Genomic and Functional Regulation of TRIB1 Contributes to Prostate Cancer Pathogenesis

 , , , ,
, , , , 
 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Animals
2.2. Histopathological Analysis and Immunohistochemistry
2.3. Cell Culture
2.4. Generation of Stable Cell Lines
2.5. Cellular Assays
2.6. Real-Time Quantitative PCR
2.7. Western Blot
2.8. Chromatin Immunoprecipitation
2.9. Dual Luciferase Reporter Assay
2.10. Bioinformatics Analysis
2.11. Statistics Analysis and Reproducibility
3. Results
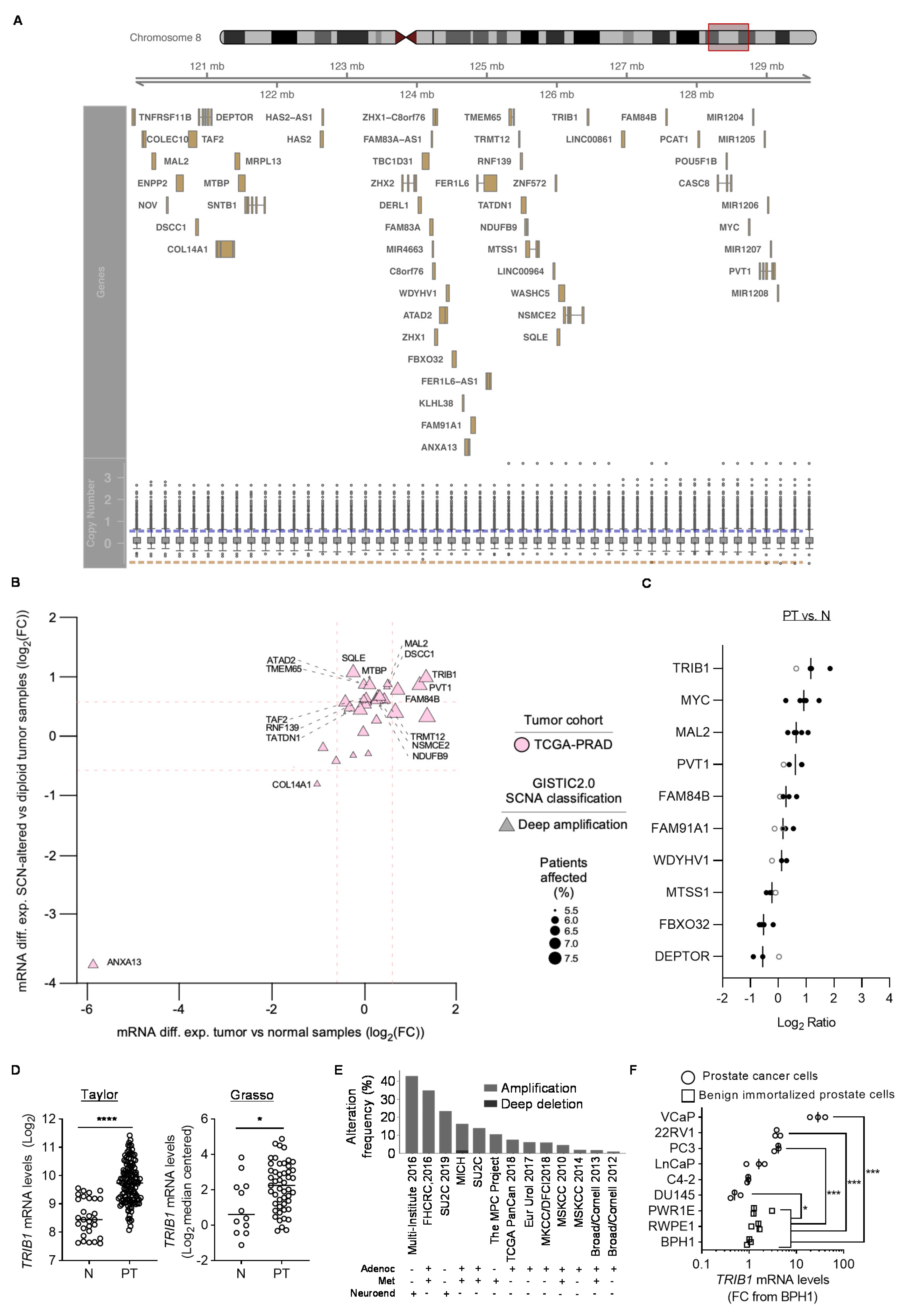
3.1. Identification of PCa-Relevant Candidate Genes in cMYC Amplicon
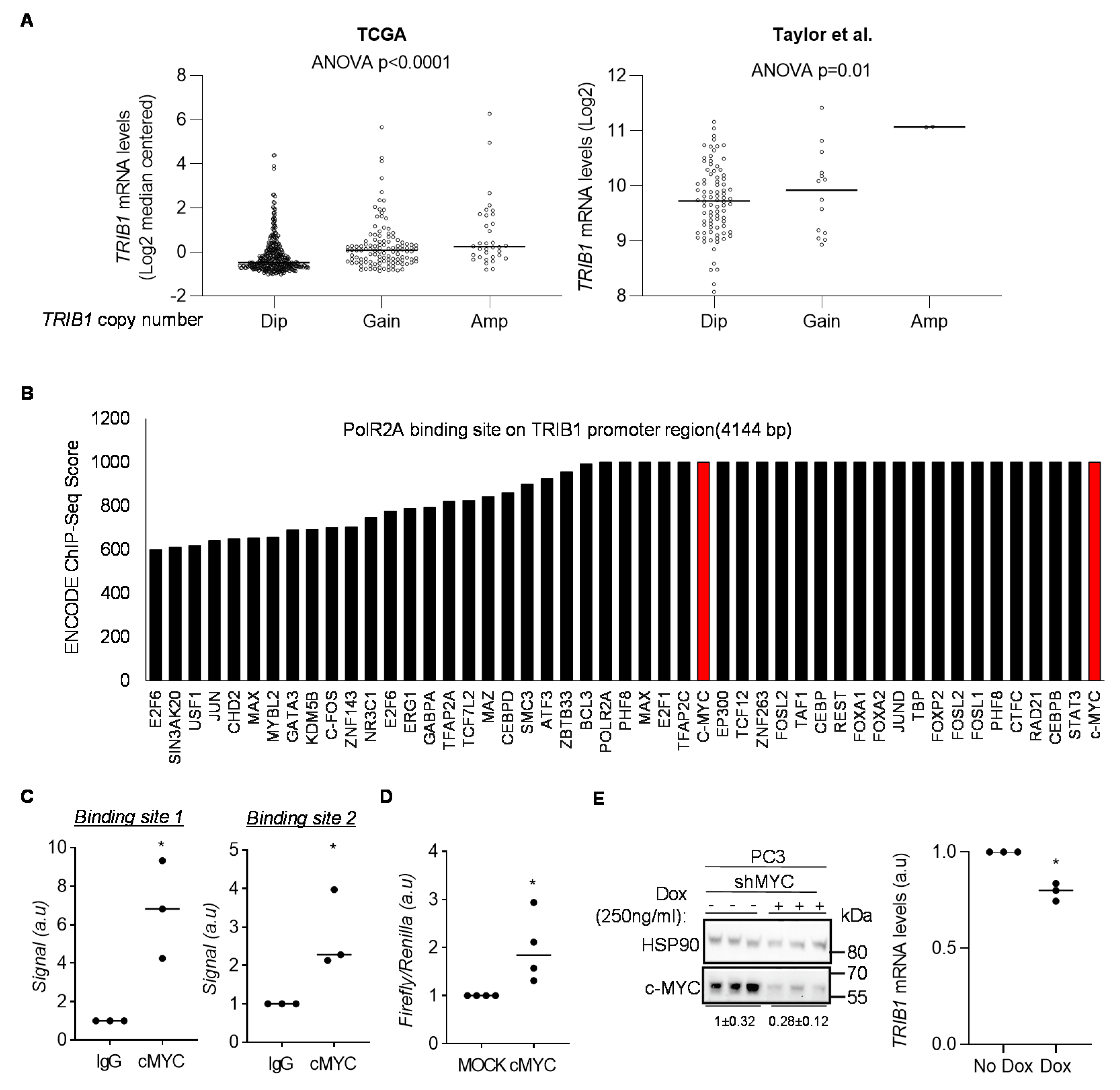
3.2. cMYC Regulates the Expression of TRIB1 in PCa
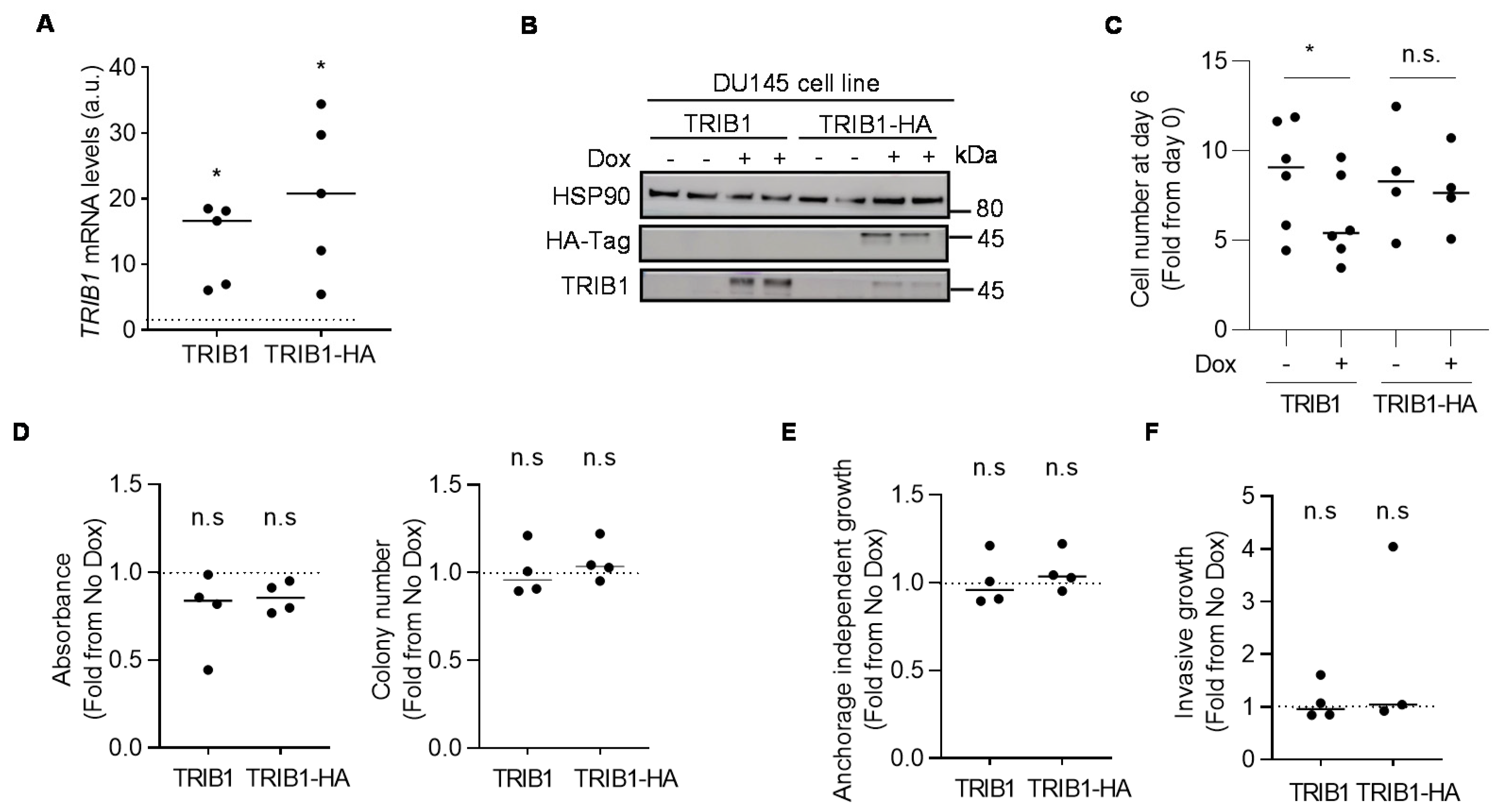
3.3. TRIB1 does not Exhibit Cell-Autonomous Tumor-Promoting Activity in PCa Cell Lines
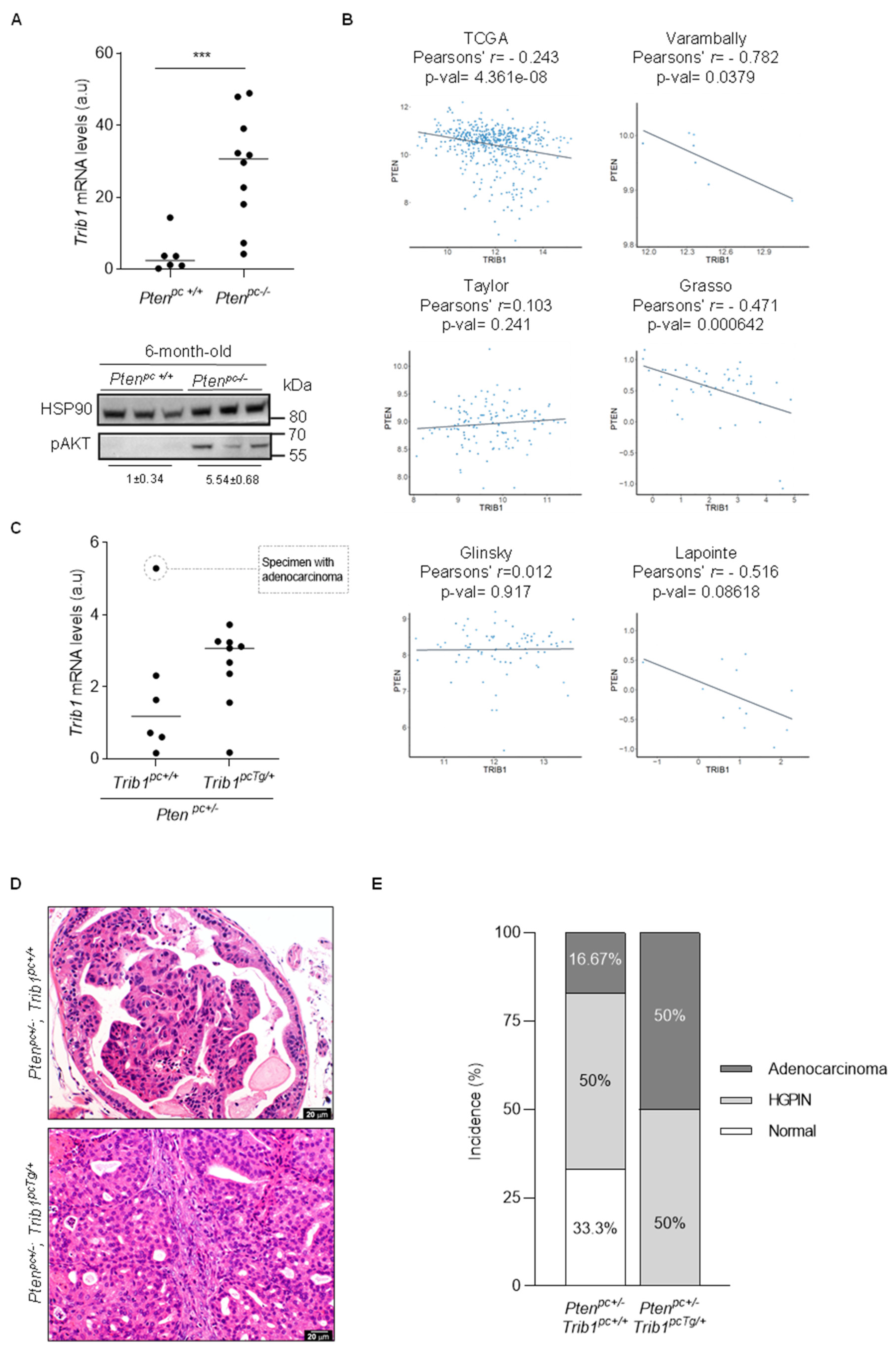
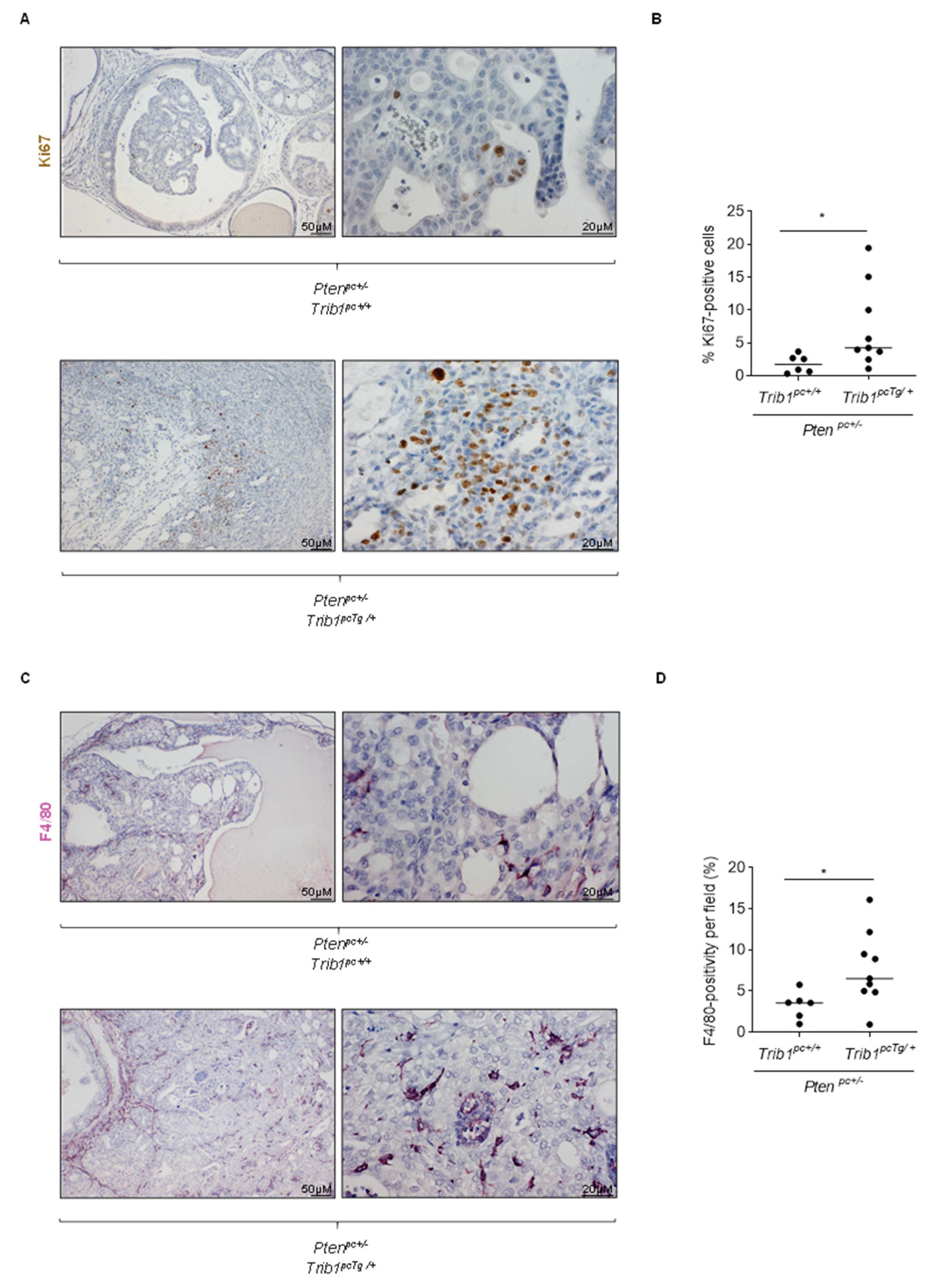
3.4. Trib1 Overexpression Cooperates with Pten Heterozygosity to Promote PCa Pathogenesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, M.F.; Mardis, E.R. The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol. 2018, 15, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grisanzio, C.; Freedman, M.L. Chromosome 8q24-Associated Cancers and MYC. Genes Cancer 2010, 1, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC Deregulation in Primary Human Cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef] [Green Version]
- Ghoussaini, M.; Song, H.; Koessler, T.; Al Olama, A.A.; Kote-Jarai, Z.; Driver, K.E.; Pooley, K.A.; Ramus, S.J.; Kjaer, S.K.; Hogdall, E.; et al. Multiple loci with different cancer specificities within the 8q24 gene desert. J. Natl. Cancer Inst. 2008, 100, 962–966. [Google Scholar] [CrossRef] [Green Version]
- Pal, P.; Xi, H.; Guha, S.; Sun, G.; Helfand, B.T.; Meeks, J.J.; Suarez, B.K.; Catalona, W.J.; Deka, R. Common variants in 8q24 are associated with risk for prostate cancer and tumor aggressiveness in men of European ancestry. Prostate 2009, 69, 1548–1556. [Google Scholar] [CrossRef] [Green Version]
- Riaz, M.; Berns, E.M.; Sieuwerts, A.M.; Ruigrok-Ritstier, K.; de Weerd, V.; Groenewoud, A.; Uitterlinden, A.G.; Look, M.P.; Klijn, J.G.; Sleijfer, S.; et al. Correlation of breast cancer susceptibility loci with patient characteristics, metastasis-free survival, and mRNA expression of the nearest genes. Breast Cancer Res. Treat. 2012, 133, 843–851. [Google Scholar] [CrossRef]
- Huppi, K.; Pitt, J.J.; Wahlberg, B.M.; Caplen, N.J. The 8q24 gene desert: An oasis of non-coding transcriptional activity. Front. Genet. 2012, 3, 69. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.B.; Maia, A.T.; O’Reilly, M.; Ghoussaini, M.; Prathalingam, R.; Porter-Gill, P.; Ambs, S.; Prokunina-Olsson, L.; Carroll, J.; Ponder, B.A. A functional variant at a prostate cancer predisposition locus at 8q24 is associated with PVT1 expression. PLoS Genet. 2011, 7, e1002165. [Google Scholar] [CrossRef] [PubMed]
- Killian, A.; Sarafan-Vasseur, N.; Sesboüé, R.; Le Pessot, F.; Blanchard, F.; Lamy, A.; Laurent, M.; Flaman, J.M.; Frébourg, T. Contribution of the BOP1 gene, located on 8q24, to colorectal tumorigenesis. Genes Chromosomes Cancer 2006, 45, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Lin, X.; Kapoor, A.; Chow, M.J.; Jiang, Y.; Zhao, K.; Tang, D. The Oncogenic Potential of the Centromeric Border Protein FAM84B of the 8q24.21 Gene Desert. Genes 2020, 11, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kypta, R.; Unda, M.; Carracedo, A. Is the bench getting closer to the bedside in the war on cancer? A quick look at prostate cancer. Front. Endocrinol. 2012, 3, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, E.D.; Schellhammer, P.F.; McLeod, D.G.; Moul, J.W.; Higano, C.S.; Shore, N.; Denis, L.; Iversen, P.; Eisenberger, M.A.; Labrie, F. Androgen Receptor Targeted Treatments of Prostate Cancer: 35 Years of Progress with Antiandrogens. J. Urol. 2018, 200, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Grimm, P.; Billiet, I.; Bostwick, D.; Dicker, A.P.; Frank, S.; Immerzeel, J.; Keyes, M.; Kupelian, P.; Lee, W.R.; Machtens, S.; et al. Comparative analysis of prostate-specific antigen free survival outcomes for patients with low, intermediate and high risk prostate cancer treatment by radical therapy. Results from the Prostate Cancer Results Study Group. BJU Int. 2012, 109 (Suppl. S1), 22–29. [Google Scholar] [CrossRef]
- Rubin, M.A.; Demichelis, F. The Genomics of Prostate Cancer: Emerging understanding with technologic advances. Mod. Pathol. 2018, 31, S1–S11. [Google Scholar] [CrossRef]
- Ellwood-Yen, K.; Graeber, T.G.; Wongvipat, J.; Iruela-Arispe, M.L.; Zhang, J.; Matusik, R.; Thomas, G.V.; Sawyers, C.L. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 2003, 4, 223–238. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Herzka, T.; Zheng, W.; Qi, J.; Wilkinson, J.E.; Bradner, J.E.; Robinson, B.D.; Castillo-Martin, M.; Cordon-Cardo, C.; Trotman, L.C. RapidCaP, a novel GEM model for metastatic prostate cancer analysis and therapy, reveals myc as a driver of Pten-mutant metastasis. Cancer Discov. 2014, 4, 318–333. [Google Scholar] [CrossRef] [Green Version]
- Pettersson, A.; Gerke, T.; Penney, K.L.; Lis, R.T.; Stack, E.C.; Pértega-Gomes, N.; Zadra, G.; Tyekucheva, S.; Giovannucci, E.L.; Mucci, L.A.; et al. MYC Overexpression at the Protein and mRNA Level and Cancer Outcomes among Men Treated with Radical Prostatectomy for Prostate Cancer. Cancer Epidemiol. Biomark. Prev. 2018, 27, 201–207. [Google Scholar] [CrossRef] [Green Version]
- Richmond, L.; Keeshan, K. Pseudokinases: A tribble-edged sword. FEBS J. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyers, P.A.; Keeshan, K.; Kannan, N. Tribbles in the 21st Century: The Evolving Roles of Tribbles Pseudokinases in Biology and Disease. Trends Cell Biol. 2017, 27, 284–298. [Google Scholar] [CrossRef] [Green Version]
- Kung, J.E.; Jura, N. The pseudokinase TRIB1 toggles an intramolecular switch to regulate COP1 nuclear export. EMBO J. 2019, 38, e99708. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, S.A.; Ruan, Z.; Burgess, A.E.; Curry, J.R.; McMillan, H.D.; Brewster, J.L.; Dunbier, A.K.; Axtman, A.D.; Kannan, N.; Mace, P.D. Substrate binding allosterically relieves autoinhibition of the pseudokinase TRIB1. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uljon, S.; Xu, X.; Durzynska, I.; Stein, S.; Adelmant, G.; Marto, J.A.; Pear, W.S.; Blacklow, S.C. Structural Basis for Substrate Selectivity of the E3 Ligase COP1. Structure 2016, 24, 687–696. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, A.; Kato, J.Y.; Nakamae, I.; Yoneda-Kato, N. COP1 targets C/EBPα for degradation and induces acute myeloid leukemia via Trib1. Blood 2013, 122, 1750–1760. [Google Scholar] [CrossRef]
- Liu, Z.Z.; Han, Z.D.; Liang, Y.K.; Chen, J.X.; Wan, S.; Zhuo, Y.J.; Cai, Z.D.; Deng, Y.L.; Lin, Z.Y.; Mo, R.J.; et al. TRIB1 induces macrophages to M2 phenotype by inhibiting IKB-zeta in prostate cancer. Cell. Signal. 2019, 59, 152–162. [Google Scholar] [CrossRef]
- Mashima, T.; Soma-Nagae, T.; Migita, T.; Kinoshita, R.; Iwamoto, A.; Yuasa, T.; Yonese, J.; Ishikawa, Y.; Seimiya, H. TRIB1 supports prostate tumorigenesis and tumor-propagating cell survival by regulation of endoplasmic reticulum chaperone expression. Cancer Res. 2014, 74, 4888–4897. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.Y.; Huang, Y.Q.; Zhang, Y.Q.; Han, Z.D.; He, H.C.; Ling, X.H.; Fu, X.; Dai, Q.S.; Cai, C.; Chen, J.H.; et al. MicroRNA-224 inhibits progression of human prostate cancer by downregulating TRIB1. Int. J. Cancer 2014, 135, 541–550. [Google Scholar] [CrossRef]
- Torrano, V.; Valcarcel-Jimenez, L.; Cortazar, A.R.; Liu, X.; Urosevic, J.; Castillo-Martin, M.; Fernandez-Ruiz, S.; Morciano, G.; Caro-Maldonado, A.; Guiu, M.; et al. The metabolic co-regulator PGC1alpha suppresses prostate cancer metastasis. Nat. Cell Biol. 2016, 18, 645–656. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotman, L.C.; Niki, M.; Dotan, Z.A.; Koutcher, J.A.; Di Cristofano, A.; Xiao, A.; Khoo, A.S.; Roy-Burman, P.; Greenberg, N.M.; Van Dyke, T.; et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003, 1, E59. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.M.; Angyal, A.; Bauer, R.C.; Hamby, S.; Suvarna, S.K.; Baidžajevas, K.; Hegedus, Z.; Dear, T.N.; Turner, M.; Wilson, H.L.; et al. Myeloid Tribbles 1 induces early atherosclerosis via enhanced foam cell expansion. Sci. Adv. 2019, 5, eaax9183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shappell, S.B.; Thomas, G.V.; Roberts, R.L.; Herbert, R.; Ittmann, M.M.; Rubin, M.A.; Humphrey, P.A.; Sundberg, J.P.; Rozengurt, N.; Barrios, R.; et al. Prostate pathology of genetically engineered mice: Definitions and classification. The consensus report from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res. 2004, 64, 2270–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiederschain, D.; Wee, S.; Chen, L.; Loo, A.; Yang, G.; Huang, A.; Chen, Y.; Caponigro, G.; Yao, Y.M.; Lengauer, C.; et al. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 2009, 8, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Valcarcel-Jimenez, L.; Macchia, A.; Crosas-Molist, E.; Schaub-Clerigue, A.; Camacho, L.; Martin-Martin, N.; Cicogna, P.; Viera-Bardon, C.; Fernandez-Ruiz, S.; Rodriguez-Hernandez, I.; et al. PGC1alpha Suppresses Prostate Cancer Cell Invasion through ERRalpha Transcriptional Control. Cancer Res. 2019, 79, 6153–6165. [Google Scholar] [CrossRef] [Green Version]
- Ugalde-Olano, A.; Egia, A.; Fernandez-Ruiz, S.; Loizaga-Iriarte, A.; Zuniga-Garcia, P.; Garcia, S.; Royo, F.; Lacasa-Viscasillas, I.; Castro, E.; Cortazar, A.R.; et al. Methodological aspects of the molecular and histological study of prostate cancer: Focus on PTEN. Methods 2015, 77–78, 25–30. [Google Scholar] [CrossRef]
- Martin-Martin, N.; Piva, M.; Urosevic, J.; Aldaz, P.; Sutherland, J.D.; Fernandez-Ruiz, S.; Arreal, L.; Torrano, V.; Cortazar, A.R.; Planet, E.; et al. Stratification and therapeutic potential of PML in metastatic breast cancer. Nat. Commun. 2016, 7, 12595. [Google Scholar] [CrossRef] [Green Version]
- Boehm, J.S.; Hession, M.T.; Bulmer, S.E.; Hahn, W.C. Transformation of human and murine fibroblasts without viral oncoproteins. Mol. Cell. Biol. 2005, 25, 6464–6474. [Google Scholar] [CrossRef] [Green Version]
- Cortazar, A.R.; Torrano, V.; Martin-Martin, N.; Caro-Maldonado, A.; Camacho, L.; Hermanova, I.; Guruceaga, E.; Lorenzo-Martin, L.F.; Caloto, R.; Gomis, R.R.; et al. CANCERTOOL: A Visualization and Representation Interface to Exploit Cancer Datasets. Cancer Res. 2018, 78, 6320–6328. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, E.L.; Meier, P. Nonparametric Estimation from Incomplete Observations. J. Am. Stat. Assoc. 1958, 53, 457–481. [Google Scholar] [CrossRef]
- Mantel, N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother. Rep. 1966, 50, 163–170. [Google Scholar] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Hahne, F.; Ivanek, R. Visualizing Genomic Data Using Gviz and Bioconductor. Methods Mol. Biol. 2016, 1418, 335–351. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Lapointe, J.; Li, C.; Higgins, J.P.; van de Rijn, M.; Bair, E.; Montgomery, K.; Ferrari, M.; Egevad, L.; Rayford, W.; Bergerheim, U.; et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 811–816. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Tomlins, S.A.; Mehra, R.; Rhodes, D.R.; Cao, X.; Wang, L.; Dhanasekaran, S.M.; Kalyana-Sundaram, S.; Wei, J.T.; Rubin, M.A.; Pienta, K.J.; et al. Integrative molecular concept modeling of prostate cancer progression. Nat. Genet. 2007, 39, 41–51. [Google Scholar] [CrossRef]
- Varambally, S.; Yu, J.; Laxman, B.; Rhodes, D.R.; Mehra, R.; Tomlins, S.A.; Shah, R.B.; Chandran, U.; Monzon, F.A.; Becich, M.J.; et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 2005, 8, 393–406. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.-Y.; Callan, C.Y.; Fang, Z.; Tung, H.-Y.; Park, J.Y. Interactions of PVT1 and CASC11 on Prostate Cancer Risk in African Americans. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1067–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyekucheva, S.; Bowden, M.; Bango, C.; Giunchi, F.; Huang, Y.; Zhou, C.; Bondi, A.; Lis, R.; Van Hemelrijck, M.; Andren, O.; et al. Stromal and epithelial transcriptional map of initiation progression and metastatic potential of human prostate cancer. Nat. Commun. 2017, 8, 420. [Google Scholar] [CrossRef] [Green Version]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef]
- Arreal, L.; Piva, M.; Fernandez, S.; Revandkar, A.; Schaub-Clerigue, A.; Villanueva, J.; Zabala-Letona, A.; Pujana, M.; Astobiza, I.; Cortazar, A.R.; et al. Targeting PML in triple negative breast cancer elicits growth suppression and senescence. Cell Death Differ. 2020, 27, 1186–1199. [Google Scholar] [CrossRef]
- The Cancer Cell Line Encyclopedia Consortium; The Genomics of Drug Sensitivity in Cancer Consortium. Pharmacogenomic agreement between two cancer cell line data sets. Nature 2015, 528, 84–87. [Google Scholar] [CrossRef]
- Di Cristofano, A.; Pesce, B.; Cordon-Cardo, C.; Pandolfi, P.P. Pten is essential for embryonic development and tumour suppression. Nat. Genet. 1998, 19, 348–355. [Google Scholar] [CrossRef]
- Nardella, C.; Carracedo, A.; Salmena, L.; Pandolfi, P.P. Faithfull modeling of PTEN loss driven diseases in the mouse. Curr. Top. Microbiol. Immunol. 2011, 347, 135–168. [Google Scholar] [CrossRef]
- Zhu, J.; Blenis, J.; Yuan, J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc. Natl. Acad. Sci. USA 2008, 105, 6584–6589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schild, C.; Wirth, M.; Reichert, M.; Schmid, R.M.; Saur, D.; Schneider, G. PI3K signaling maintains c-myc expression to regulate transcription of E2F1 in pancreatic cancer cells. Mol. Carcinog. 2009, 48, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Swords, R.T.; Schenk, T.; Stengel, S.; Gil, V.S.; Petrie, K.R.; Perez, A.; Ana, R.; Watts, J.M.; Vargas, F.; Elias, R.; et al. Inhibition of the PI3K/AKT/mTOR Pathway Leads to Down-Regulation of c-Myc and Overcomes Resistance to ATRA in Acute Myeloid Leukemia. Blood 2015, 126, 1363–1363. [Google Scholar] [CrossRef]
- Liu, P.; Ge, M.; Hu, J.; Li, X.; Che, L.; Sun, K.; Cheng, L.; Huang, Y.; Pilo, M.G.; Cigliano, A.; et al. A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology 2017, 66, 167–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csibi, A.; Lee, G.; Yoon, S.O.; Tong, H.; Ilter, D.; Elia, I.; Fendt, S.M.; Roberts, T.M.; Blenis, J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr. Biol. 2014, 24, 2274–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardsen, E.; Andersen, S.; Al-Saad, S.; Rakaee, M.; Nordby, Y.; Pedersen, M.I.; Ness, N.; Grindstad, T.; Movik, I.; Dønnem, T.; et al. Evaluation of the proliferation marker Ki-67 in a large prostatectomy cohort. PLoS ONE 2017, 12, e0186852. [Google Scholar] [CrossRef]
- Labbé, D.P.; Brown, M. Transcriptional Regulation in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030437. [Google Scholar] [CrossRef]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, J.R.; Beaulieu, M.E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Umemori, M.; Kurata, M.; Yamamoto, A.; Yamamoto, K.; Ishibashi, S.; Ikeda, M.; Tashiro, K.; Kimura, T.; Sato, S.; Takahashi, H.; et al. The expression of MYC is strongly dependent on the circular PVT1 expression in pure Gleason pattern 4 of prostatic cancer. Med. Mol. Morphol. 2020, 53, 156–167. [Google Scholar] [CrossRef]
- Yang, J.; Li, C.; Mudd, A.; Gu, X. LncRNA PVT1 predicts prognosis and regulates tumor growth in prostate cancer. Biosci. Biotechnol. Biochem. 2017, 81, 2301–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Huang, L.; Zhu, Q.; Wang, Z.; Tang, Z. MTSS1 hypermethylation is associated with prostate cancer progression. J. Cell. Physiol. 2020, 235, 2687–2697. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhong, J.; Guo, B.; Zhu, Q.; Liang, H.; Wen, N.; Yun, W.; Zhang, L. miR-96 promotes the growth of prostate carcinoma cells by suppressing MTSS1. Tumour Biol. 2016, 37, 12023–12032. [Google Scholar] [CrossRef] [PubMed]
- Moya, L.; Lai, J.; Hoffman, A.; Srinivasan, S.; Panchadsaram, J.; Chambers, S.; Clements, J.A.; Batra, J. Association Analysis of a Microsatellite Repeat in the TRIB1 Gene With Prostate Cancer Risk, Aggressiveness and Survival. Front. Genet. 2018, 9, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, S.; Boonvisut, S.; Makishima, S.; Ishizuka, Y.; Watanabe, K.; Nakayama, K. The role of TRIB1 in lipid metabolism; from genetics to pathways. Biochem. Soc. Trans. 2015, 43, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, N.; Pang, B.; Tong, D.; Sun, D.; Sun, H.; Zhang, C.; Sun, W.; Meng, X.; Bai, J.; et al. TRIB1 promotes colorectal cancer cell migration and invasion through activation MMP-2 via FAK/Src and ERK pathways. Oncotarget 2017, 8, 47931–47942. [Google Scholar] [CrossRef] [Green Version]
- Gendelman, R.; Xing, H.; Mirzoeva, O.K.; Sarde, P.; Curtis, C.; Feiler, H.S.; McDonagh, P.; Gray, J.W.; Khalil, I.; Korn, W.M. Bayesian Network Inference Modeling Identifies TRIB1 as a Novel Regulator of Cell-Cycle Progression and Survival in Cancer Cells. Cancer Res. 2017, 77, 1575–1585. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Carracedo, A.; Lin, H.K.; Koutcher, J.A.; Behrendt, N.; Egia, A.; Alimonti, A.; Carver, B.S.; Gerald, W.; Teruya-Feldstein, J.; et al. Differential p53-independent outcomes of p19(Arf) loss in oncogenesis. Sci. Signal. 2009, 2, ra44. [Google Scholar] [CrossRef] [Green Version]
- Hermanova, I.; Zuniga-Garcia, P.; Caro-Maldonado, A.; Fernandez-Ruiz, S.; Salvador, F.; Martin-Martin, N.; Zabala-Letona, A.; Nunez-Olle, M.; Torrano, V.; Camacho, L.; et al. Genetic manipulation of LKB1 elicits lethal metastatic prostate cancer. J. Exp. Med. 2020, 217, e20191787. [Google Scholar] [CrossRef] [Green Version]
- Mack, E.A.; Stein, S.J.; Rome, K.S.; Xu, L.; Wertheim, G.B.; Melo, R.C.N.; Pear, W.S. Trib1 regulates eosinophil lineage commitment and identity by restraining the neutrophil program. Blood 2019, 133, 2413–2426. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahrouzi, P.; Astobiza, I.; Cortazar, A.R.; Torrano, V.; Macchia, A.; Flores, J.M.; Niespolo, C.; Mendizabal, I.; Caloto, R.; Ercilla, A.; et al. Genomic and Functional Regulation of TRIB1 Contributes to Prostate Cancer Pathogenesis. Cancers 2020, 12, 2593. https://doi.org/10.3390/cancers12092593
Shahrouzi P, Astobiza I, Cortazar AR, Torrano V, Macchia A, Flores JM, Niespolo C, Mendizabal I, Caloto R, Ercilla A, et al. Genomic and Functional Regulation of TRIB1 Contributes to Prostate Cancer Pathogenesis. Cancers. 2020; 12(9):2593. https://doi.org/10.3390/cancers12092593
Chicago/Turabian StyleShahrouzi, Parastoo, Ianire Astobiza, Ana R. Cortazar, Verónica Torrano, Alice Macchia, Juana M. Flores, Chiara Niespolo, Isabel Mendizabal, Ruben Caloto, Amaia Ercilla, and et al. 2020. "Genomic and Functional Regulation of TRIB1 Contributes to Prostate Cancer Pathogenesis" Cancers 12, no. 9: 2593. https://doi.org/10.3390/cancers12092593
APA StyleShahrouzi, P., Astobiza, I., Cortazar, A. R., Torrano, V., Macchia, A., Flores, J. M., Niespolo, C., Mendizabal, I., Caloto, R., Ercilla, A., Camacho, L., Arreal, L., Bizkarguenaga, M., Martinez-Chantar, M. L., Bustelo, X. R., Berra, E., Kiss-Toth, E., Velasco, G., Zabala-Letona, A., & Carracedo, A. (2020). Genomic and Functional Regulation of TRIB1 Contributes to Prostate Cancer Pathogenesis. Cancers, 12(9), 2593. https://doi.org/10.3390/cancers12092593