Novel Somatic Genetic Variants as Predictors of Resistance to EGFR-Targeted Therapies in Metastatic Colorectal Cancer Patients

 ,
,  ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Patients and Methods
2.1. Patient Population
2.2. Gene Selection and Primer Design
2.3. Isolation and Quantification of DNA
2.4. Library Preparation, Sequencing Runs, and NGS Analysis
2.5. Statistical Analyses
3. Results
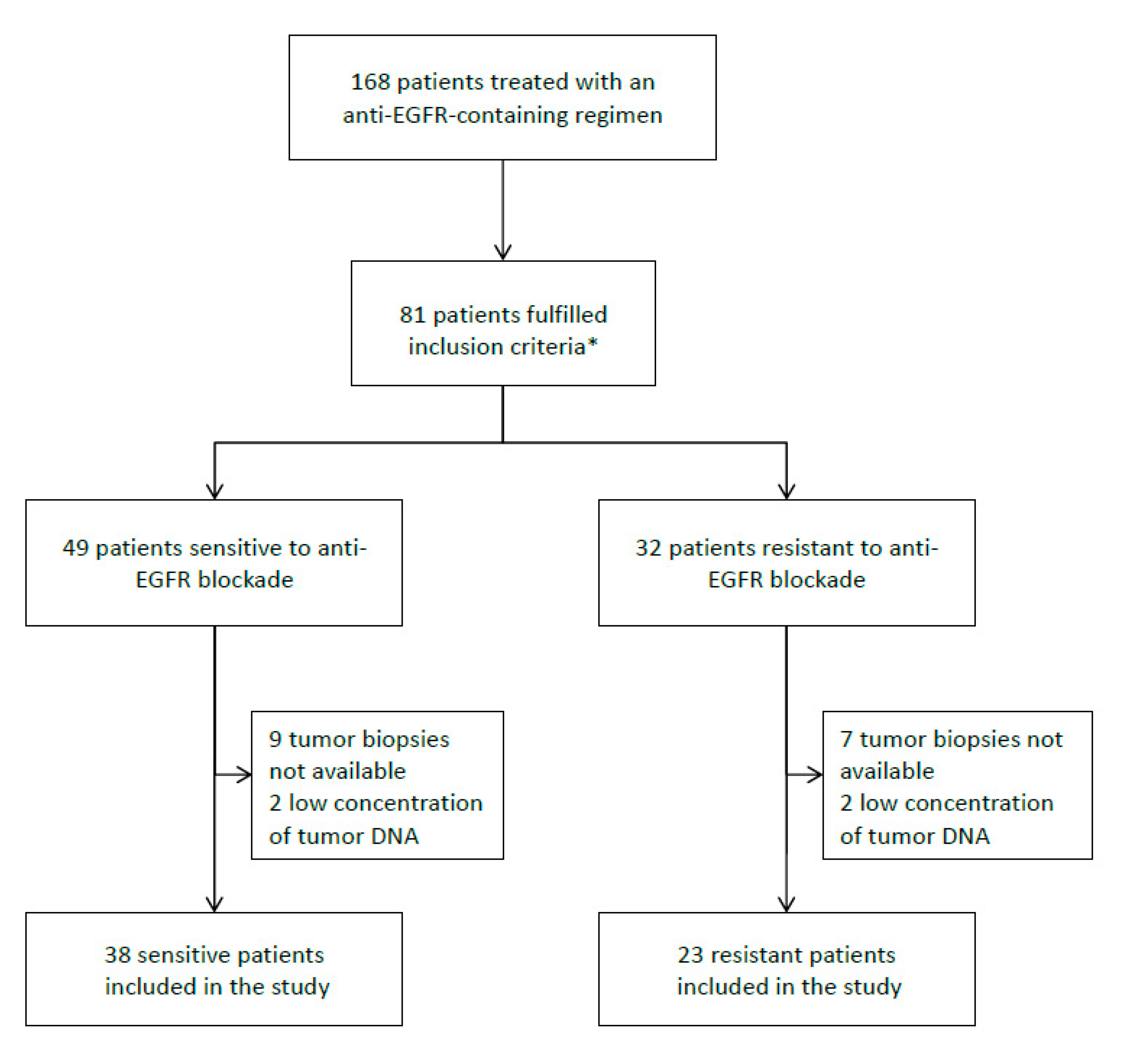
3.1. Patient Population
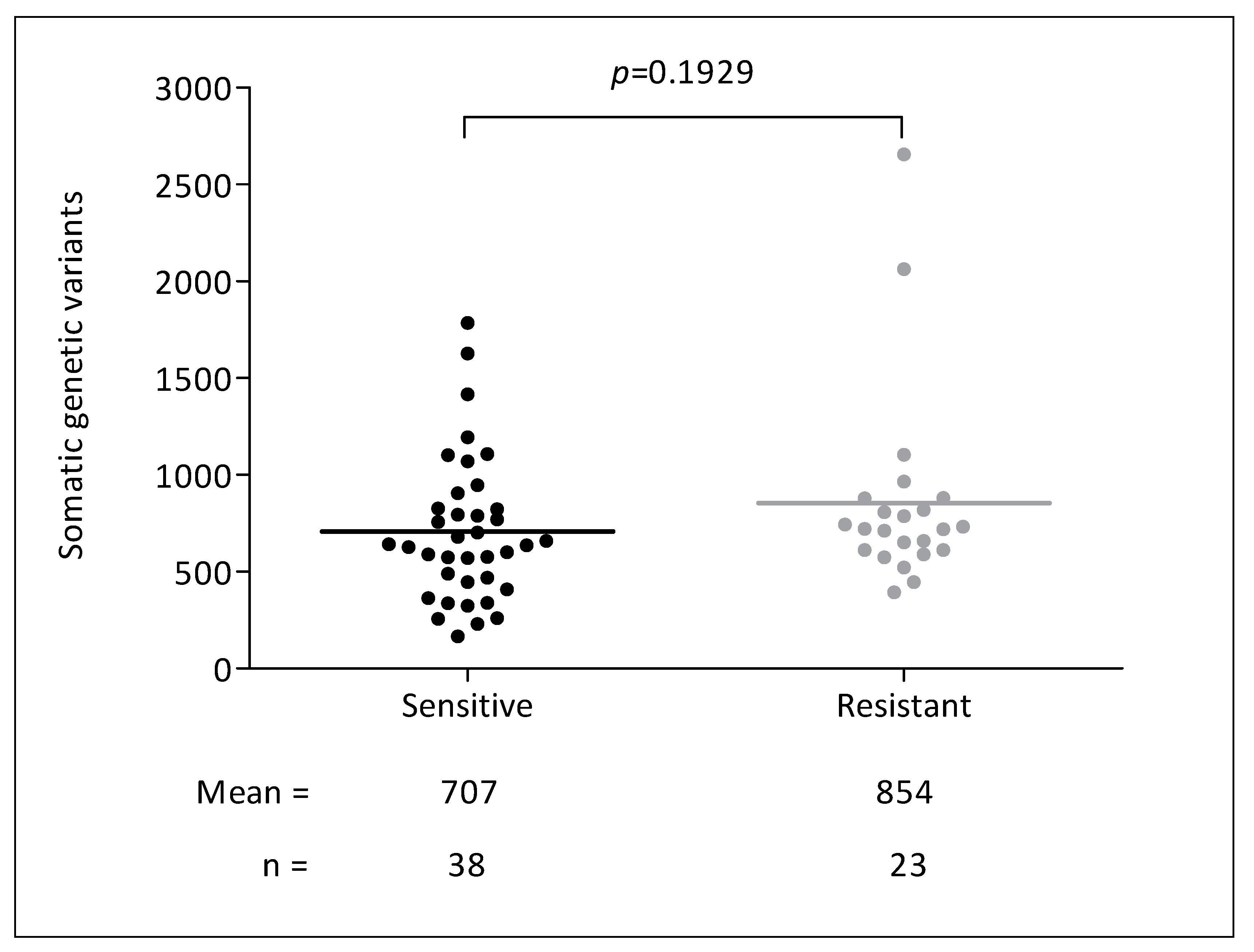
3.2. Genetic Analyses
3.3. KRAS, NRAS, BRAF, and PIK3CA Assessment and Patient Selection
3.4. Identification of Novel Genetic Variants Related to Anti-EGFR Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saif, M.W. Colorectal cancer in review: The role of the EGFR pathway. Expert Opin. Investig. Drugs 2010, 19, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Yarden, Y. EGF-ERBB signalling: Towards the systems level. Nat. Rev. Mol. Cell Biol 2006, 7, 505–516. [Google Scholar] [CrossRef]
- Avraham, R.; Yarden, Y. Feedback regulation of EGFR signalling: Decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 2011, 12, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cutsem, E.; Köhne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Allegra, C.J.; Rumble, R.B.; Hamilton, S.R.; Mangu, P.B.; Roach, N.; Hantel, A.; Schilsky, R.L. Extended RAS Gene Mutation Testing in Metastatic Colorectal Carcinoma to Predict Response to Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015. J. Clin. Oncol. 2015, 34, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Sorich, M.J.; Wiese, M.D.; Rowland, A.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: A meta-analysis of randomized, controlled trials. Ann. Oncol. 2015, 26, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Kaltenbrun, E.; Anderson, G.R.; Stephens, S.J.; Arena, S.; Bardelli, A.; Counter, C.M.; Wood, K.C. Codon bias imposes a targetable limitation on KRAS-driven therapeutic resistance. Nat. Commun. 2017, 8, 15617. [Google Scholar] [CrossRef] [Green Version]
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; Di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef]
- Rowland, A.; Dias, M.M.; Wiese, M.D.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S.; Sorich, M.J. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br. J. Cancer 2015, 112, 1888–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Brummelen, E.M.J.; de Boer, A.; Beijnen, J.H.; Schellens, J.H.M. BRAF Mutations as Predictive Biomarker for Response to Anti-EGFR Monoclonal Antibodies. Oncologist 2017, 22, 864–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Therkildsen, C.; Bergmann, T.K.; Henrichsen-Schnack, T.; Ladelund, S.; Nilbert, M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol. 2014, 53, 852–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karapetis, C.S.; Jonker, D.; Daneshmand, M.; Hanson, J.E.; O’Callaghan, C.J.; Marginean, C.; Zalcberg, J.R.; Simes, J.; Moore, M.J.; Tebbutt, N.C.; et al. PIK3CA, BRAF, and PTEN status and benefit from cetuximab in the treatment of advanced colorectal cancer—results from NCIC CTG/AGITG CO.17. Clin. Cancer Res. 2014, 20, 744–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, D.; Lueza, B.; Douillard, J.Y.; Peeters, M.; Lenz, H.J.; Venook, A.; Heinemann, V.; Van Cutsem, E.; Pignon, J.P.; Tabernero, J.; et al. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann. Oncol. 2017, 28, 1713–1729. [Google Scholar] [CrossRef] [PubMed]
- Moretto, R.; Cremolini, C.; Rossini, D.; Pietrantonio, F.; Battaglin, F.; Mennitto, A.; Bergamo, F.; Loupakis, F.; Marmorino, F.; Berenato, R.; et al. Location of Primary Tumor and Benefit From Anti-Epidermal Growth Factor Receptor Monoclonal Antibodies in Patients With RAS and BRAF Wild-Type Metastatic Colorectal Cancer. Oncologist 2016, 21, 988–994. [Google Scholar] [CrossRef] [Green Version]
- De Sousa EMelo, F.; Wang, X.; Jansen, M.; Fessler, E.; Trinh, A.; de Rooij, L.P.; de Jong, J.H.; de Boer, O.J.; van Leersum, R.; Bijlsma, M.F.; et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat. Med. 2013, 19, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Linnekamp, J.F.; Wang, X.; Medema, J.P.; Vermeulen, L. Colorectal cancer heterogeneity and targeted therapy: A case for molecular disease subtypes. Cancer Res. 2015, 75, 245–249. [Google Scholar] [CrossRef] [Green Version]
- Khattak, M.A.; Martin, H.; Davidson, A.; Phillips, M. Role of first-line anti-epidermal growth factor receptor therapy compared with anti-vascular endothelial growth factor therapy in advanced colorectal cancer: A meta-analysis of randomized clinical trials. Clin. Colorectal Cancer 2015, 14, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kopetz, S. Current and Future Approaches to Target the Epidermal Growth Factor Receptor and Its Downstream Signaling in Metastatic Colorectal Cancer. Clin. Colorectal Cancer 2015, 14, 203–218. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Sassi, F. Molecular Pathways: Sensitivity and Resistance to Anti-EGFR Antibodies. Clin. Cancer Res. 2015, 21, 3377–3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertotti, A.; Papp, E.; Jones, S.; Adleff, V.; Anagnostou, V.; Lupo, B.; Sausen, M.; Phallen, J.; Hruban, C.A.; Tokheim, C.; et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015, 526, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Tamborero, D.; Rubio-Perez, C.; Deu-Pons, J.; Schroeder, M.P.; Vivancos, A.; Rovira, A.; Tusquets, I.; Albanell, J.; Rodon, J.; Tabernero, J.; et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018, 10, 25. [Google Scholar] [CrossRef]
- Jones, J.C.; Renfro, L.A.; Al-Shamsi, H.O.; Schrock, A.B.; Rankin, A.; Zhang, B.Y.; Kasi, P.M.; Voss, J.S.; Leal, A.D.; Sun, J.; et al. Non-V600BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J. Clin. Oncol. 2017, 35, 2624–2630. [Google Scholar] [CrossRef]
- Cremolini, C.; Di Bartolomeo, M.; Amatu, A.; Antoniotti, C.; Moretto, R.; Berenato, R.; Perrone, F.; Tamborini, E.; Aprile, G.; Lonardi, S.; et al. BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Ann. Oncol. 2015, 26, 2092–2097. [Google Scholar] [CrossRef]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabour, L.; Sabour, M.; Ghorbian, S. Clinical Applications of Next-Generation Sequencing in Cancer Diagnosis. Pathol. Oncol. Res. 2017, 23, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.; Azuara, D.; Viéitez, J.M.; Páez, D.; Falcó, E.; Élez, E.; López-López, C.; Valladares, M.; Robles-Díaz, L.; García-Alfonso, P.; et al. Phase II study of high-sensitivity genotyping of KRAS, NRAS, BRAF and PIK3CA to ultra-select metastatic colorectal cancer patients for panitumumab plus FOLFIRI: The ULTRA trial. Ann. Oncol. 2019, 30, 796–803. [Google Scholar] [CrossRef] [Green Version]
- Vidal, J.; Bellosillo, B.; Santos Vivas, C.; García-Alfonso, P.; Carrato, A.; Cano, M.T.; García-Carbonero, R.; Élez, E.; Losa, F.; Massutí, B.; et al. Ultra-selection of metastatic colorectal cancer patients using next-generation sequencing to improve clinical efficacy of anti-EGFR therapy. Ann. Oncol. 2019, 30, 439–446. [Google Scholar] [CrossRef]
- Isnaldi, E.; Garuti, A.; Cirmena, G.; Scabini, S.; Rimini, E.; Ferrando, L.; Lia, M.; Murialdo, R.; Tixi, L.; Carminati, E.; et al. Clinico-pathological associations and concomitant mutations of the RAS/RAF pathway in metastatic colorectal cancer. J. Transl. Med. 2019, 17, 137. [Google Scholar] [CrossRef]
- Salem, M.E.; Weinberg, B.A.; Xiu, J.; El-Deiry, W.S.; Hwang, J.J.; Gatalica, Z.; Philip, P.A.; Shields, A.F.; Lenz, H.J.; Marshall, J.L. Comparative molecular analyses of left-sided colon, right-sided colon, and rectal cancers. Oncotarget 2017, 8, 86356–86368. [Google Scholar] [CrossRef] [Green Version]
- Prenen, H.; De Schutter, J.; Jacobs, B.; De Roock, W.; Biesmans, B.; Claes, B.; Lambrechts, D.; Van Cutsem, E.; Tejpar, S. PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin. Cancer Res. 2009, 15, 3184–3188. [Google Scholar] [CrossRef] [Green Version]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Lee, J.; Jain, A.; Kim, P.; Lee, T.; Kuller, A.; Princen, F.; Gu, I.; Kim, S.H.; Park, J.O.; Park, Y.S.; et al. Activated cMET and IGF1R-driven PI3K signaling predicts poor survival in colorectal cancers independent of KRAS mutational status. PLoS ONE 2014, 9, e103551. [Google Scholar] [CrossRef] [PubMed]
- Dallas, N.A.; Xia, L.; Fan, F.; Gray, M.J.; Gaur, P.; van Buren, G., 2nd; Samuel, S.; Kim, M.P.; Lim, S.J.; Ellis, L.M.; et al. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009, 69, 1951–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppola, D.; Ferber, A.; Miura, M.; Sell, C.; D’Ambrosio, C.; Rubin, R.; Baserga, R. A functional insulin-like growth factor I receptor is required for the mitogenic and transforming activities of the epidermal growth factor receptor. Mol. Cell. Biol. 1994, 14, 4588–4595. [Google Scholar] [CrossRef] [Green Version]
- Alonso, V.; Escudero, P.; Fernández-Martos, C.; Salud, A.; Méndez, M.; Gallego, J.; Rodriguez, J.R.; Martín-Richard, M.; Fernández-Plana, J.; Manzano, H.; et al. Coexpression of p-IGF-1R and MMP-7 Modulates Panitumumab and Cetuximab Efficacy in RAS Wild-Type Metastatic Colorectal Cancer Patients. Neoplasia 2018, 20, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Bardsley, M.R.; Toyomasu, Y.; Milosavljevic, S.; Gajdos, G.B.; Choi, K.M.; Reid-Lombardo, K.M.; Kendrick, M.L.; Bingener-Casey, J.; Tang, C.M.; et al. Platelet-Derived Growth Factor Receptor-α Regulates Proliferation of Gastrointestinal Stromal Tumor Cells With Mutations in KIT by Stabilizing ETV1. Gastroenterology 2015, 149, 420–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, M.C.; Corless, C.L.; Duensing, A.; McGreevey, L.; Chen, C.J.; Joseph, N.; Singer, S.; Griffith, D.J.; Haley, A.; Town, A.; et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003, 299, 708–810. [Google Scholar] [CrossRef]
- Rafidi, H.; Mercado, F., 3rd; Astudillo, M.; Fry, W.H.; Saldana, M.; Carraway, K.L., 3rd; Sweeney, C. Leucine-rich repeat and immunoglobulin domain-containing protein-1 (Lrig1) negative regulatory action toward ErbB receptor tyrosine kinases is opposed by leucine-rich repeat and immunoglobulin domain-containing protein 3 (Lrig3). J. Biol. Chem. 2013, 288, 21593–21605. [Google Scholar] [CrossRef] [Green Version]
- Faraz, M.; Herdenberg, C.; Holmlund, C.; Henriksson, R.; Hedman, H. A protein interaction network centered on leucine-rich repeats and immunoglobulin-like domains 1 (LRIG1) regulates growth factor receptors. J. Biol. Chem. 2018, 293, 3421–3435. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Poulin, E.J.; Coffey, R.J. LRIG1 is a triple threat: ERBB negative regulator, intestinal stem cell marker and tumour suppressor. Br. J. Cancer 2013, 108, 1765–1770. [Google Scholar] [CrossRef]
- Wong, V.W.; Stange, D.E.; Page, M.E.; Buczacki, S.; Wabik, A.; Itami, S.; van de Wetering, M.; Poulsom, R.; Wright, N.A.; Trotter, M.W.; et al. Lrig1 controls intestinal stem-cell homeostasis by negative regulation of ErbB signalling. Nat. Cell. Biol. 2012, 14, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, A.E.; Wang, Y.; Li, Y.; Poulin, E.J.; Means, A.L.; Washington, M.K.; Higginbotham, J.N.; Juchheim, A.; Prasad, N.; Levy, S.E.; et al. The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell 2012, 149, 146–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Han, L.; Chen, R.; Cai, M.; Han, F.; Lei, T.; Guo, D. Downregulation of LRIG2 expression by RNA interference inhibits glioblastoma cell (GL15) growth, causes cell cycle redistribution, increases cell apoptosis and enhances cell adhesion and invasion in vitro. Cancer Biol. Ther. 2009, 8, 1018–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.-K.; Chen, H.; Mao, F.; Xiao, Q.-G.; Xie, R.-F.; Lei, T. Downregulation of LRIG2 expression inhibits angiogenesis of glioma via EGFR/VEGF-A pathway. Oncol. Lett. 2017, 14, 4021–4028. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Hedman, H.; Bergqvist, M.; Bergström, S.; Henriksson, R.; Gullbo, J.; Lennartsson, J.; Hesselius, P.; Ekman, S. Expression of EGFR and LRIG proteins in oesophageal carcinoma with emphasis on patient survival and cellular chemosensitivity. Acta Oncol. 2012, 51, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Yang, H.; Guo, Y.; Xiao, Q.; Mao, F.; Tan, Y.; Wan, X.; Wang, B.; Lei, T. LRIG3 modulates proliferation, apoptosis and invasion of glioblastoma cells as a potent tumor suppressor. J. Neurol. Sci. 2015, 350, 61–68. [Google Scholar] [CrossRef]
- Gelfo, V.; Pontis, F.; Mazzeschi, M.; Sgarzi, M.; Mazzarini, M.; Solmi, R.; D’Uva, G.; Lauriola, M. Glucocorticoid Receptor Modulates EGFR Feedback upon Acquisition of Resistance to Monoclonal Antibodies. J. Clin. Med. 2019, 8, 600. [Google Scholar] [CrossRef] [Green Version]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Hammond, W.A.; Swaika, A.; Mody, K. Pharmacologic resistance in colorectal cancer: A review. Ther. Adv. Med. Oncol. 2016, 8, 57–84. [Google Scholar] [CrossRef] [Green Version]
- Bignucolo, A.; De Mattia, E.; Cecchin, E.; Roncato, R.; Toffoli, G. Pharmacogenomics of Targeted Agents for Personalization of Colorectal Cancer Treatment. Int. J. Mol. Sci. 2017, 18, 1522. [Google Scholar] [CrossRef] [Green Version]
- Cremolini, C.; Morano, F.; Moretto, R.; Berenato, R.; Tamborini, E.; Perrone, F.; Rossini, D.; Gloghini, A.; Busico, A.; Zucchelli, G.; et al. Negative hyper-selection of metastatic colorectal cancer patients for anti-EGFR monoclonal antibodies: The PRESSING case–control study. Ann. Oncol. 2017, 28, 3009–3014. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Martin, A.; Cufí, S.; Oliveras-Ferraros, C.; Torres-Garcia, V.Z.; Corominas-Faja, B.; Cuyàs, E.; Bonavia, R.; Visa, J.; Martin-Castillo, B.; Barrajón-Catalán, E.; et al. IGF-1R/epithelial-to-mesenchymal transition (EMT) crosstalk suppresses the erlotinib-sensitizing effect of EGFR exon 19 deletion mutations. Sci. Rep. 2013, 3, 2560. [Google Scholar] [CrossRef] [PubMed]
- Sclafani, F.; Kim, T.Y.; Cunningham, D.; Kim, T.W.; Tabernero, J.; Schmoll, H.J.; Roh, J.K.; Kim, S.Y.; Park, Y.S.; Guren, T.K.; et al. A Randomized Phase II/III Study of Dalotuzumab in Combination With Cetuximab and Irinotecan in Chemorefractory, KRAS Wild-Type, Metastatic Colorectal Cancer. J. Natl. Cancer Inst. 2015, 107, djv258. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Ligands | Receptors | Intracellular Downstream Effectors | Proteins Involved in EGFR Turnover | Others |
|---|---|---|---|---|
| AREG | EGFR (ERBB1 or HER1) | AKT1 | AGR2 | TP53 |
| BTC | ERBB2 (HER2) | BRAF | CBL | YAP1 |
| EGF | ERBB3 (HER3) | HRAS | LRIG1 | |
| EPGN | ERBB4 (HER4) | IRS2 | LRIG2 | |
| EREG | FGFR1 | KRAS | LRIG3 | |
| HBEGF | IGF1R | MAP2K1 | NEDD8 | |
| HGF | MET | NRAS | ERRFI1 (RALT or MIG6) | |
| IGF1-2 | PDGFRA | PIK3CA | SOCS4 | |
| NRG1-4 | PTEN | SOCS5 | ||
| TGFα | SPRY2 |
| Characteristic | Study Population (n = 61) | Sensitive Patients (n = 38) | Resistant Patients (n = 23) | p-Value |
|---|---|---|---|---|
| N (%) | N (%) | N (%) | ||
| Sex | ||||
| Male | 40 (65.6%) | 32 (84%) | 8 (34.8%) | <0.001 |
| Female | 21 (34.4%) | 6 (16%) | 15 (65.2%) | |
| Age | ||||
| <75 | 43 (70.5%) | 26 (68.4%) | 17 (73.9%) | 0.649 |
| ≥75 | 18 (29.5%) | 12 (31.6%) | 6 (26.1%) | |
| Mean Age | 67.7 | 66.9 | ||
| Performance status (ECOG) | ||||
| 0 | 31 (50.8%) | 24 (63.2%) | 7 (30.4%) | 0.013 |
| 1–2 | 30 (49.2%) | 14 (36.8%) | 16 (69.6%) | |
| Smoking habit | ||||
| Never smokers | 24 (39.3%) | 13 (34.2%) | 11 (47.8%) | 0.291 |
| Current or former smokers | 37 (60.7%) | 25 (65.8%) | 12 (52.2%) | |
| Tumor side | ||||
| Right | 24 (39.4%) | 15 (39.5%) | 9 (39.2%) | |
| Left | 20 (32.7%) | 13 (34.2%) | 7 (30.4 %) | 0.634 |
| Rectal | 16 (26.3%) | 10 (26.3%) | 6 (26.1%) | |
| Jejunum | 1 (1.6%) | 0 | 1 (4.3%) | |
| Number of metastatic sites | ||||
| 1 | 30 (49.2%) | 18 (47.4%) | 12 (52.3%) | 0.716 |
| ≥2 | 31 (50.8%) | 20 (52.6%) | 11 (47.7%) | |
| Time to metastases | ||||
| Synchronous | 38 (62.3%) | 27 (71.1%) | 11 (47.8%) | 0.070 |
| Metachronous | 23 (37.7%) | 11 (28.9%) | 12 (52.2%) | |
| Primary resected | ||||
| Yes | 49 (80.3%) | 32 (84.2%) | 17 (73.9%) | 0.327 |
| No | 12 (19.7%) | 6 (15.8%) | 6 (26.1%) | |
| Previous lines of treatment | ||||
| 0 | 24 (39.3%) | 18 (47.4%) | 6 (26.1%) | |
| 1 | 32 (52.5%) | 17 (44.7%) | 15 (65.2%) | 0.246 |
| ≥2 | 5 (8.2%) | 3 (7.9%) | 2 (8.7%) | |
| Type of anti-EGFR | ||||
| Cetuximab | 33 (54.1%) | 21 (55.3%) | 12 (52.2%) | 0.814 |
| Panitumumab | 28 (45.9%) | 17 (44.7%) | 11 (47.8%) | |
| Combination QT | ||||
| FOLFOX | 18 (29.5%) | 14 (36.8 %) | 4 (17.4%) | |
| Irinotecan scheme | 40 (65.6%) | 23 (60.6%) | 17 (73.9%) | 0.192 |
| Monotherapy | 3 (4.9%) | 1 (2.6 %) | 2 (8.7%) | |
| PFS (months) | 18.8 | 4.7 | ||
| OS (months) | 41.2 | 17.2 |
| Treatment Outcome | Gene | Mutation | Patient | % of Mutation | Coverage | Mutational Status Prior to Anti-EGFR Prescription |
|---|---|---|---|---|---|---|
| Resistant | KRAS | G12C | P11 | 45.7% | 2316 | KRAS wild-type |
| KRAS | Q61L | P39 | 0.8% | 14,637 | KRAS exon 3 not tested | |
| NRAS | G12S | P51 | 2.6% | 5093 | NRAS wild-type | |
| NRAS | G13D | P55 | 8.1% | 12,265 | NRAS not tested | |
| NRAS | G13D | P57 | 0.5% | 41,758 | NRAS wild-type | |
| BRAF | V600E | P3 | 24.4% | 10,995 | BRAF V600E mutated | |
| BRAF | V600E | P39 | 10.6% | 7906 | BRAF V600E mutated | |
| BRAF | V600E | P55 | 20.8% | 22,185 | BRAF V600E mutated | |
| BRAF | V600E | P64 | 12.1% | 1316 | BRAF V600E wild-type | |
| PIK3CA | E545K | P63 | 10.1% | 17,051 | PIK3CA not tested | |
| Sensitive | KRAS | A146V | P28 | 5.0% | 2743 | KRAS wild-type |
| BRAF | D594N | P45 | 19.7% | 23,584 | BRAF codon 594 not tested | |
| BRAF | G466A | P59 | 21.0% | 10,234 | BRAF codon 466 not tested | |
| PIK3CA | E545K | P66 | 6.1% | 12,888 | PIK3CA not tested |
| Gene | Genetic Variant | Patients with the Variant | % of the Somatic Variant | Coverage | p- Value * | Presence in COSMIC Cancer Database v91 |
|---|---|---|---|---|---|---|
| IGF1R | NM_001291858.1: c.2003T > A; p.(I668N) | P4 | 3.5% | 8008 | 0.029 | Not described |
| P9 | 4.2% | 1745 | ||||
| P10 | 10.6% | 3093 | ||||
| NM_001291858.1: c.3652G > A; p.(E1218K) | P1 | 6.3% | 2740 | 0.008 | Mutation Id: 6919417 Patient with a leiomyosarcoma (n = 1) [33] | |
| P2 | 11.8% | 1866 | ||||
| P9 | 2.4% | 1238 | ||||
| P61 | 1.5% | 10,017 | ||||
| IRS2 | NM_003749.2: c.3467C > T; p.(T1156M) | P12 | 7.0% | 743 | 0.029 | Mutation Id: 6974893 Patient with colon cancer (n = 1) [33] |
| P14 | 5.1% | 1093 | ||||
| P57 | 3.4% | 11,118 | ||||
| LRIG1 | NM_015541: c.456G > A; p.(T152T) | P12 | 89.3% | 196 | 0.008 | Mutation Id: 4005617 Patient with colon cancer (n = 1) Patient with bladder cancer (n = 1) [34] |
| P57 | 2.7% | 12,073 | ||||
| P63 | 7.9% | 6106 | ||||
| P67 | 4.0% | 3218 | ||||
| LRIG2 | NM_014813.2: c.2090C > T; p.(S697L) | P1 | 2.2% | 8802 | 0.029 | Not described |
| P10 | 4.2% | 1464 | ||||
| P12 | 7.9% | 1794 | ||||
| LRIG3 | NM_001136051.2: c.2434G > A; p.(V812M) | P1 | 1.7% | 12,378 | 0.029 | Not described |
| P2 | 4.8% | 4477 | ||||
| P9 | 4.6% | 2266 | ||||
| NRAS | NM_002524.3: c.344del; p.(G115Efs*46) | P9 | 1.6% | 2273 | 0.029 | Not described |
| P10 | 3.1% | 3140 | ||||
| P63 | 2.2% | 20,507 | ||||
| PDGFRA | NM_001347828: c.903G > A; p.(T301T) | P52 | 22.0% | 19,078 | 0.029 | Not described |
| P63 | 3.8% | 26,774 | ||||
| P67 | 1.2% | 55,158 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riera, P.; Rodríguez-Santiago, B.; Lasa, A.; Gonzalez-Quereda, L.; Martín, B.; Salazar, J.; Sebio, A.; Virgili, A.C.; Minguillón, J.; Camps, C.; et al. Novel Somatic Genetic Variants as Predictors of Resistance to EGFR-Targeted Therapies in Metastatic Colorectal Cancer Patients. Cancers 2020, 12, 2245. https://doi.org/10.3390/cancers12082245
Riera P, Rodríguez-Santiago B, Lasa A, Gonzalez-Quereda L, Martín B, Salazar J, Sebio A, Virgili AC, Minguillón J, Camps C, et al. Novel Somatic Genetic Variants as Predictors of Resistance to EGFR-Targeted Therapies in Metastatic Colorectal Cancer Patients. Cancers. 2020; 12(8):2245. https://doi.org/10.3390/cancers12082245
Chicago/Turabian StyleRiera, Pau, Benjamín Rodríguez-Santiago, Adriana Lasa, Lidia Gonzalez-Quereda, Berta Martín, Juliana Salazar, Ana Sebio, Anna C. Virgili, Jordi Minguillón, Cristina Camps, and et al. 2020. "Novel Somatic Genetic Variants as Predictors of Resistance to EGFR-Targeted Therapies in Metastatic Colorectal Cancer Patients" Cancers 12, no. 8: 2245. https://doi.org/10.3390/cancers12082245
APA StyleRiera, P., Rodríguez-Santiago, B., Lasa, A., Gonzalez-Quereda, L., Martín, B., Salazar, J., Sebio, A., Virgili, A. C., Minguillón, J., Camps, C., Surrallés, J., & Páez, D. (2020). Novel Somatic Genetic Variants as Predictors of Resistance to EGFR-Targeted Therapies in Metastatic Colorectal Cancer Patients. Cancers, 12(8), 2245. https://doi.org/10.3390/cancers12082245