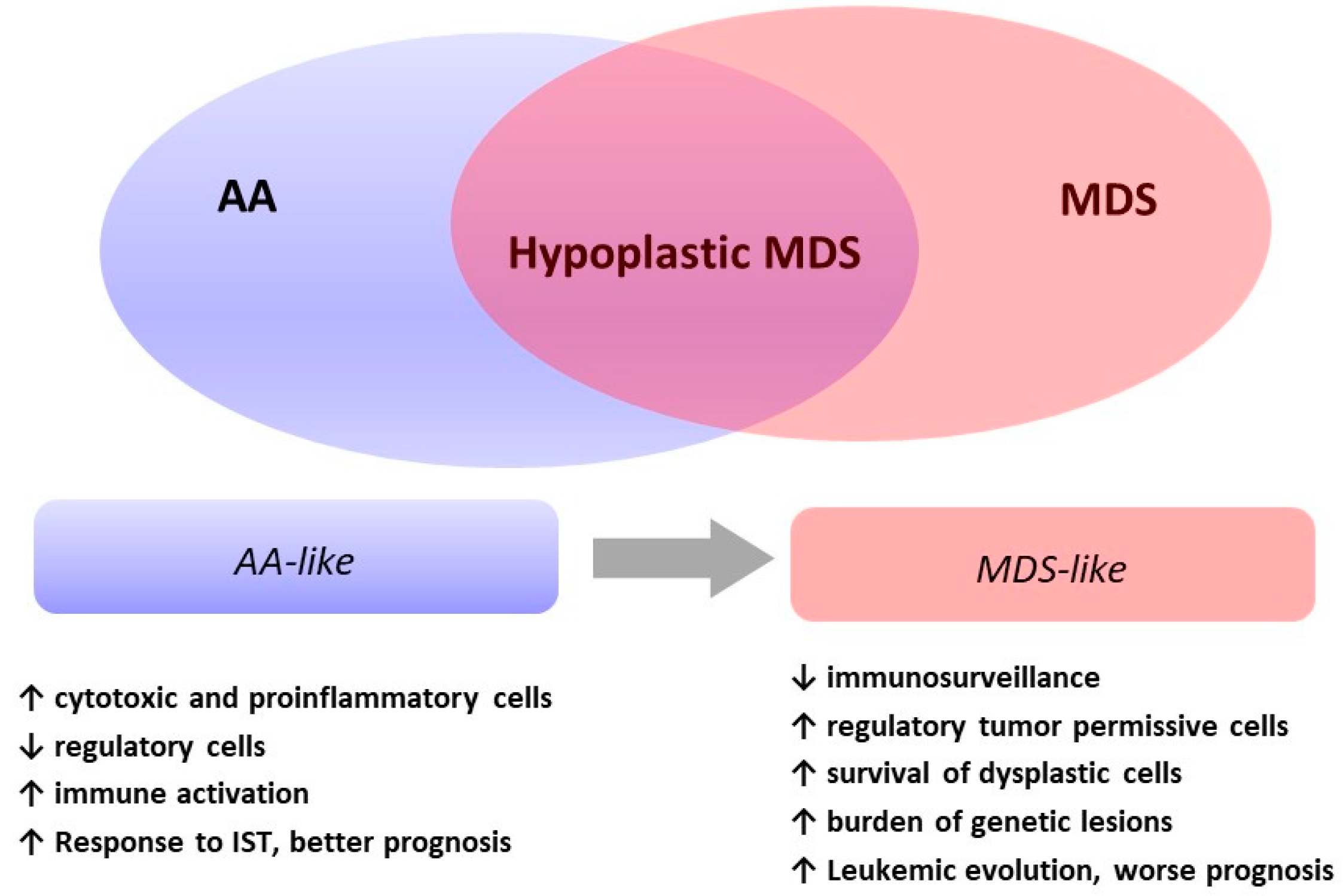
Hypoplastic Myelodysplastic Syndromes: Just an Overlap Syndrome?



Abstract
:Simple Summary
Abstract
1. Introduction
2. Clinical Features
3. Pathogenesis
3.1. Genomic Landscape
3.2. Immunological Features
3.2.1. Cytotoxic T Cells (CTLs)
3.2.2. T-CD4-Positive Cells
3.2.3. Large Granular Lymphocytes (LGL)
3.2.4. B Lymphocytes
3.2.5. Autoantibodies
3.2.6. Monocytes
3.2.7. Mastocytes (MCs)
3.2.8. Mesenchymal Stem Cells (MSCs)
3.2.9. Cytokine Dysregulation
3.2.10. PNH Clone
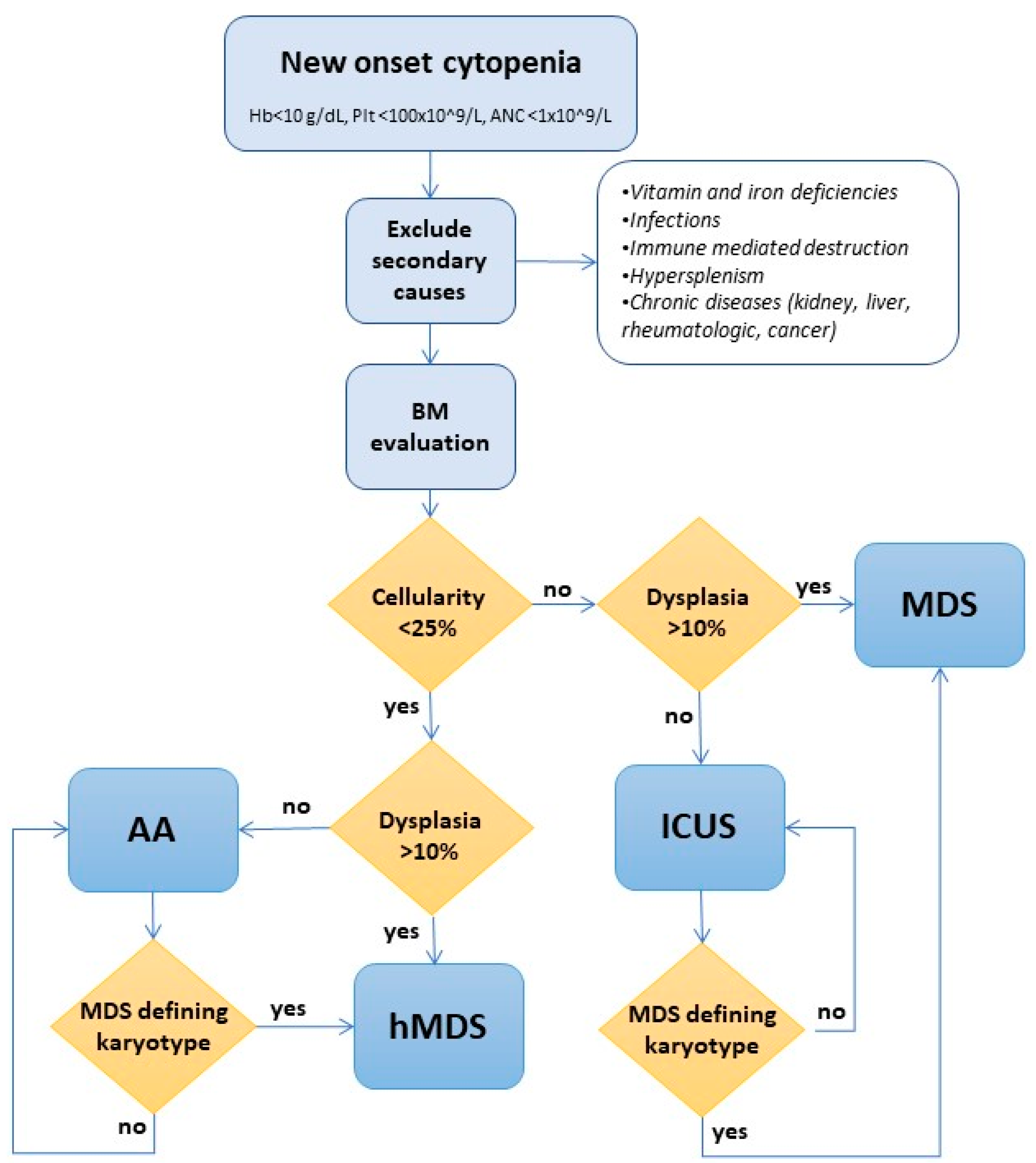
4. Diagnosis of Hypoplastic MDS
5. Therapeutic Approaches
5.1. MDS-Like Treatment
5.2. AA-Like Treatment
6. Clonal Evolution
7. Survival and Prognostic Factors
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Sekeres, M.A. The Epidemiology of Myelodysplastic Syndromes. Hematol. Clin. North Am. 2010, 24, 287–294. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.F.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Papaemmanuil, E.; Ambaglio, I.; Elena, C.; Gallì, A.; Della Porta, M.G.; Travaglino, E.; Pietra, D.; Pascutto, C.; Ubezio, M.; et al. Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood 2014, 124, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Montalban-Bravo, G.; Garcia-Manero, G. Myelodysplastic syndromes: 2018 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2018, 93, 129–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bono, E.; McLornan, D.; Travaglino, E.; Gandhi, S.; Gallì, A.; Khan, A.A.; Kulasekararaj, A.G.; Boveri, E.; Raj, K.; Elena, C.; et al. Clinical, histopathological and molecular characterization of hypoplastic myelodysplastic syndrome. Leukemia 2019, 33, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Calado, R.T. Immunologic Aspects of Hypoplastic Myelodysplastic Syndrome. Semin. Oncol. 2011, 38, 667–672. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.M.; Orazi, A. Diagnostic criteria to distinguish hypocellular acute myeloid leukemia from hypocellular myelodysplastic syndromes and aplastic anemia: Recommendations for a standardized approach. Haematologica 2009, 94, 264–268. [Google Scholar] [CrossRef]
- Marisavljević, D.; Čemerikić, V.; Rolović, Z.; Bošković, D.; Čolović, M. Hypocellular myelodysplastic syndromes: Clinical and biological significance. Med. Oncol. 2005, 22, 169–175. [Google Scholar] [CrossRef]
- Sloand, E.M. Hypocellular Myelodysplasia. Hematol. Clin. North Am. 2009, 23, 347–360. [Google Scholar] [CrossRef]
- Koh, Y.; Lee, H.R.; Song, E.Y.; Kim, H.K.; Kim, I.; Park, S.; Park, M.H.; Kim, B.K.; Yoon, S.-S.; Lee, D.S. Hypoplastic myelodysplastic syndrome (h-MDS) is a distinctive clinical entity with poorer prognosis and frequent karyotypic and FISH abnormalities compared to aplastic anemia (AA). Leuk. Res. 2010, 34, 1344–1350. [Google Scholar] [CrossRef]
- Huang, T.-C.; Ko, B.-S.; Tang, J.-L.; Hsu, C.; Chen, C.-Y.; Tsay, W.; Huang, S.-Y.; Yao, M.; Chen, Y.-C.; Shen, M.-C.; et al. Comparison of hypoplastic myelodysplastic syndrome (MDS) with normo-/hypercellular MDS by International Prognostic Scoring System, cytogenetic and genetic studies. Leukemia 2007, 22, 544–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Sokol, L.; Bennett, J.M.; Moscinski, L.C.; List, A.; Zhang, L. T-cell large granular lymphocyte proliferation in myelodysplastic syndromes: Clinicopathological features and prognostic significance. Leuk. Res. 2016, 43, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-F.; Huang, Z.-D.; Wu, X.-R.; Li, Q.; Yu, Z.-F. Comparison of T lymphocyte subsets in aplastic anemia and hypoplastic myelodysplastic syndromes. Life Sci. 2017, 189, 71–75. [Google Scholar] [CrossRef]
- Yao, C.-Y.; Hou, H.-A.; Lin, T.-Y.; Lin, C.-C.; Chou, W.-C.; Tseng, M.-H.; Chiang, Y.-C.; Liu, M.-C.; Liu, C.-W.; Kuo, Y.-Y.; et al. Distinct mutation profile and prognostic relevance in patients with hypoplastic myelodysplastic syndromes (h-MDS). Oncotarget 2016, 7, 63177–63188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazha, A.; Seastone, D.; Radivoyevitch, T.; Przychodzen, B.; Carraway, H.E.; Patel, B.J.; Carew, J.; Makishima, H.; Sekeres, M.A.; Maciejewski, J.P. Genomic patterns associated with hypoplastic compared to hyperplastic myelodysplastic syndromes. Haematologica 2015, 100, e434–e437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malcovati, L.; Gallì, A.; Travaglino, E.; Ambaglio, I.; Rizzo, E.; Molteni, E.; Elena, C.; Ferretti, V.V.; Catricalà, S.; Bono, E.; et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017, 129, 3371–3378. [Google Scholar] [CrossRef]
- Wang, C.; Yang, Y.; Gao, S.; Chen, J.; Yu, J.; Zhang, H.; Li, M.; Zhan, X.; Li, W. Immune dysregulation in myelodysplastic syndrome: Clinical features, pathogenesis and therapeutic strategies. Crit. Rev. Oncol. 2018, 122, 123–132. [Google Scholar] [CrossRef]
- Yang, L.; Qian, Y.; Eksioglu, E.; Epling-Burnette, P.K.; Wei, S. The inflammatory microenvironment in MDS. Cell. Mol. Life Sci. 2015, 72, 1959–1966. [Google Scholar] [CrossRef]
- Kordasti, S.; Afzali, B.; Lim, Z.; Ingram, W.; Hayden, J.; Barber, L.; Matthews, K.; Chelliah, R.; Guinn, B.-A.; Lombardi, G.; et al. IL-17-producing CD4+T cells, pro-inflammatory cytokines and apoptosis are increased in low risk myelodysplastic syndrome. Br. J. Haematol. 2009, 145, 64–72. [Google Scholar] [CrossRef]
- Bizymi, N.; Bjelica, S.; Kittang, A.O.; Mojsilovic, S.; Velegraki, M.; Pontikoglou, C.; Roussel, M.; Ersvær, E.; Santibañez, J.F.; Lipoldová, M.; et al. Myeloid-Derived Suppressor Cells in Hematologic Diseases: Promising Biomarkers and Treatment Targets. HemaSphere 2019, 3, e168. [Google Scholar] [CrossRef] [PubMed]
- Risitano, A.M.; Maciejewski, J.P.; Green, S.; Plasilova, M.; Zeng, W.; Young, N.S. In-vivo dominant immune responses in aplastic anaemia: Molecular tracking of putatively pathogenetic T-cell clones by TCR β-CDR3 sequencing. Lancet 2004, 364, 355–364. [Google Scholar] [CrossRef]
- Maciejewski, J.P.; O’Keefe, C.; Gondek, L.; Tiu, R.V. Immune-mediated bone marrow failure syndromes of progenitor and stem cells: Molecular analysis of cytotoxic T cell clones. Folia Histochem. Cytobiol. 2007, 45, 5–14. [Google Scholar] [PubMed]
- Wlodarski, M.W.; Gondek, L.P.; Nearman, Z.P.; Plasilova, M.; Kalaycio, M.; Hsi, E.D.; Maciejewski, J.P. Molecular strategies for detection and quantitation of clonal cytotoxic T-cell responses in aplastic anemia and myelodysplastic syndrome. Blood 2006, 108, 2632–2641. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Kobayashi, S.; Wieder, E.D.; Su, C.; Molldrem, J.J. Loss of T-lymphocyte clonal dominance in patients with myelodysplastic syndrome responsive to immunosuppression. Blood 2002, 100, 3639–3645. [Google Scholar] [CrossRef] [Green Version]
- Giudice, V.; Feng, X.; Lin, Z.; Hu, W.; Zhang, F.; Qiao, W.; Ibanez, M.D.P.F.; Rios, O.; Young, N.S. Deep sequencing and flow cytometric characterization of expanded effector memory CD8 + CD57 + T cells frequently reveals T-cell receptor Vβ oligoclonality and CDR3 homology in acquired aplastic anemia. Haematologica 2018, 103, 759–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risitano, A.; Maciejewski, J.; Selleri, C.; Rotoli, B.M.; Risitano, A.P.; Maciejewski, J. Function and Malfunction of Hematopoietic Stem Cells in Primary Bone Marrow Failure Syndromes. Curr. Stem Cell Res. Ther. 2007, 2, 39–52. [Google Scholar] [CrossRef] [Green Version]
- Sloand, E.M.; Kim, S.; Fuhrer, M.; Risitano, A.M.; Nakamura, R.; Maciejewski, J.P.; Barrett, A.J.; Young, N.S. Fas-mediated apoptosis is important in regulating cell replication and death in trisomy 8 hematopoietic cells but not in cells with other cytogenetic abnormalities. Blood 2002, 100, 4427–4432. [Google Scholar] [CrossRef]
- Gyan, E.; Frisan, E.; Beyne-Rauzy, O.; Deschemin, J.-C.; Pierre-Eugene, C.; Randriamampita, C.; Dubart-Kupperschmitt, A.; Garrido, C.; Dreyfus, F.; Mayeux, P.; et al. Spontaneous and Fas-induced apoptosis of low-grade MDS erythroid precursors involves the endoplasmic reticulum. Leukemia 2008, 22, 1864–1873. [Google Scholar] [CrossRef]
- Deeg, H.J.; Beckham, C.; Loken, M.R.; Bryant, E.; Lesnikova, M.; Shulman, H.M.; Gooley, T. Negative Regulators of Hemopoiesis and Stroma Function in Patients with Myelodysplastic Syndrome. Leuk. Lymphoma 2000, 37, 405–414. [Google Scholar] [CrossRef]
- Selleri, C.; Sato, T.; Raiola, A.M.; Rotoli, B.; Young, N.S.; Maciejewski, J.P. Induction of nitric oxide synthase is involved in the mechanism of Fas-mediated apoptosis in haemopoietic cells. Br. J. Haematol. 1997, 99, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Claessens, Y.-E.; Bouscary, D.; Dupont, J.-M.; Picard, F.; Melle, J.; Gisselbrecht, S.; Lacombe, C.; Dreyfus, F.; Mayeux, P.; Fontenay, M. In vitro proliferation and differentiation of erythroid progenitors from patients with myelodysplastic syndromes: Evidence for Fas-dependent apoptosis. Blood 2002, 99, 1594–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettou, S.; Audureau, E.; Humbrecht, C.; Benet, B.; Jammes, H.; Clozel, T.; Bardet, V.; Lacombe, C.; Dreyfus, F.; Mayeux, P.; et al. Fas expression at diagnosis as a biomarker of azacitidine activity in high-risk MDS and secondary AML. Leukemia 2012, 26, 2297–2299. [Google Scholar] [CrossRef] [PubMed]
- Selleri, C.; Maciejewski, J.P.; Catalano, L.; Ricci, P.; Andretta, C.; Luciano, L.; Rotoli, B. Effects of cyclosporine on hematopoietic and immune functions in patients with hypoplastic myelodysplasia. Cancer 2002, 95, 1911–1922. [Google Scholar] [CrossRef]
- Sloand, E.M.; Pfannes, L.; Chen, G.; Shah, S.; Solomou, E.E.; Barrett, J.; Young, N.S. CD34 cells from patients with trisomy 8 myelodysplastic syndrome (MDS) express early apoptotic markers but avoid programmed cell death by up-regulation of antiapoptotic proteins. Blood 2006, 109, 2399–2405. [Google Scholar] [CrossRef] [Green Version]
- Barrett, A.J.; Sloand, E. Autoimmune mechanisms in the pathophysiology of myelodysplastic syndromes and their clinical relevance. Haematologica 2009, 94, 449–451. [Google Scholar] [CrossRef]
- Chen, G.; Zeng, W.; Miyazato, A.; Billings, E.; Maciejewski, J.P.; Kajigaya, S.; Sloand, E.M.; Young, N.S. Distinctive gene expression profiles of CD34 cells from patients with myelodysplastic syndrome characterized by specific chromosomal abnormalities. Blood 2004, 104, 4210–4218. [Google Scholar] [CrossRef] [Green Version]
- Sloand, E.M.; Melenhorst, J.J.; Tucker, Z.C.G.; Pfannes, L.; Brenchley, J.M.; Yong, A.S.; Visconte, V.; Wu, C.; Gostick, E.; Scheinberg, P.; et al. T-cell immune responses to Wilms tumor 1 protein in myelodysplasia responsive to immunosuppressive therapy. Blood 2011, 117, 2691–2699. [Google Scholar] [CrossRef] [Green Version]
- Sloand, E.M.; Mainwaring, L.; Fuhrer, M.; Ramkissoon, S.; Risitano, A.M.; Keyvanafar, K.; Lu, J.; Basu, A.; Barrett, A.J.; Young, N.S. Preferential suppression of trisomy 8 compared with normal hematopoietic cell growth by autologous lymphocytes in patients with trisomy 8 myelodysplastic syndrome. Blood 2005, 106, 841–851. [Google Scholar] [CrossRef]
- Li, X.; Xu, F.; He, Q.; Wu, L.; Zhang, Z.; Chang, C. Comparison of Immunological Abnormalities of Lymphocytes in Bone Marrow in Myelodysplastic Syndrome (MDS) and Aplastic Anemia (AA). Intern. Med. 2010, 49, 1349–1355. [Google Scholar] [CrossRef] [Green Version]
- Fozza, C.; Longinotti, M. Are T-cell dysfunctions the other side of the moon in the pathogenesis of myelodysplastic syndromes? Eur. J. Haematol. 2012, 88, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Bouchliou, I.; Miltiades, P.; Nakou, E.; Spanoudakis, E.; Goutzouvelidis, A.; Vakalopoulou, S.; Garypidou, V.; Kotoula, V.; Bourikas, G.; Tsatalas, C.; et al. Th17 and Foxp3+ T regulatory cell dynamics and distribution in myelodysplastic syndromes. Clin. Immunol. 2011, 139, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Adam, W.M.; Epling-Burnette, P.K. Effector memory regulatory T-cell expansion marks a pivotal point of immune escape in myelodysplastic syndromes. OncoImmunology 2013, 2, e22654. [Google Scholar]
- Mailloux, A.W.; Sugimori, C.; Komrokji, R.S.; Yang, L.; Maciejewski, J.P.; Sekeres, M.A.; Paquette, R.; Loughran, T.P.; List, A.; Epling-Burnette, P.K. Expansion of effector memory regulatory T cells represents a novel prognostic factor in lower risk myelodysplastic syndrome. J. Immunol. 2012, 189, 3198–3208. [Google Scholar] [CrossRef] [Green Version]
- Fozza, C.; Longu, F.; Contini, S.; Galleu, A.; Virdis, P.; Bonfigli, S.; Murineddu, M.; Gabbas, A.; Longinotti, M. Patients with early-stage myelodysplastic syndromes show increased frequency of CD4?high?low Regulatory T Cells. Acta Haematol. 2012, 128, 178–182. [Google Scholar] [CrossRef]
- Alfinito, F.; Sica, M.; Luciano, L.; Della Pepa, R.; Palladino, C.; Ferrara, I.; Giani, U.; Ruggiero, G.; Terrazzano, G. Immune dysregulation and dyserythropoiesis in the myelodysplastic syndromes. Br. J. Haematol. 2010, 148, 90–98. [Google Scholar] [CrossRef]
- Feng, X.-M.; Xu, H.-Z.; Zhang, J.; Yin, D.-M.; Liu, M.-M.; Sui, X.-H. Expressive changes of CD4(+)T cell subset transcription factors in patients with aplastic anemia, myelodysplastic syndrome and acute myeloid leukemia and their clinical significances. J. Exp. Hematol. 2014, 22, 1038–1042. [Google Scholar]
- Quintanilla-Martinez, L. The 2016 updated WHO classification of lymphoid neoplasias. Hematol. Oncol. 2017, 35, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teramo, A.; Barilà, G.; Calabretto, G.; Vicenzetto, C.; Gasparini, V.R.; Semenzato, G.; Zambello, R. Insights Into Genetic Landscape of Large Granular Lymphocyte Leukemia. Front. Oncol. 2020, 10, 152. [Google Scholar] [CrossRef] [Green Version]
- Jerez, A.; Clemente, M.J.; Makishima, H.; Rajala, H.; Gómez-Seguí, I.; Olson, T.; McGraw, K.; Przychodzen, B.; Kulasekararaj, A.; Afable, M.; et al. STAT3 mutations indicate the presence of subclinical T-cell clones in a subset of aplastic anemia and myelodysplastic syndrome patients. Blood 2013, 122, 2453–2459. [Google Scholar] [CrossRef]
- Sato, T.; Selleri, C.; Young, N.S.; Maciejewski, J.P. Hematopoietic inhibition by interferon-γ is partially mediated through interferon regulatory factor-1. Blood 1995, 86, 3373–3380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Selleri, C.; Young, N.S.; Maciejewski, J.P. Inhibition of interferon regulatory factor-1 expression results in predominance of cell growth stimulatory effects of interferon-γ due to phosphorylation of Stat1 and Stat3. Blood 1997, 90, 4749–4758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejewski, J.P.; Selleri, C.; Sato, T.; Cho, H.J.; Keefer, L.K.; Nathan, C.F.; Young, N.S. Nitric oxide suppression of human hematopoiesis in vitro: Contribution to inhibitory action of interferon-γ and tumor necrosis factor-α. J. Clin. Investig. 1995, 96, 1085–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejewski, J.; Selleri, C.; Anderson, S.; Young, N.S. Fas antigen expression on CD34+ human marrow cells is induced by interferon γ and tumor necrosis factor α and potentiates cytokine-mediated hematopoietic suppression in vitro. Blood 1995, 85, 3183–3190. [Google Scholar] [CrossRef]
- Ogata, K.; Kishikawa, Y.; Satoh, C.; Tamura, H.; Dan, K.; Hayashi, A. Diagnostic application of flow cytometric characteristics of CD34+ cells in low-grade myelodysplastic syndromes. Blood 2006, 108, 1037–1044. [Google Scholar] [CrossRef] [Green Version]
- Sternberg, A.; Killick, S.; Littlewood, T.; Hatton, C.; Peniket, A.; Seidl, T.; Soneji, S.; Leach, J.; Bowen, D.; Chapman, C.; et al. Evidence for reduced B-cell progenitors in early (low-risk) myelodysplastic syndrome. Blood 2005, 106, 2982–2991. [Google Scholar] [CrossRef] [Green Version]
- Zaimoku, Y.; Patel, B.A.; Kajigaya, S.; Feng, X.; Alemu, L.; Quinones, R.D.; Groarke, E.M.; Young, N.S. Deficit of circulating CD19+CD24hiCD38hi regulatory B cells in severe aplastic anaemia. Br. J. Haematol. 2020, 190, 610–617. [Google Scholar] [CrossRef] [Green Version]
- Mufti, G.J.; Figes, A.; Hamblin, T.J.; Oscier, D.G.; Copplestoni, J.A. Immunological abnormalities in myelodysplastic syndromes I. Serum Immunoglobulins and Autoantibodies. Br. J. Haematol. 1986, 63, 143–147. [Google Scholar] [CrossRef]
- Martín Vega, C.; Vallespí, T.; Juliá, A.; Zuazu, J.; Torrabadella, M. Anemia hemolítica autoinmune y síndromes mielodisplásicos. Sangre 1989. [Google Scholar]
- Hebbar, M.; Kaplan, C.; Caulier, M.T.; Wattel, E.; Morel, P.; Wetterwald, M.; Bauters, F.; Fenaux, P. Low incidence of specific anti-platelet antibodies detected by the MAIPA assay in the serum of thrombocytopenic MDS patients and lack of correlation between platelet autoantibodies, platelet lifespan and response to danazol therapy. Br. J. Haematol. 1996, 94, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Novaretti, M.C.; Sopelete, C.; Velloso, E.R.P.; Rosa, M.; Dorlhiac-Llacer, P.E.; Chamone, D. Immunohematological findings in myelodysplastic syndrome. Acta Haematol. 2001, 105, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Arriaga, F.; Bonanad, S.; Larrea, L.; De La Rubia, J.; López, F.; Sanz, M.A.; Sanz, G.; Marty, M.L. Immunohematologic study in 112 patients with myelodysplastic syndromes: 10-year analysis. Sangre 1995, 40, 177. [Google Scholar] [PubMed]
- Okamoto, T.; Okada, M.; Mori, A.; Saheki, K.; Takatsuka, H.; Wada, H.; Tamura, A.; Fujimori, Y.; Takemoto, Y.; Kanamaru, A.; et al. Correlation between immunological abnormalities and prognosis in myelodysplastic syndrome patients. Int. J. Hematol. 1997, 66, 345–351. [Google Scholar] [CrossRef]
- Barcellini, W.; Zaninoni, A.; Imperiali, F.G.; Boschetti, C.; Colombi, M.; Iurlo, A.; Zanella, A. Anti-erythroblast autoimmunity in early myelodysplastic syndromes. Haematologica 2007, 92, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allampallam, K.; Shetty, V.; Mundle, S.; Dutt, D.; Kravitz, H.; Reddy, P.L.; Alvi, S.; Galili, N.; Saberwal, G.S.; Anthwal, S.; et al. Biological Significance of Proliferation, Apoptosis, Cytokines, and Monocyte/Macrophage Cells in Bone Marrow Biopsies of 145 Patients With Myelodysplastic Syndrome. Int. J. Hematol. 2002, 75, 289–297. [Google Scholar] [CrossRef]
- Velegraki, M.; Papakonstanti, E.; Mavroudi, I.; Psyllaki, M.; Tsatsanis, C.; Oulas, A.; Iliopoulos, I.; Katonis, P.; Papadaki, H.A. Impaired clearance of apoptotic cells leads to HMGB1 release in the bone marrow of patients with myelodysplastic syndromes and induces TLR4-mediated cytokine production. Haematologica 2013, 98, 1206–1215. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Wu, Z.; Lin, Z.; Hollinger, M.; Chen, J.; Feng, X.; Young, N.S. Macrophage TNF-a licenses donor T cells in murine bone marrow failure and can be implicated in human aplastic anemia. Blood 2018, 132, 2730–2743. [Google Scholar] [CrossRef] [Green Version]
- McCabe, A.; Smith, J.N.; Costello, A.; Maloney, J.; Katikaneni, D.; MacNamara, K.C. Hematopoietic stem cell loss and hematopoietic failure in severe aplastic anemia is driven by macrophages and aberrant podoplanin expression. Haematologica 2018, 103, 1451–1461. [Google Scholar] [CrossRef] [Green Version]
- Ozdemir, O.; Savaşan, S. The role of mast cells in bone marrow diseases. J. Clin. Pathol. 2004, 57, 108–109. [Google Scholar] [CrossRef] [Green Version]
- Leskinen, M.; Wang, Y.; Leszczynski, D.; Lindstedt, K.A.; Kovanen, P.T. Mast cell chymase induces apoptosis of vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 516–522. [Google Scholar] [CrossRef]
- Chien, M.; Abella, E.R.R. Mast cell persistence is associated with poor outcome in childhood severe aplastic anemia following immune suppression. Pediatr. Res. 2003, 53, 292A. [Google Scholar]
- Fattizzo, B.; Giannotta, J.A.; Barcellini, W. Mesenchymal Stem Cells in Aplastic Anemia and Myelodysplastic Syndromes: The “Seed and Soil” Crosstalk. Int. J. Mol. Sci. 2020, 21, 5438. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-H.; Gao, C.-J.; Da, W.-M.; Cao, Y.-B.; Wang, Z.-H.; Xu, L.-X.; Wu, Y.-M.; Liu, B.; Liu, Z.-Y.; Yan, B.; et al. Reduced Intensity Conditioning, Combined Transplantation of Haploidentical Hematopoietic Stem Cells and Mesenchymal Stem Cells in Patients with Severe Aplastic Anemia. PLoS ONE 2014, 9, e89666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Zhang, Y.; Xiao, H.; Yao, Z.; Zhang, H.; Liu, Q.; Wu, B.; Nie, D.; Li, Y.; Pang, Y.; et al. Cotransplantation of bone marrow-derived mesenchymal stem cells in haploidentical hematopoietic stem cell transplantation in patients with severe aplastic anemia: An interim summary for a multicenter phase II trial results. Bone Marrow Transplant. 2017, 52, 704–710. [Google Scholar] [CrossRef]
- Xu, L.; Liu, Z.; Wu, Y.; Yang, X.; Cao, Y.; Li, X.; Yan, B.; Li, S.; Da, W.; Wu, X. Clinical evaluation of haploidentical hematopoietic combined with human umbilical cord-derived mesenchymal stem cells in severe aplastic anemia. Eur. J. Med Res. 2018, 23, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allampallam, K.; Shetty, V.T.; Raza, A. Cytokines and MDS. Cancer Treat. Res. 2001, 108, 93–100. [Google Scholar] [PubMed]
- Kitagawa, M.; Saito, I.; Kuwata, T.; Yoshida, S.; Yamaguchi, S.; Takahashi, M.; Tanizawa, T.; Kamiyama, R.; Hirokawa, K. Overexpression of tumor necrosis factor (TNF)-α and interferon (IFN)-γ by bone marrow cells from patients with myelodysplastic syndromes. Leukemia 1997, 11, 2049–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stifter, G.; Heiss, S.; Gastl, G.; Tzankov, A.; Stauder, R. Over-expression of tumor necrosis factor-alpha in bone marrow biopsies from patients with myelodysplastic syndromes: Relationship to anemia and prognosis. Eur. J. Haematol. 2005, 75, 485–491. [Google Scholar] [CrossRef]
- Selleri, C.; Maciejewski, J.P. Impact of Immunogenetic Polymorphisms in Bone Marrow Failure Syndromes. Mini-Rev. Med. Chem. 2011, 11, 544–552. [Google Scholar]
- Benesch, M.; Platzbecker, U.; Ward, J.; Deeg, H.J.; Leisenring, W. Expression of FLIPLong and FLIPShort in bone marrow mononuclear and CD34+ cells in patients with myelodysplastic syndrome: Correlation with apoptosis. Leukemia 2003, 17, 2460–2466. [Google Scholar] [CrossRef]
- Secchiero, P.; Zauli, G. Tumor necrosis factor-related apoptosis-inducing ligand and the regulation of hematopoiesis. Curr. Opin. Hematol. 2008, 15, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Kim, S.; Selleri, C.; Young, N.S.; Maciejewski, J.P. Measurement of secondary colony formation after 5 weeks in long-term cultures in patients with myelodysplastic syndrome. Leukemia 1998, 12, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, R.F.; Metze, K.; Silva, M.R.R.; Chauffai, M.D.L.L.F. The ambiguous role of interferon regulatory factor-1 (IRF-1) immunoexpression in myelodysplastic syndrome. Leuk. Res. 2009, 33, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, X.; Guo, J.; Xu, F.; He, Q.; Zhao, Y.; Yang, Y.; Gu, S.; Zhang, Y.; Wu, L.; et al. Interleukin-17 enhances the production of interferon-γ and tumour necrosis factor-α by bone marrow T lymphocytes from patients with lower risk myelodysplastic syndromes. Eur. J. Haematol. 2013, 90, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Navas, T.A.; Mohindru, M.; Estes, M.; Ma, J.Y.; Sokol, L.; Pahanish, P.; Parmar, S.; Haghnazari, E.; Zhou, L.; Collins, R.; et al. Inhibition of overactivated p38 MAPK can restore hematopoiesis in myelodysplastic syndrome progenitors. Blood 2006, 108, 4170–4177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, L.; Cripe, L.; Kantarjian, H.; Sekeres, M.A.; Parmar, S.; Greenberg, P.; Goldberg, S.L.; Bhushan, V.; Shammo, J.; Hohl, R.; et al. Randomized, dose-escalation study of the p38α MAPK inhibitor SCIO-469 in patients with myelodysplastic syndrome. Leukemia 2012, 27, 977–980. [Google Scholar] [CrossRef] [Green Version]
- Fattizzo, B.; Dunlop, A.; Ireland, R.; Kassam, S.; Yallop, D.; Mufti, G.; Marsh, J.; Kulasekararaj, A. Prevalence of small PNH clones and their prognostic significance in patients tested for unusual indications: A single center experience. Br. J. Haematol. 2019, 185, 125. [Google Scholar]
- Meletis, J.; Loukopoulos, D.; Mavrogianni, D.; Komninaka, V.; Meletis, C.; Korovesis, K.; Konstantopoulos, K.; Variami, E.; Apostolidou, E.; Terpos, E.; et al. Detection of CD55 and/or CD59 deficient red cell populations in patients with aplastic anaemia, myelodysplastic syndromes and myeloproliferative disorders. Haematologia 2001, 31, 7–16. [Google Scholar] [CrossRef]
- Passweg, J.R.; Giagounidis, A.A.N.; Simcock, M.; Aul, C.; Dobbelstein, C.; Stadler, M.; Ossenkoppele, G.; Hofmann, W.; Schilling, K.; Tichelli, A.; et al. Immunosuppressive therapy for patients with myelodysplastic syndrome: A prospective randomized multicenter phase III trial comparing antithymocyte globulin plus cyclosporine with best supportive care—SAKK 33/99. J. Clin. Oncol. 2011, 29, 303–309. [Google Scholar] [CrossRef] [Green Version]
- Lim, Z.Y.; Killick, S.; Germing, U.; Cavenagh, J.; Culligan, D.; Bacigalupo, A.; Marsh, J.; Mufti, G.J. Low IPSS score and bone marrow hypocellularity in MDS patients predict hematological responses to antithymocyte globulin. Leukemia 2007, 21, 1436–1441. [Google Scholar] [CrossRef]
- Maschek, H.; Kaloutsi, V.; Rodriguez-Kaiser, M.; Werner, M.; Choritz, H.; Mainzer, K.; Dietzfelbinger, M.; Georgii, A. Hypoplastic myelodysplastic syndrome: Incidence, morphology, cytogenetics, and prognosis. Ann. Hematol. 1993, 66, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Orazi, A.; Maher, A.; Heerema, N.A.; Haskins, M.S.; Neiman, R.S. Hypoplastic Myelodysplastic Syndromes Can Be Distinguished From Acquired Aplastic Anemia by CD34 and PCNA Immunostaining of Bone Marrow Biopsy Specimens. Am. J. Clin. Pathol. 1997, 107, 268–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, J.; Saunthararajah, Y.; Molldrem, J. Myelodysplastic syndrome and aplastic anemia: Distinct entities or diseases linked by a common pathophysiology? Semin. Hematol. 2000, 37, 15–29. [Google Scholar] [CrossRef]
- Calado, R.T.; Young, N.S. Telomere Diseases. N. Engl. J. Med. 2009, 361, 2353–2365. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Cazzola, M. The shadowlands of MDS: Idiopathic cytopenias of undetermined significance (ICUS) and clonal hematopoiesis of indeterminate potential (CHIP). Hematology 2015, 2015, 299–307. [Google Scholar] [CrossRef]
- Brown, A.L.; Churpek, J.E.; Malcovati, L.; Döhner, H.; Godley, L.A. Recognition of familial myeloid neoplasia in adults. Semin. Hematol. 2017, 54, 60–68. [Google Scholar] [CrossRef]
- Choi, J.W.; Fujino, M.; Ito, M. F-blast is a useful marker for differentiating hypocellular refractory anemia from aplastic anemia. Int. J. Hematol. 2002, 75, 257–260. [Google Scholar] [CrossRef]
- Kawankar, N.; Vundinti, B.R. Cytogenetic abnormalities in myelodysplastic syndrome: An overview. Hematology 2011, 16, 131–138. [Google Scholar] [CrossRef] [PubMed]
- DeZern, A.E.; Sekeres, M.A. The Challenging World of Cytopenias: Distinguishing Myelodysplastic Syndromes from Other Disorders of Marrow Failure. Oncologist 2014, 19, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Durrani, J.; Maciejewski, J.P. Idiopathic aplastic anemia vs hypocellular myelodysplastic syndrome. Hematology 2019, 2019, 97–104. [Google Scholar] [CrossRef]
- Blombery, P.; Fox, L.; Ryland, G.L.; Thompson, E.R.; Lickiss, J.; McBean, M.; Yerneni, S.; Hughes, D.; Greenway, A.; Mechinaud, F.; et al. Utility of clinical comprehensive genomic characterisation for diagnostic categorisation in patients presenting with hypocellular bone marrow failure syndromes. Haematologica 2020, 106, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.N.; Bejar, R. MDS overlap disorders and diagnostic boundaries. Blood 2019, 133, 1086–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, V. Treatment of low-risk myelodysplastic syndromes. Hematology 2016, 2016, 462–469. [Google Scholar] [CrossRef] [Green Version]
- Santini, V.; Schemenau, J.; Levis, A.; Balleari, E.; Sapena, R.; Adès, L.; Guerci, A.; Beyne-Rauzy, O.; Gourin, M.-P.; Cheze, S.; et al. Can the revised IPSS predict response to erythropoietic-stimulating agents in patients with classical IPSS low or intermediate-1 MDS? Blood 2013, 122, 2286–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobiasson, M.; Dybedahl, I.; Holm, M.S.; Karimi, M.; Brandefors, L.; Garelius, H.; Grövdal, M.; Høgh-Dufva, I.; Grønbæk, K.; Jansson, M.; et al. Limited clinical efficacy of azacitidine in transfusion-dependent, growth factor-resistant, low- and Int-1-risk MDS: Results from the nordic NMDSG08A phase II trial. Blood Cancer J. 2014, 4, e189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musto, P.; Maurillo, L.; Spagnoli, A.; Gozzini, A.; Rivellini, F.; Lunghi, M.; Villani, O.; Aloe-Spiriti, M.; Venditti, A.; Santini, V. Azacitidine for the treatment of lower risk myelodysplastic syndromes: A retrospective study of 74 patients enrolled in an Italian named patient program. Cancer 2010, 116, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.S.; Kim, Y.J.; Kim, Y.K.; Park, S.K.; Kim, H.G.; Kim, S.J.; Park, J.; Choi, C.W.; Do, Y.R.; Kim, I.; et al. Clinical Outcomes of Decitabine Treatment for Patients With Lower-Risk Myelodysplastic Syndrome on the Basis of the International Prognostic Scoring System. Clin. Lymphoma Myeloma Leuk. 2019, 19, 656–664. [Google Scholar] [CrossRef]
- Lee, B.; Kang, K.-W.; Jeon, M.J.; Yu, E.S.; Kim, D.S.; Choi, H.; Lee, S.R.; Sung, H.J.; Kim, B.S.; Choi, C.W.; et al. Comparison between 5-day decitabine and 7-day azacitidine for lower-risk myelodysplastic syndromes with poor prognostic features: A retrospective multicentre cohort study. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Seymour, J.F.; Bennett, J.M.; List, A.F.; Mufti, G.J.; Gore, S.D.; Fenaux, P.; Santini, V.; Hetzer, J.; Songer, S.; Skikne, B.S.; et al. Bone marrow hypocellularity does not affect tolerance or efficacy of azacitidine in patients with higher-risk myelodysplastic syndromes. Br. J. Haematol. 2014, 165, 49–56. [Google Scholar] [CrossRef]
- Nazha, A.; Narkhede, M.; Radivoyevitch, T.; Seastone, D.J.; Patel, B.J.; Gerds, A.T.; Mukherjee, S.; Kalaycio, M.; Advani, A.; Przychodzen, B.; et al. Incorporation of molecular data into the Revised International Prognostic Scoring System in treated patients with myelodysplastic syndromes. Leukemia 2016, 30, 2214–2220. [Google Scholar] [CrossRef]
- Zhou, M.; Wu, L.; Zhang, Y.; Mo, W.; Li, Y.; Chen, X.; Wang, C.; Pan, S.; Xu, S.; Zhou, W.; et al. Outcome of allogeneic hematopoietic stem cell transplantation for hypoplastic myelodysplastic syndrome. Int. J. Hematol. 2020, 112, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Risitano, A.M. Immunosuppressive therapies in the management of acquired immune-mediated marrow failures. Curr. Opin. Hematol. 2012, 19, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Deveaux, M.; De Witte, T.; Neukirchen, J.; Sekeres, M.A.; Brunner, A.M.; Roboz, G.J.; Steensma, D.P.; Bhatt, V.R.; Platzbecker, U.; et al. The use of immunosuppressive therapy in MDS: Clinical outcomes and their predictors in a large international patient cohort. Blood Adv. 2018, 2, 1765–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixit, A.; Chatterjee, T.; Mishra, P.; Choudhary, D.R.; Mahapatra, M.; Saxena, R.; Choudhry, V.P. Cyclosporin A in myelodysplastic syndrome: A preliminary report. Ann. Hematol. 2005, 84, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Gologan, R.; Ostroveanu, D.; Dobrea, C.; Gioadă, L. Hypoplastic myelodysplastic syndrome transformed in acute myeloid leukemia after androgens and cyclosporin. A treatment. Rom. J. Intern. Med. 2003, 41, 447–455. [Google Scholar] [PubMed]
- Cutler, C.; Lee, S.J.; Greenberg, P.; Deeg, H.J.; Pérez, W.S.; Anasetti, C.; Bolwell, B.J.; Cairo, M.S.; Gale, R.P.; Klein, J.P.; et al. A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: Delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood 2004, 104, 579–585. [Google Scholar] [CrossRef]
- Molldrem, J.J.; Caples, M.; Mavroudis, D.; Plante, M.; Young, N.S.; Barrett, A.J. Antithymocyte globulin for patients with myelodysplastic syndrome. Br. J. Haematol. 1997, 99, 699–705. [Google Scholar] [CrossRef] [Green Version]
- Jonásová, A.; Nnuwirtova, R.; Cermák, J.; Vozobulová, V.; Mocikovä, K.; Siaková, M.; Hocuová, I. Cyclosporin a therapy in hypoplastic MDS patients and certain refractory anaemias without hypoplastic bone marrow. Br. J. Haematol. 1998, 100, 304–309. [Google Scholar] [CrossRef]
- Raza, A.; Meyer, P.; Dutt, D.; Zorat, F.; Lisak, L.; Nascimben, F.; Du Randt, M.; Kaspar, C.; Goldberg, C.; Loew, J.; et al. Thalidomide produces transfusion independence in long-standing refractory anemias of patients with myelodysplastic syndromes. Blood 2001, 98, 958–965. [Google Scholar] [CrossRef] [Green Version]
- Molldrem, J.J.; Leifer, E.; Bahceci, E.; Saunthararajah, Y.; Rivera, M.; Dunbar, C.E.; Liu, J.; Nakamura, R.; Young, N.S.; Barrett, A.J. Antithymocyte globulin for treatment of the bone marrow failure associated with myelodysplastic syndromes. Ann. Intern. Med. 2002, 137, 156–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazji, S.; Giles, F.J.; Tsimberidou, A.M.; Estey, E.H.; Kantarjian, H.M.; O’Brien, S.A.; Kurzrock, R. Antithymocyte of globulin (ATG)-based therapy in patients with myelodysplastic syndromes. Leukemia 2003, 17, 2101–2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killick, S.B.; Mufti, G.; Cavenagh, J.D.; Mijovic, A.; Peacock, J.L.; Gordon-Smith, E.C.; Bowen, D.; Marsh, J.C.W. A pilot study of antithymocyte globulin (ATG) in the treatment of patients with ‘low-risk’ myelodysplasia. Br. J. Haematol. 2003, 120, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Shimamoto, T.; Tohyama, K.; Okamoto, T.; Uchiyama, T.; Mori, H.; Tomonaga, M.; Asano, Y.; Niho, Y.; Teramura, M.; Mizoguchi, H.; et al. Cyclosporin A therapy for patients with myelodysplastic syndrome: Multicenter pilot studies in Japan. Leuk. Res. 2003, 27, 783–788. [Google Scholar] [CrossRef]
- Joachim Deeg, H.; Jiang, P.Y.Z.; Holmberg, L.A.; Scott, B.; Petersdorf, E.W.; Appelbaum, F.R. Hematologic responses of patients with MDS to antithymocyte globulin plus etanercept correlate with improved flow scores of marrow cells. Leuk. Res. 2004, 28, 1177–1180. [Google Scholar] [CrossRef]
- Raza, A.; Candoni, A.; Khan, U.; Lisak, L.; Tahir, S.; Silvestri, F.; Billmeier, J.; Alvi, M.I.; Mumtaz, M.; Gezer, S.; et al. Remicade as TNF Suppressor in Patients with Myelodysplastic Syndromes. Leuk. Lymphoma 2004, 45, 2099–2104. [Google Scholar] [CrossRef]
- Stadler, M.B.; Germing, U.; Kliche, K.-O.; Josten, K.M.; Kuse, R.; Hofmann, W.-K.; Schrezenmeier, H.; Novotny, J.E.; Anders, O.; Eimermacher, H.; et al. A prospective, randomised, phase II study of horse antithymocyte globulin vs rabbit antithymocyte globulin as immune-modulating therapy in patients with low-risk myelodysplastic syndromes. Leukemia 2004, 18, 460–465. [Google Scholar] [CrossRef]
- Broliden, P.A.; Dahl, I.-M.; Hast, R.; Johansson, B.; Juvonen, E.; Kjeldsen, L.; Porwit-MacDonald, A.; Sjoo, M.; Tangen, J.-M.; Uggla, B.; et al. Antithymocyte globulin and cyclosporine A as combination therapy for low-risk non-sideroblastic myelodysplastic syndromes. Haematologica 2006, 91, 667–770. [Google Scholar]
- Sloand, E.M.; Wu, C.O.; Greenberg, P.; Young, N.; Barrett, J. Factors Affecting Response and Survival in Patients With Myelodysplasia Treated With Immunosuppressive Therapy. J. Clin. Oncol. 2008, 26, 2505–2511. [Google Scholar] [CrossRef]
- Garg, R.; Faderl, S.; Garcia-Manero, G.; Cortes, J.; Koller, C.; Huang, X.; York, S.; Pierce, S.; Brandt, M.; Beran, M.; et al. Phase II study of rabbit anti-thymocyte globulin, cyclosporine and granulocyte colony-stimulating factor in patients with aplastic anemia and myelodysplastic syndrome. Leukemia 2009, 23, 1297–1302. [Google Scholar] [CrossRef] [Green Version]
- Scott, B.L.; Ramakrishnan, A.; Fosdal, M.; Storer, B.; Becker, P.; Petersdorf, S.; Deeg, H.J. Anti-thymocyte globulin plus etanercept as therapy for myelodysplastic syndromes (MDS): A phase II study. Br. J. Haematol. 2010, 149, 706–710. [Google Scholar] [CrossRef] [Green Version]
- Sloand, E.M.; Olnes, M.J.; Shenoy, A.; Weinstein, B.; Boss, C.; Loeliger, K.; Wu, C.O.; More, K.; Barrett, A.J.; Scheinberg, P.; et al. Alemtuzumab Treatment of Intermediate-1 Myelodysplasia Patients Is Associated With Sustained Improvement in Blood Counts and Cytogenetic Remissions. J. Clin. Oncol. 2010, 28, 5166–5173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.-F.; Qin, T.-J.; Zhang, Y.; Liu, K.-Q.; Hao, Y.-S.; Xiao, Z.J. Cyclosporine A in combination with thalidomide for the treatment of patients with myelodysplastic syndromes. J. Exp. Hematol. 2010, 31, 451–455. [Google Scholar]
- Li, X.; Qi, Z.; Qiusheng, C.; Li, X.; Luxi, S.; Lingyun, W. The use of selective immunosuppressive therapy on myelodysplastic syndromes in targeted populations results in good response rates and avoids treatment-related disease progression. Am. J. Hematol. 2012, 87, 26–31. [Google Scholar]
- Kadia, T.M.; Borthakur, G.; Garcia-Manero, G.; Faderl, S.; Jabbour, E.; Estrov, Z.; York, S.; Huang, X.; Pierce, S.; Brandt, M.; et al. Final results of the phase II study of rabbit anti-thymocyte globulin, ciclosporin, methylprednisone, and granulocyte colony-stimulating factor in patients with aplastic anaemia and myelodysplastic syndrome. Br. J. Haematol. 2012, 157, 312–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haider, M.; Al Ali, N.; Padron, E.; Epling-Burnette, P.; Lancet, J.; List, A.; Komrokji, R. Immunosuppressive Therapy: Exploring an Underutilized Treatment Option for Myelodysplastic Syndrome. Clin. Lymphoma Myeloma Leuk. 2016, 16, S44–S48. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Fenaux, P.; Sekeres, M.A.; Becker, P.S.; Boruchov, A.; Bowen, D.; Hellstrom-Lindberg, E.; Larson, R.A.; Lyons, R.M.; Muus, P.; et al. Safety and Efficacy of Romiplostim in Patients With Lower-Risk Myelodysplastic Syndrome and Thrombocytopenia. J. Clin. Oncol. 2010, 28, 437–444. [Google Scholar] [CrossRef] [Green Version]
- Sekeres, M.A.; Kantarjian, H.; Fenaux, P.; Becker, P.; Boruchov, A.; Guerci-Bresler, A.; Hu, K.; Franklin, J.; Wang, Y.-M.C.; Berger, D. Subcutaneous or intravenous administration of romiplostim in thrombocytopenic patients with lower risk myelodysplastic syndromes. Cancer 2010, 117, 992–1000. [Google Scholar] [CrossRef]
- Oliva, E.N.; Alati, C.; Santini, V.; Poloni, A.; Molteni, A.; Niscola, P.; Salvi, F.; Sanpaolo, G.; Balleari, E.; Germing, U.; et al. Eltrombopag versus placebo for low-risk myelodysplastic syndromes with thrombocytopenia (EQoL-MDS): Phase 1 results of a single-blind, randomised, controlled, phase 2 superiority trial. Lancet Haematol. 2017, 4, e127–e136. [Google Scholar] [CrossRef]
- Wang, E.S.; Lyons, R.M.; Larson, R.A.; Gandhi, S.; Liu, D.; Matei, C.; Scott, B.L.; Hu, K.; Yang, A.S. A randomized, double-blind, placebo-controlled phase 2 study evaluating the efficacy and safety of romiplostim treatment of patients with low or intermediate-1 risk myelodysplastic syndrome receiving lenalidomide. J. Hematol. Oncol. 2012, 5, 71. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, P.L.; Garcia-Manero, G.; Moore, M.; Damon, L.; Roboz, G.; Hu, K.; Yang, A.S.; Franklin, J. A randomized controlled trial of romiplostim in patients with low- or intermediate-risk myelodysplastic syndrome receiving decitabine. Leuk. Lymphoma 2012, 54, 321–328. [Google Scholar] [CrossRef]
- Fenaux, P.; Muus, P.; Kantarjian, H.; Lyons, R.M.; Larson, R.A.; Sekeres, M.A.; Becker, P.S.; Orejudos, A.; Franklin, J. Romiplostim monotherapy in thrombocytopenic patients with myelodysplastic syndromes: Long-term safety and efficacy. Br. J. Haematol. 2017, 178, 906–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente, A.; Patel, B.A.; Gutierrez-Rodrigues, F.; Groarke, E.M.; Giudice, V.; Lotter, J.; Feng, X.; Kajigaya, S.; Weinstein, B.; Barranta, E.; et al. Eltrombopag monotherapy can improve hematopoiesis in patients with low to intermediate risk-1 myelodysplastic syndrome. Haematologica 2020, 105, 2785–2794. [Google Scholar] [CrossRef] [PubMed]
- Townsley, D.M.; Dumitriu, B.; Liu, D.; Biancotto, A.; Weinstein, B.; Chen, C.; Hardy, N.; Mihalek, A.D.; Lingala, S.; Kim, Y.J.; et al. Danazol Treatment for Telomere Diseases. N. Engl. J. Med. 2016, 374, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Colunga-Pedraza, P.R.; Colunga-Pedraza, J.E.; Garza-Ledezma, M.A.; Jaime-Pérez, J.C.; Cantú-Rodríguez, O.G.; Gutiérrez-Aguirre, C.H.; Rendón-Ramírez, E.J.; López-García, Y.K.; Lozano-Morales, R.E.; Gómez-De León, A.; et al. Danazol as First-Line Therapy for Myelodysplastic Syndrome. Clin. Lymphoma, Myeloma Leuk. 2018, 18, e109–e113. [Google Scholar] [CrossRef]
- Chan, G.; DiVenuti, G.; Miller, K. Danazol for the treatment of thrombocytopenia in patients with myelodysplastic syndrome. Am. J. Hematol. 2002, 71, 166–171. [Google Scholar] [CrossRef]
- Andrew, M.; Brunner, J.E.; Kimmo, P.; Steve, K.; Norbert, V.; Sebastian, S.; Guillermo, G.-M.; Martin, W.; Jeroen, J.; Elie, T.; et al. Efficacy and Safety of Sabatolimab (MBG453) in Combination with Hypomethylating Agents (HMAs) in Patients with Acute Myeloid Leukemia (AML) and High-Risk Myelodysplastic Syndrome (HR-MDS): Updated Results from a Phase 1b Study. In Proceedings of the Oral Present 62nd ASH Annu Meet Expo (Online meeting), 5–8 December 2020. [Google Scholar]
- Sallman, D.A.; Donnellan, W.B.; Asch, A.S.; Lee, D.J.; Al Malki, M.; Marcucci, G.; Pollyea, D.A.; Kambhampati, S.; Komrokji, R.S.; Van Elk, J.; et al. The first-in-class anti-CD47 antibody Hu5F9-G4 is active and well tolerated alone or with azacitidine in AML and MDS patients: Initial phase 1b results. J. Clin. Oncol. 2019, 37, 7009. [Google Scholar] [CrossRef]
- Kobayashi, T.; Nannya, Y.; Ichikawa, M.; Oritani, K.; Kanakura, Y.; Tomita, A.; Kiyoi, H.; Kobune, M.; Kato, J.; Kawabata, H.; et al. A nationwide survey of hypoplastic myelodysplastic syndrome (a multicenter retrospective study). Am. J. Hematol. 2017, 92, 1324–1332. [Google Scholar] [CrossRef] [Green Version]
- Jerez, A.; Sugimoto, Y.; Makishima, H.; Verma, A.; Jankowska, A.M.; Przychodzen, B.; Visconte, V.; Tiu, R.V.; O’Keefe, C.L.; Mohamedali, A.M.; et al. Loss of heterozygosity in 7q myeloid disorders: Clinical associations and genomic pathogenesis. Blood 2012, 119, 6109–6117. [Google Scholar] [CrossRef]
- Hunt, A.A.; Khan, A.B.; Potter, V.T.; McLornan, D.; Raj, K.; De Lavallade, H.; Pagliuca, A.; Smith, A.E.; Marsh, J.C.; Kulasekararaj, A.; et al. Hypoplastic MDS Is a Distinct Clinico-Pathological Entity with Somatic Mutations Frequent in Patients with Prior Aplastic Anaemia with Favorable Clinical Outcome. Blood 2014, 124, 3269. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Marsh, J.C.W.; Gutierrez-Rodrigues, F.; Cooper, J.; Jiang, J.; Gandhi, S.; Kajigaya, S.; Feng, X.; Ibanez, M.D.P.F.; Donaires, F.S.; Da Silva, J.P.L.; et al. Heterozygous RTEL1 variants in bone marrow failure and myeloid neoplasms. Blood Adv. 2018, 2, 36–48. [Google Scholar] [CrossRef]
- Borges, D.D.P.; Dos Santos, A.W.A.; Paier, C.R.K.; Ribeiro, H.L.; Costa, M.B.; Farias, I.R.; De Oliveira, R.T.G.; França, I.G.D.F.; Cavalcante, G.M.; Magalhães, S.M.M.; et al. Prognostic importance of Aurora Kinases and mitotic spindle genes transcript levels in Myelodysplastic syndrome. Leuk. Res. 2018, 64, 61–70. [Google Scholar] [CrossRef]
- Ribeiro, H.L.; Maia, A.R.S.; De Oliveira, R.T.G.; Costa, M.B.; Farias, I.R.; De Paula Borges, D.; De Sousa, J.C.; Magalhães, S.M.M.; Pinheiro, R.F. DNA repair gene expressions are related to bone marrow cellularity in myelodysplastic syndrome. J. Clin. Pathol. 2017, 70, 970–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heredia, F.F.; De Sousa, J.C.; Júnior, H.L.R.; Carvalho, A.F.; Magalhães, S.M.M.; Pinheiro, R.F. Proteins related to the spindle and checkpoint mitotic emphasize the different pathogenesis of hypoplastic MDS. Leuk. Res. 2014, 38, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Van De Loosdrecht, A.A.; Alhan, C.; Ossenkoppele, G.J.; Westers, T.M.; Bontkes, H.J. Role of immune responses in the pathogenesis of low-risk MDS and high-risk MDS: Implications for immunotherapy. Br. J. Haematol. 2011, 153, 568–581. [Google Scholar] [CrossRef]
- Glenthøj, A.; Ørskov, A.D.; Hansen, J.W.; Hadrup, S.R.; O’Connell, C.; Grønbæk, K. Immune Mechanisms in Myelodysplastic Syndrome. Int. J. Mol. Sci. 2016, 17, 944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nand, S.; Godwin, J.E. Hypoplastic myelodysplastic syndrome. Cancer 1988, 62, 958–964. [Google Scholar] [CrossRef]
- Yue, G.; Hao, S.; Fadare, O.; Baker, S.P.; Pozdnyakova, O.; Galili, N.; Woda, B.A.; Raza, A.; Wang, S. Hypocellularity in myelodysplastic syndrome is an independent factor which predicts a favorable outcome. Leuk. Res. 2008, 32, 553–558. [Google Scholar] [CrossRef]
- Tong, W.G.; Quintás-Cardama, A.; Kadia, T.; Borthakur, G.; Jabbour, E.; Ravandi, F.; Faderl, S.; Wierda, W.; Pierce, S.; Shan, J.; et al. Predicting survival of patients with hypocellular myelodysplastic syndrome: Development of a disease-specific prognostic score system. Cancer 2012, 118, 4462–4470. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Properties | Normo/Hypercellular MDS | hMDS | AA |
|---|---|---|---|
| Clinical Features | |||
| Median Age | +++ | ++ | +/− |
| Male/Female Ratio | >1 | = | =/<1 |
| Bleeding | +/− | + | ++ |
| Transfusion-Dependence | +/− | + | ++ |
| Infections | +/− | +/− | ++ |
| Laboratory Features | |||
| Cytopenia and Macrocytosis | + | ++ | ++ |
| LDH | +/− | + | ++ |
| BM Blasts | =/+ | − | −− |
| Cytogenetic/Molecular Alterations | ++ | +/− | rare |
| Associated Conditions | |||
| PNH Clone | +/− | + | ++ |
| LGL Clone | + | ++ | +/− |
| Extrahematologic Autoimmunity | − | ++ | +/- |
| Prognosis | |||
| Leukemic Evolution | + | +/− | − |
| Survival | −− | +/− | +/− |
| Mutations | Normo/Hypercellular MDS | hMDS | AA |
|---|---|---|---|
| Cytogenetic Abnormalities, n | ++ | +/− | rare |
| Cytogenetic Abnormalities, Type | Low to high risk | Mostly low risk | Mostly low risk |
| Somatic Mutations, n | ++ | +/− | +/− |
| Somatic Mutations, Clone Size (VAF) | +++ | ++ | + |
| Somatic Mutations, Type | |||
| Splicing SF3B1, SRSF2, U2AF1, ZRSR2 | +++ | + | +/− |
| DNA Methylation DNMT3A, TET2, IDH1, IDH2 | ++ | + | +/− |
| Chromatin Modification ASXL1, EZH2, KDM6A | ++ | + | +/− |
| Cohesin STAG2 | + | +/− | rare |
| Tumor Suppressor TP53 | + | +/− | rare |
| Signaling CBL, FLT3, JAK2, KIT, NRAS, KRAS | +/− | +/− | rare |
| Transcription RUNX1, CEBPA, ETV6, GATA2, NPM1 | RUNX1 =/++; others = +/− | +/− | rare |
| Pathogenic germline RTEL1 mutations | − | +/− | + |
| Types | Normocellular/Hypercellular MDS | hMDS | AA | References |
|---|---|---|---|---|
| T-cytotoxic cells (CTLs) | Increased and oligoclonal. In high-risk patients, IFN-γ-producing CTLs decrease favoring leukemia evolution. | Increased and clonal; produce interferon-gamma (IFN-γ) and decrease after response to IST. | Highly increased and polyclonal. Higher in patients not responding to IST and at relapse. | [14,22,23,24] |
| T-CD4+ cells Th and Tregs | Increased T regs collaborate in the suppression of immune surveillance and leukemic evolution. | Increased and polyclonal Th cells producing IFN-γ. Tregs are reduced and correlate with dyserythropoiesis. | Reduced Tregs and effector memory Tregs. | [23,25,26] |
| LGL clones | Increased polyclonal and oligoclonal (both T-LGL and NK-LGL) more than in AA. | Increased in AA; STAT3 mutation correlates with better response to IST. | [14,27,28] | |
| B-cells and autoantibodies | Reduced B-cells and B-related gene expression, possibly recovering after therapy. Autoantibodies are present in up to 20–34.4% of patients) but true autoimmunity only in 4%. | B-cells and IL-10 producing B-regs are reduced and correlate with severity and response to IST. | [14,28,29,30,31,32,33,34] | |
| Macrophages | Increased number and activity and upregulation of TLR4, correlating with apoptosis. Increased number of TNFα producing intermediate monocytes (CD14bright/CD16−, proinflammatory cells). Impaired capacity to engulf the apoptotic dysplastic cells. | Increased TNFα producing macrophages. Their depletion induced recovery and reduced mortality in murine models. | [35,36] | |
| Mastocytes | Increased tryptase-producing-MCs; tryptase is a mitogen that contributes to hypercellularity. | Increased chymase-producing MCs. Chymase induces apoptosis. | Increased polyclonal benign MCs. Their persistence after IST correlates with poor outcomes. | [37,38] |
| Mesenchymal stem cells (MSCs) | Reduced expression of immunomodulatory cytokines. MSCs show an ineffective production of osteopontin, angiopoietin, Jagged1, and stromal-derived factor 1-CXCL-12, failing to support hematopoiesis. MDS-MSCs display genetic abnormalities associated with the 5q- syndrome or with a high-risk karyotype. | Reduced angiogenic and osteogenic potential, and adipogenicity is increased. AA-MSCs contribute to Treg/Th17 imbalance. | [39] | |
| PNH clone | Present in 20% of patients and correlates with better survival and response to HSCT | Present in up to 40% of patients and correlates with higher LDH levels, deeper cytopenias, better response to IST, and survival. | Present in up to 60% of patients and correlates with higher LDH levels, better response to IST, and better survival. | [6,40] |
| Cytokine levels | Proinflammatory cytokines (IFN-γ and TNFα) are increased. In high-risk patients, IL-10 is increased and contributes to the suppression of leukemic evolution. | Proinflammatory cytokines and TGF-b are increased inducing bone marrow failure. IL-10 is reduced and fails to suppress inflammation. | Proinflammatory cytokines and TGF-b are highly increased. IL-10 is increased as a rebound effect. | [7,10,22,41,42,43,44,45,46,47,48,49,50,51,52,53] |
| Reference | N | Study Design | Treatment | hMDS % | ORR | Time to Response (m) | Response Duration (m) |
|---|---|---|---|---|---|---|---|
| [117] | 25 | Phase II trial | ATG | - | 44 | - | 10 |
| [118] | 17 | Retrospective | CyA | 53 | 82 | 3 | - |
| [119] | 83 | Pilot study | Thalidomide | 15 | 19 | 4 | 10 |
| [120] | 61 | Phase II trial | ATG | 38 | 34 | 2.5 | 36 |
| [34] | 11 | Phase I/II trial | CyA | 100 | 73 | 2.3 | 58 |
| [121] | 32 | Phase II trial | ATG + CyA | - | 26 | 2.5 | 12 |
| [122] | 30 | Pilot study | ATG | 27 | 33 | - | 15 |
| [123] | 50 | Retrospective | CyA | 20 | 60 | 1.8 | - |
| [124] | 15 | Phase II trial | ATG + etanercept | 7 | 46 | - | 24–36 |
| [125] | 37 | Pilot study | Infliximab | - | 22 | - | 6-12 |
| [126] | 35 | Phase II trial | ATG | 11 | 34 | 3 | 9 |
| [114] | 19 | Phase II trial | CyA | 21 | 58 | 2.5 | - |
| [127] | 25 | Phase II trial | ATG + CyA | 20 | 24 | 2 | 7 |
| [128] | 129 | Retrospective | ATG/CyA/ATG + CyA | 33 | 30 (24/8/48) | 4 | 36 |
| [129] | 15 | Phase II trial | ATG + CyA | - | 33 | 3.7 | - |
| [130] | 25 | Phase II trial | ATG + etanercept | - | 56 | 2 | 5–36 |
| [131] | 31 | Phase I/II trial | Alemtuzumab | 35 | 68 | 3 | - |
| [89] | 45 | Phase III trial | ATG + CyA | 20 | 29 | - | 16 |
| [132] | 37 | Phase II trial | CyA + thalidomide | 14 | 57 | 1.8 | 22 |
| [133] | 71 | Phase II trial | CyA | 48 | 77 | 1.5 | 24 |
| [134] | 24 | Phase II trial | ATG + CyA | - | 25 | 4 | - |
| [135] | 66 | Retrospective | ATG/CyA/ATG + CyA | - | 42 | - | 12 |
| [113] | 207 | Retrospective | Any | 26 | 49 | 2.5 | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fattizzo, B.; Serpenti, F.; Barcellini, W.; Caprioli, C. Hypoplastic Myelodysplastic Syndromes: Just an Overlap Syndrome? Cancers 2021, 13, 132. https://doi.org/10.3390/cancers13010132
Fattizzo B, Serpenti F, Barcellini W, Caprioli C. Hypoplastic Myelodysplastic Syndromes: Just an Overlap Syndrome? Cancers. 2021; 13(1):132. https://doi.org/10.3390/cancers13010132
Chicago/Turabian StyleFattizzo, Bruno, Fabio Serpenti, Wilma Barcellini, and Chiara Caprioli. 2021. "Hypoplastic Myelodysplastic Syndromes: Just an Overlap Syndrome?" Cancers 13, no. 1: 132. https://doi.org/10.3390/cancers13010132
APA StyleFattizzo, B., Serpenti, F., Barcellini, W., & Caprioli, C. (2021). Hypoplastic Myelodysplastic Syndromes: Just an Overlap Syndrome? Cancers, 13(1), 132. https://doi.org/10.3390/cancers13010132