reveal inter-sample heterogeneity [
13,
14]. Although several approaches have explored the possibility of reconstructing sample-specific networks [
15,
16], their application may be impaired by an incapacity to code a complete set of bidirectional, signed, and weighted interactions into a graph. Recovering such fully informative networks essentially requires the dynamic fitting of densely spaced temporal data [
17], which are extremely difficult or even impossible to collect in practice; for example, multiple sampling and monitoring are not logistically permitted for cancer single-cell analysis and human gut microbiota studies. A majority of genomic studies conducted for cancer dissection in recent decades only have static data available.
More recently, Chen et al. [
18] developed an interdisciplinary framework for reconstructing informative, dynamic, omnidirectional, and personalized networks (idopNetworks) using steady-state gene expression data from regular genomic experiments. The idopNetwork model can maximize the utility efficiency of genomic data to reconstruct biologically meaningful, context-specific, and large-scale but sparse networks. In clinical studies of graft remodeling, the use of idopNetworks has provided new insight into the genomic mechanisms of gene co-regulation that determine the success and failure of surgical operation. In this article, we apply and modify the idopNetwork model to reconstruct co-regulation networks of immune genes whose function may be related to neuroblastoma formation and identify which gene interactions determine high vs. low risk of this disease. As the proof of concept, idopNetworks characterize previously unknown gene interactions that can better serve as the biomarkers of neuroblastoma risk.
3. Discussion
Differentiated expression of genes has been widely used as an approach for stratifying neuroblastoma risk. Although single gene analysis has proven its power for risk stratification, we argue that neuroblastoma risk includes multiple genes that interact with each other through intricate but coordinated interaction networks. Traditional approaches for inferring informative networks, i.e., those coded by bidirectional, signed, and weighted interactions, rely on high-density temporal data which are hardly available in most cancer studies. These approaches can also only reconstruct context-agnostic networks, failing to reveal the change in network structure in response to environmental and developmental signals. We implement and modify Chen et al.’s [
18] networking model to recover informative, dynamic, omnidirectional, and personalized networks (idopNetworks) from static expression data and compare how idopNetworks vary structurally and organizationally over biological and environmental regimes. By incorporating functional clustering and variable selection, idopNetworks can be identified at any dimension and scale and with any topological complexity. All these advantages are especially essential and of the utmost importance for neuroblastoma-gene networking because this disease involves multitudinous genes interacting in a complex, dynamic manner.
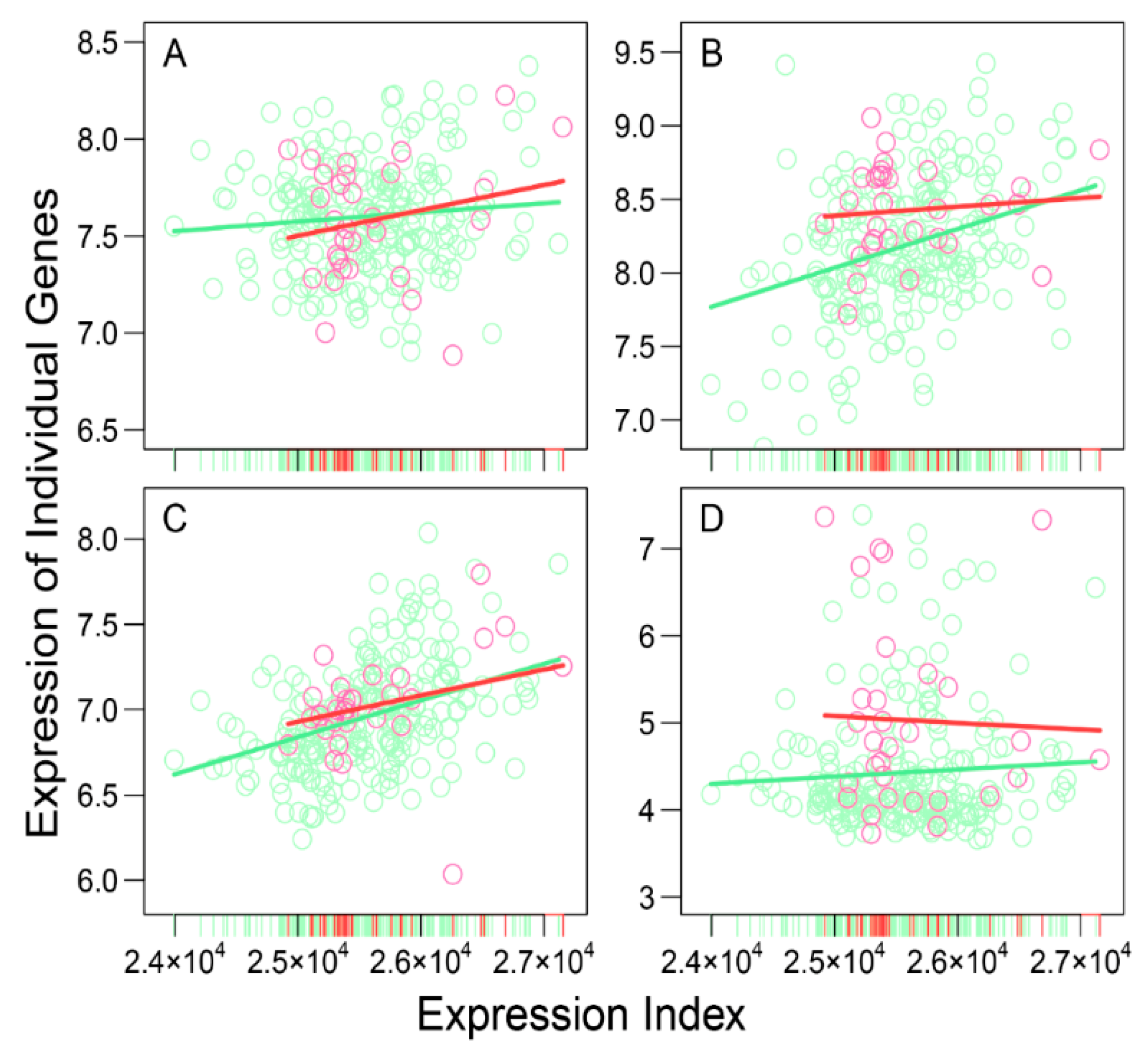
As the proof of concept, we analyzed publicly available data containing 3439 genes expressed in neuroblastoma patients to show the potential of idopNetworks as a predictive biomarker of genomic differences between high- and low-risk patients. In reconstructing idopNetworks, we found that neuroblastoma risk can be predicted at three levels. First, the premise of reconstructing idopNetworks is to define the concept of expression index defined as the total amount of gene expression in a sample. We found that the expression index of low-risk patients spans the upper two-third of high-risk patients (
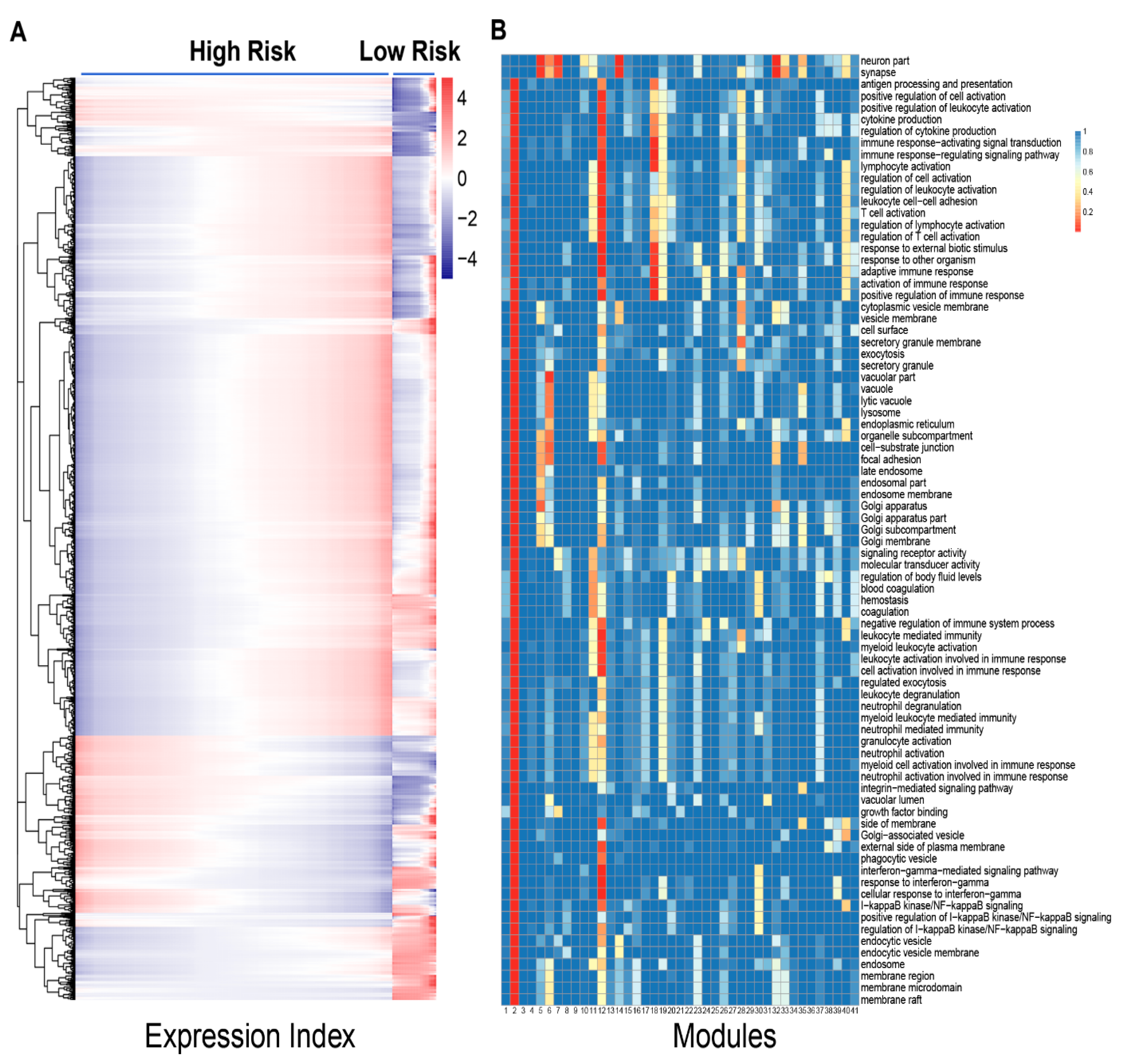
Figure 1), showing the possibility to assess those in the lower one third of expression index as high-risk patients. Second, among a total of 41 distinct gene modules classified by functional clustering, we found that some modules change their expression pattern over expression index differently between two groups of patients (
Figure 2). As thus, these modules can be potentially used to predict neuroblastoma risk.
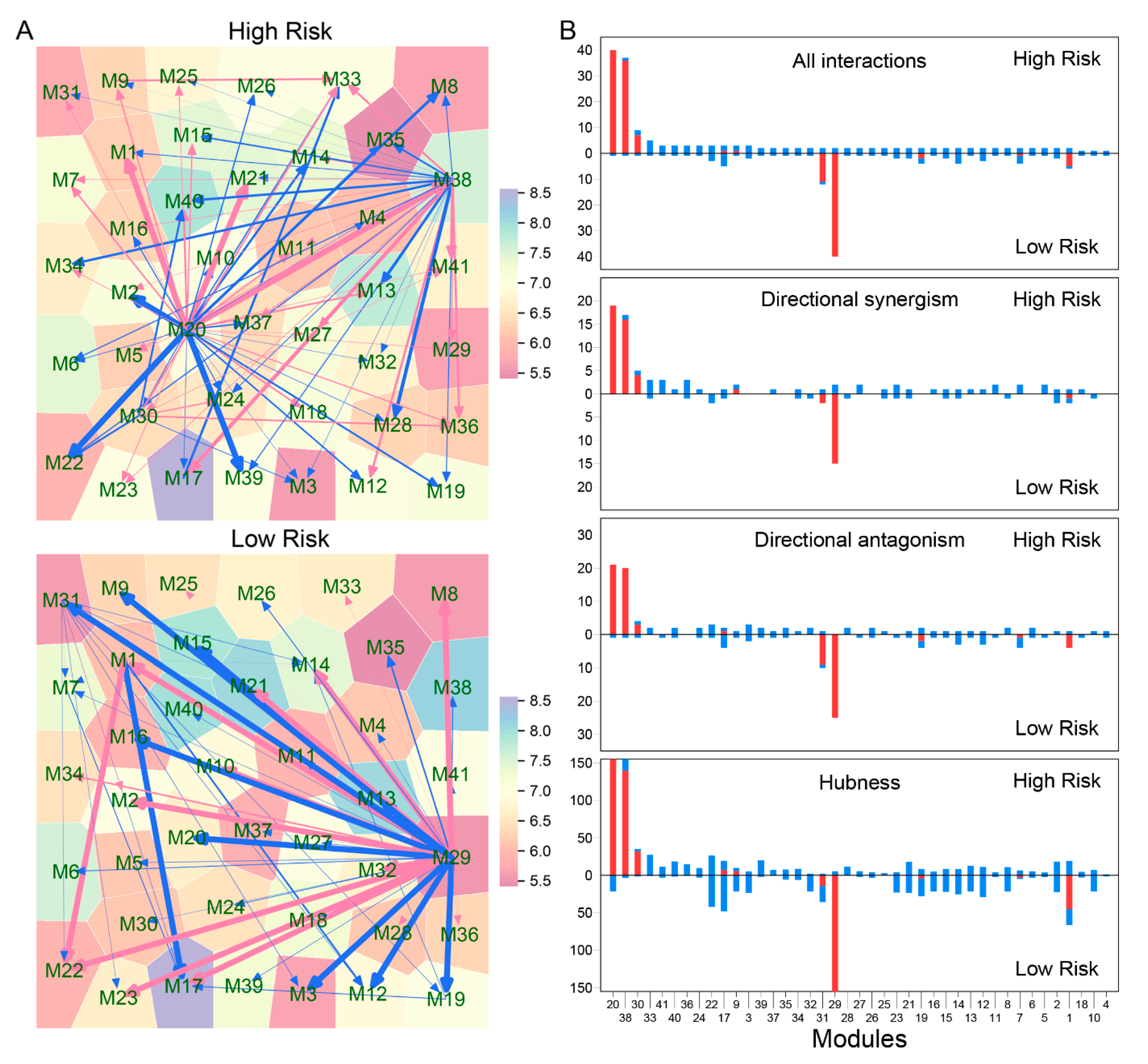
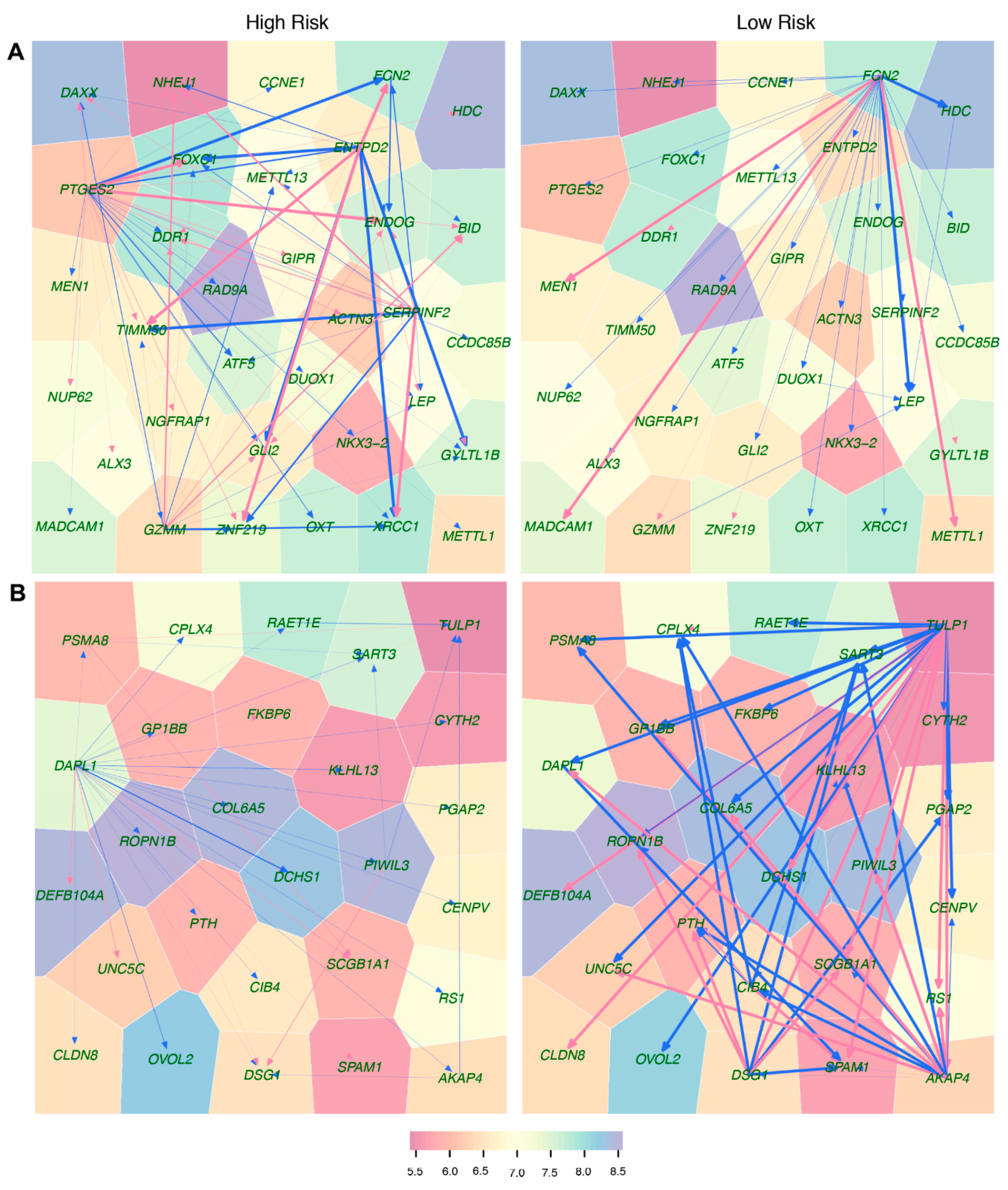
Third, relative to the two biomarkers above, idopNetworks provide a more precise predictor of neuroblastoma risk. We dissect a 3439-node network into 41 subnetworks based on module classification. The network of these subnetworks reflects how gene modules interact with each other to form a coarse-grained network, while each subnetwork characterizes interactions among individual genes within a fine-grained network. We found that directional synergism and directional antagonism overwhelmingly dominate both coarse- and fine-grained networks. Evolutionary studies suggest that commensalism (analogous to directional synergism) and amensalism (analogous to directional antagonism) are the two most economic strategies for animals to interact with others in nature [
20]. We speculate that this phenomenon may also occur for genes to choose their interaction strategies in a regulatory network.
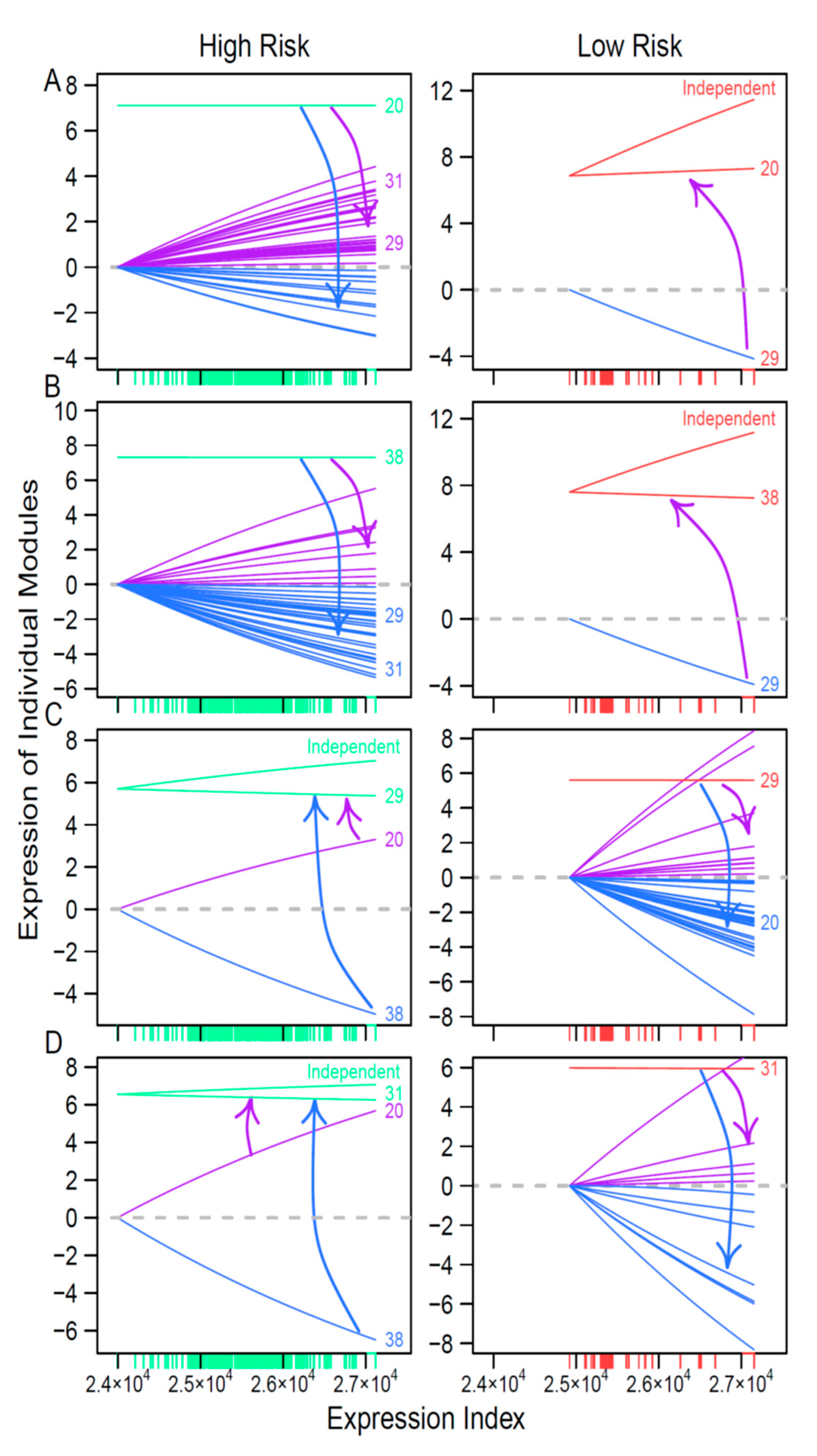
We found that both coarse- and fine-grained networks show dramatic structural and organization differences between low- and high-risk patients. We characterized the role of key hub regulators in driving how high-risk patients differ from low-risk patients. These regulators promote or inhibit the expression of numerous genes that are directly involved in immunity processes towards neuroblastoma pathogenesis. We identified different hubs that mediate low- and high-risk networks. For example, modules 29 and 31 are hubs in the low-risk network, whereas hubs in the high-risk network become modules 20 and 38. It is interesting to find that hubs in one coarse-grained network are reciprocally regulated by those in the other coarse-grained network, showing that the leadership change may be an important driver of neuroblastoma risk. The detailed biological functions of these hubs deserve further investigation.
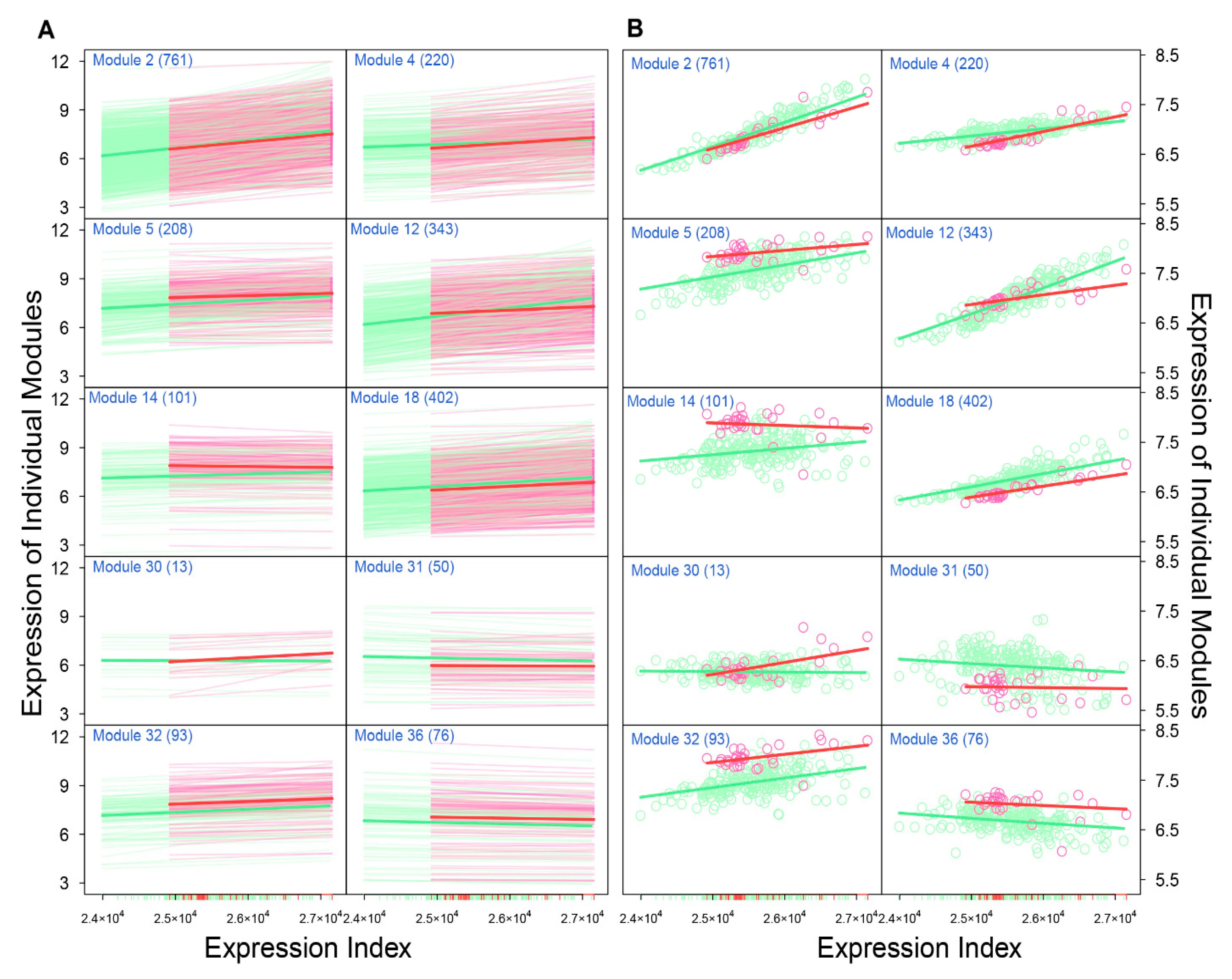
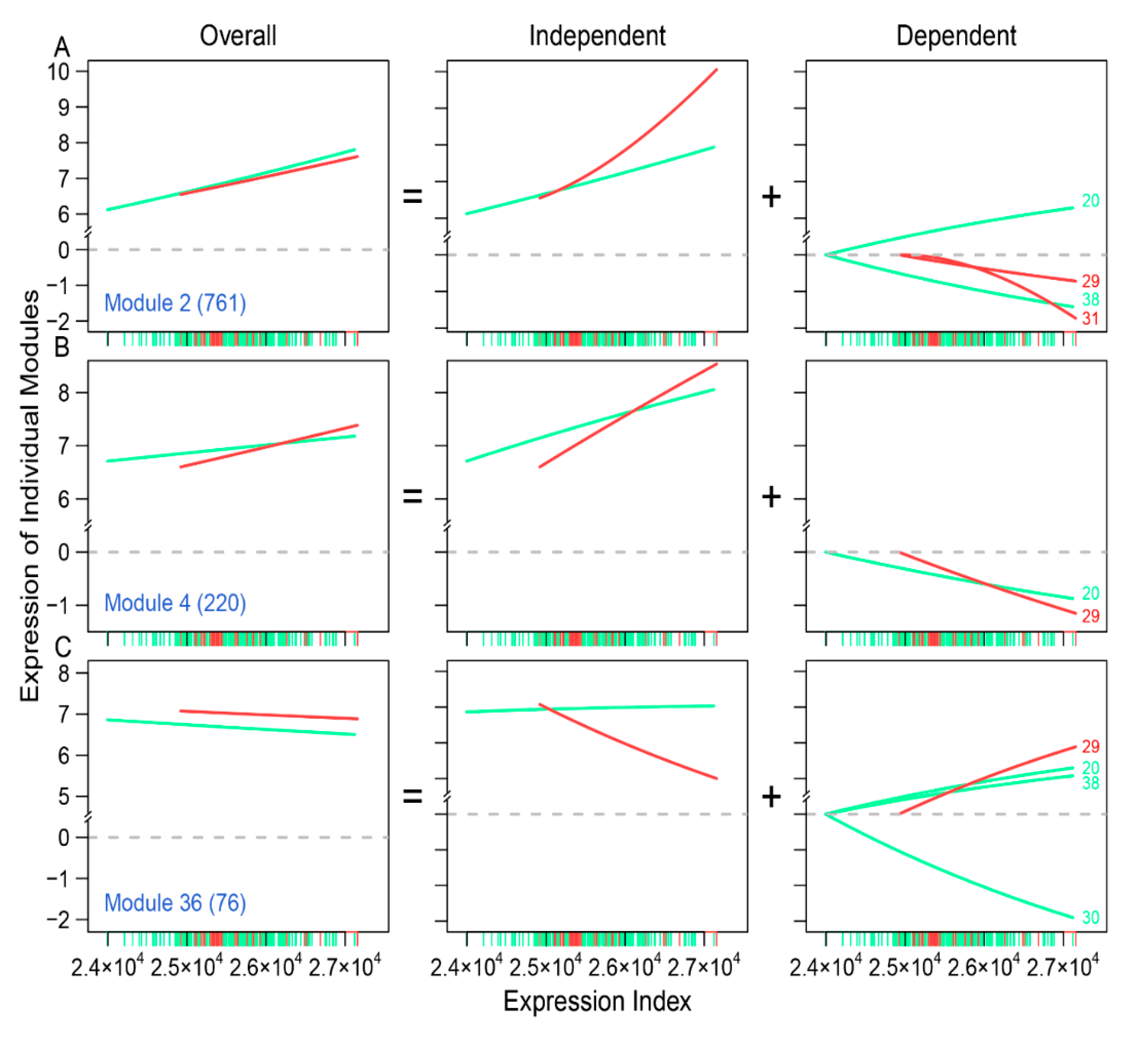
Through idopNetworks, we can determine how gene–gene interactions can be used to serve as a predictor of neuroblastoma risk. For example, module 2 shows no difference in the dynamic pattern of gene expression between two groups of patients (
Figure 5A), suggesting that this module cannot serve as a biomarker. Yet, by decomposing the overall expression of this module into its independent and dependent components, we found that the independent component increases its expression with expression index much more strikingly for low-risk than for high-risk patients. Thus, the dynamic pattern of the independent component can be potentially used to predict neuroblastoma risk. In practical clinics, neuroblastoma risk can be reduced by designing target interventions to obstruct the expression of modules 29 and 31 that inhibit module 2. Similarly, the independent component of module 36 is associated with reduced neuroblastoma risk, which can be strengthened by artificially inhibiting the expression of module 29 that activates module 36. Taken together, the multiplayer game model derived from idopNetworks may provide a more precise predictive means of neuroblastoma risk and solid scientific guidance on drug design for neuroblastoma control.
We reconstructed neuroblastoma-related idopNetworks based on gene expression data. These networks may serve as a starting point to explore the biological and functional relevance of key gene-gene interactions for neuroblastomas. Although GO analysis shows the biological relevance of some gene interactions detected by the model, close collaboration with experimental oncologists is crucial for not only further justifying the model but also making novel discoveries. In addition, mounting evidence indicates that, in addition to transcriptional dysregulation, cancer may also be predicted by epigenetic, protein, or metabolite biomarkers [
27,
28]. By incorporating these omics data, idopNetworks can be extended to infer multiscale and hierarchic networks, which can better interrogate the biological mechanisms that drive neuroblastoma. This work focuses on the comparison of idopNetworks between different neuroblastoma risks, but it should be expanded to compare network differences determined by other biological or environmental regimes, such as death vs. survival. In idopNetwork inference, we combine patients from the same group, thus neglecting genetic differences among patients. As cancer cells hosted on patients, neuroblastoma may not only be controlled by host genetics, but also somatic mutations of tumor cells. While many studies have identified a number of host loci for cancer based on GWAS, Sang et al. [
29] developed a micro-GWAS model to detect mutation loci that affect tumor phenotypes. The integration of idopNetwork and Sang et al.’s [
29] micro-GWAS will shed light on why and how neuroblastoma is formed, knowledge that can greatly facilitate the design of personalized neuroblastoma therapies.
4. Methods
4.1. Design of Genomic Experiment for Neuroblastoma Risk
Consider a set of n sampled neuroblastoma patients, sorted into two prognostic subgroups with expected high and low risk of death from disease. These samples are stratified according to demographical and clinical factors, such as age, gender, MYCN status, ploidy, stage, and overall (OS) and event-free survival (OS). Suppose a total of m genes are profiled and measured for their expression amounts on all samples. Let denote the expression level of gene j on sample i (i = 1, …, n). We consider each sample as an ecosystem composed of m interacting genes. Thus, we define the total expression of all genes on a sample, i.e., , as its expression index (EI) that reflects the sample’s capacity to carry essential resources for the simultaneous expression of all genes. Based on the definition, and establish a part-whole relationship across samples and, thus, can be understood as a function of , which is equivalently denoted as . This part-whole relationship theory forms an analytical basis for reconstructing regulatory networks for high- vs. low-risk neuroblastoma.
4.2. Modularity Detection by Power Law-Based Functional Clustering
Modular organization is a generic property of gene networks, recognized as a design principle of biological systems. A module or community is a subset of nodes that are more densely connected with each other than with those nodes from the remaining network [
30]. Modules, as function units, are pervasive in a range of biological processes, such as cell differentiation, metabolism, cell cycle, and signal transduction. Evidence shows that modularity structure enhances the adaptability and robustness of biological systems to perturbations. The existence of gene modules alerts us not to attempt to link each pair of genes in the network, but to detect gene subnetworks or communities. The modularity theory avoids computational prohibition when the number of genes considered is very large.
We develop a top-down approach for modularity detection by breaking down a whole network into its compositional subnetworks in a reverse engineering fashion. This approach is based on the functional clustering of all genes into different modules according to their EI-varying functional similarity. As stated above,
is part of
, whose relationship is thought to obey the power law, as universally observed in nature [
31,
32,
33]. Thus, how
scales with
across samples is mathematically described as
where
and
are the constant and allometric exponent for gene j that changes its expression level with EI. We incorporate Equation (1) into a mixture functional clustering algorithm [
19] by which we cluster all m gene into different modules. The EI-varying pattern of gene expression is more similar within than between modules, leading genes within modules to be densely linked but those between modules to be sparsely linked. We implement an information criterion, such as AIC or BIC, to determine the optimal number of modules (denoted as
L) for a given set of genes.
With the information about gene modules, we will reconstruct coarse-grained regulatory networks among genes from between modules, and fine-grained regulatory networks among genes within modules. We cluster genes based on how they are expressed differently across a full range of samples from both risk groups. It is possible that these two groups change their expression across samples in a different manner. Thus, we need to reconstruct different types of networks each for a risk group.
4.3. Game-Theoretic Modeling of Gene Interactions
The integration of the part-whole relationship theory and the mathematical aspect of evolutionary game theory allows us to characterize how each gene interacts with every other gene across samples by formulating a system of ordinary differential equations (ODEs). Suppose we want to reconstruct
L-node coarse-grained networks at the module level. For a particular group of neuroblastoma risk
k (
k = for low and 2 for high), such an
L-dimensional system of ODEs can be written as
where
is the expression level of gene
j on sample
i (here
i = 1, …,
nk) from risk group
k, and the derivative of expression of each gene
j is partitioned into the
independent component that occurs if this gene is assumed to be in isolation and
dependent component that is the aggregated effect of the influence of all other genes on this gene. Function
, specifies how gene
j is expressed independently as a function of EI and is determined by parameters
, whereas function
, describes how gene
affects the expression of gene
j as a function of EI and is determined by parameters
(
= 1, …,
j − 1,
j + 1, …,
L). Unlike classic ODEs with respect to time, ODEs in Equation (1) are specified by the EI derivative, which are thus called quasi-dynamic ODEs (qdODEs) [
18].
4.4. Variable Selection
Network theory states that there is a cognitive limit to the number of links an individual can stably maintain in a cohesive network [
34]. This so-called Dunbar’s law, originally observed in primate societies, can be explained to be due to the limit of the volume of the neocortex. We argue that Dunbar’s law may also be at play in the network constituted by other biological entities, such as cells and genes. The limit to the number of gene–gene interactions exists in living cells, because it is unlikely that each gene interacts with all other genes to form a completely linked network.
To detect a subset of the most significant genes that interact with a focal gene, we implement LASSO-based variable selection. Let
denote the observed expression level of gene
j on sample
i from risk group
k. A regression model of gene
j on other genes as predictors, implemented with qdODEs of Equation (2), can be formulated as
where the first two terms at the right side characterize the independent and dependent expression components of gene
j as a function of
for risk group
k, and
is the residual error of gene
j on sample
i from risk group
k, obeying a multivariate normal distribution with mean vector
0 and sample-dependent covariance matrix
for gene
j. We assume that the residual errors of gene expression are independent among samples so that
is structured as
where
is the residual variance of gene
j at the same sample and
is the identity matrix.
We use power Equation (1) to model the independent expression component, specified by parameter vector
, and a non-parametric approach to model the dependent expression component, specified by parameter vector
. We implement group LASSO [
35] and adaptive group LASSO [
36] to select the most significant genes that link with gene
j for each risk group. After variable selection, the number of genes that are involved in the dependent component of gene
j will reduce from
L to
, making full qdODEs of Equation (2) become sparse ones. We pose a constraint on the number of regulated genes by a regulator but no constraint on the number of regulators. Through reconstructing high-dimensional but sparse networks by the sparse qdODEs, this gives us a full capacity to identify all possible regulators.
4.5. Likelihood and Test
We formulate a likelihood of expression data of
L modules from two risk groups, as a function of qdODE parameters and residual (co)variances. Assuming that residual errors are independent between the two risk groups, this likelihood is written as
where vector
, …,
denotes the observed expression levels of gene j (j = 1, …, L) on
samples from risk group k, and
is the multivariate normal probability density function with mean vector (
) (with
, …,
) and covariance matrix
for risk group k. Here, we assume that microarray gene expression data follow a normal distribution, but other forms of distribution, such as Poisson or negative binomial for RNA-seq expression data, can also be considered. By implementing sparse qdODEs to model the mean vector, we obtain the maximum likelihood estimates (MLEs) of risk-specific ODE parameters
and
(j = 1, …, L;
= 1, …, j − 1, j + 1, …,
) for each gene for risk group k.
We develop a statistical procedure for testing whether gene networks can explain differences between high and low neuroblastoma risks. Under the assumption of no risk-specific difference, we formulate the likelihood as follows
where vector
denotes the observed expression levels of gene
j (
j = 1, …,
m) on all
n mixed samples from two risk groups. Similarly, by implementing sparse qdODEs to model the mean vector, we obtain the MLEs of risk-agnostic ODE parameters
and
(
j = 1, …,
L;
= 1, …,
j – 1,
j + 1, …,
) for each gene. Note that
is the number of regulated genes by gene
j as a regulator, which is determined through variable selection on a regression model of gene
j on all other
L – 1 genes across
n samples.
By plugging in the MLEs of model parameters into likelihoods (4) and (5), we obtain the likelihood values
(assuming that there is a risk-specific difference) and
(assuming that there is no risk-specific difference), respectively. We further estimate the log-likelihood ratio
as a statistic used to test if
n samples should be sorted into
C contexts. By reshuffling
n samples randomly into two risk groups, we calculate the LR value. If this permutation procedure is repeated 1000 times, we obtain the 95th percentile from 1000 LR values and use it as a critical threshold.
4.6. idopNetwork Recovery
If the risk group is tested to be significantly different from one another, we use the MLEs of and to estimate the integrals of and , denoted as and (j = 1, …, L; = 1, …, j – 1, j + 1, …, ), respectively. We encapsulate as nodes and as edges into a graph as an L-dimensional gene regulatory network G(). This network can capture all three possible features of gene interactions—bidirectional, weighted, and signed—because can characterize the strength and sign (promotion vs. inhibition) with which gene affects j, and also because and can describe and compare how genes j and are reciprocally affected. Relative to most existing networks that do not meet these three features simultaneously, G() is regarded as being fully informative.
G(
) is a function of
, suggesting that we can reconstruct a network for each sample, i.e., patient. To the end, we can reconstruct
n personalized networks and compare how the networks vary structurally and functionally from one patient to next. If the same patient is transcriptionally monitored at multiple timepoints and/or under multiple treatments, we can reconstruct spatiotemporal network for this specific patient. Increasing evidence shows that a complex disease is controlled by a full set of genome-wide genes [
37], indicating the necessity of reconstructing an omnigenic network. Although it is highly challenging to reconstruct large networks, Chen et al. [
18] has integrated developmental modularity and Dunbar’s law, which enables them to reconstruct G(
) from high- or even ultrahigh-dimensional data of genes. Taken together, we will reconstruct informative, dynamic, omnidirectional, and personalized networks (idopNetworks).
4.7. Neuroblastoma Expression Data
We downloaded 23,434-gene expression data of neuroblastoma patients from TARGET website [
38]. The data include 247 patients, of whom 217 and 30 belong to high- and low-risk groups, respectively. For each patient, age, gender, MYCN status, PLOIDY, stage, race, risk, overall survival and event free survival are provided. In this study, we focus on the investigation of how immune genes interact with each other to cause risk-specific discrepancies. Although the model can analyze any number of genes, as the proof of concept, we chose 3439 genes that are shown to display various immune-related functions from InnateDB [
39]. Using these genes, we reconstructed immune-related idopNetworks for neuroblastoma risk.
4.8. Data and Code Availability

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






