
Targeting CSF1R Alone or in Combination with PD1 in Experimental Glioma

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
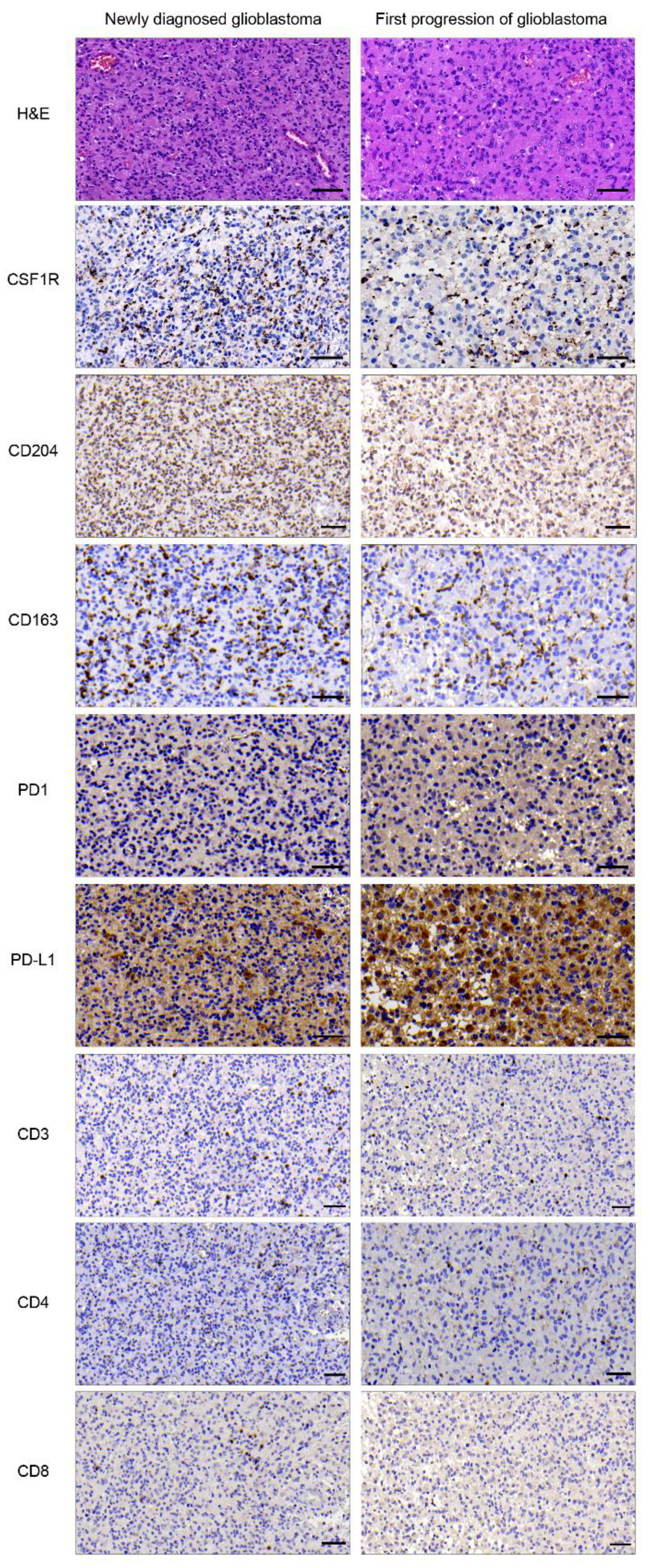
2.1. Expression of CSF1R, CD204, CD163, PD1, and PD-L1 in Primary and Progressive Glioblastoma
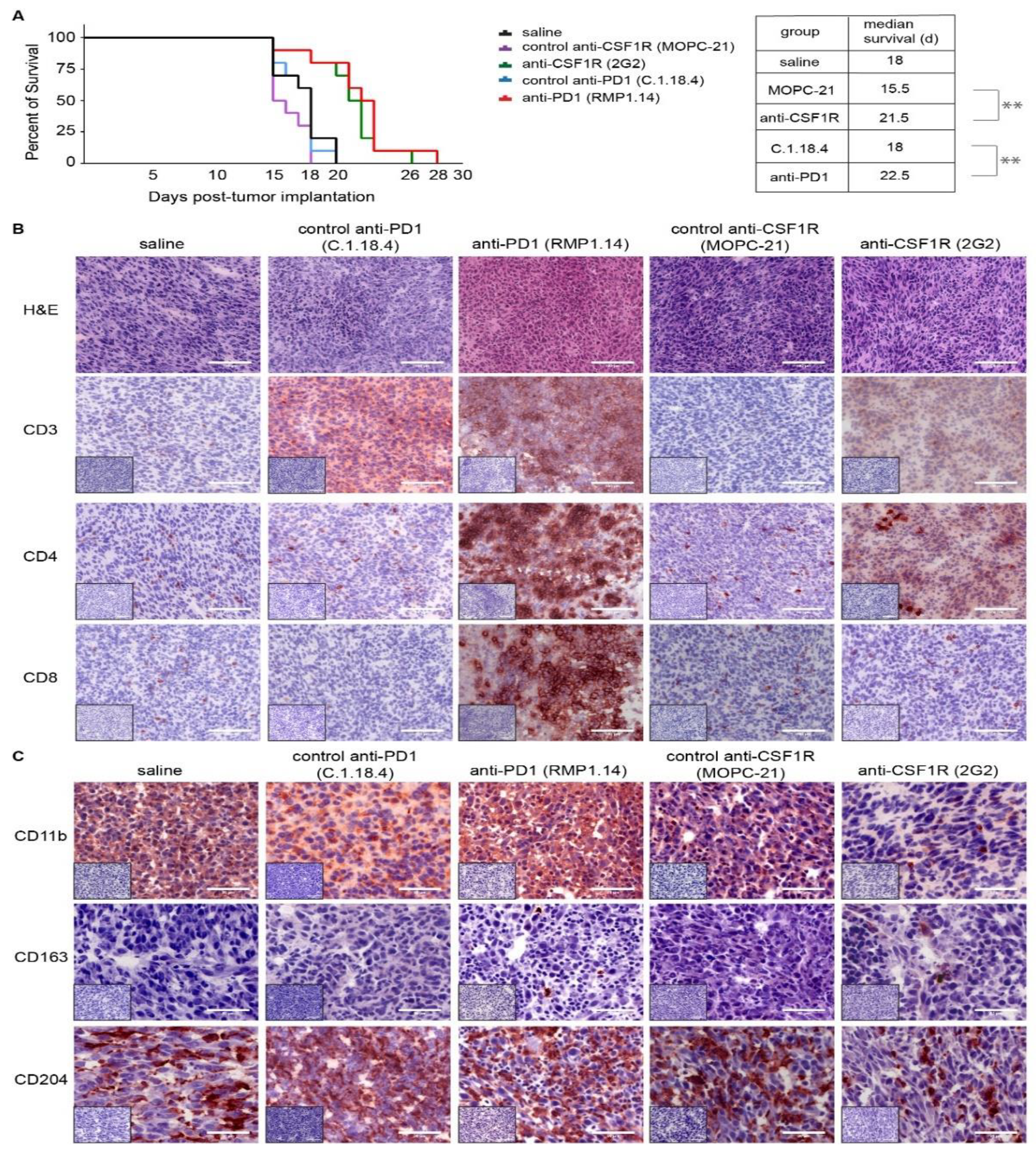
2.2. Monotherapies with PD1 Antibody and CSF1R Antibody Prolong the Latency until the Onset of Neurological Symptoms In Vivo and Lead to an Altered Immune Signature in the Microenvironment
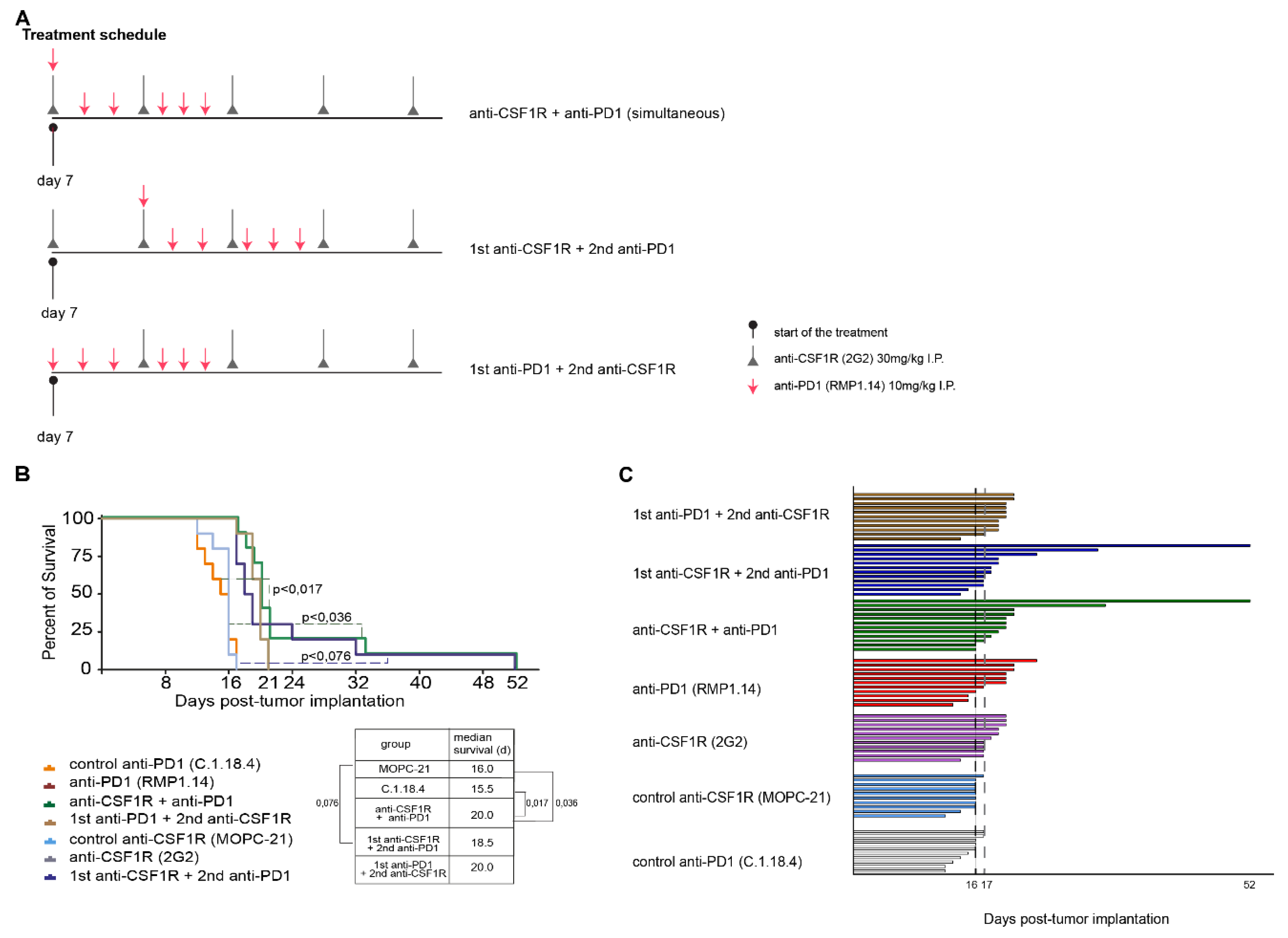
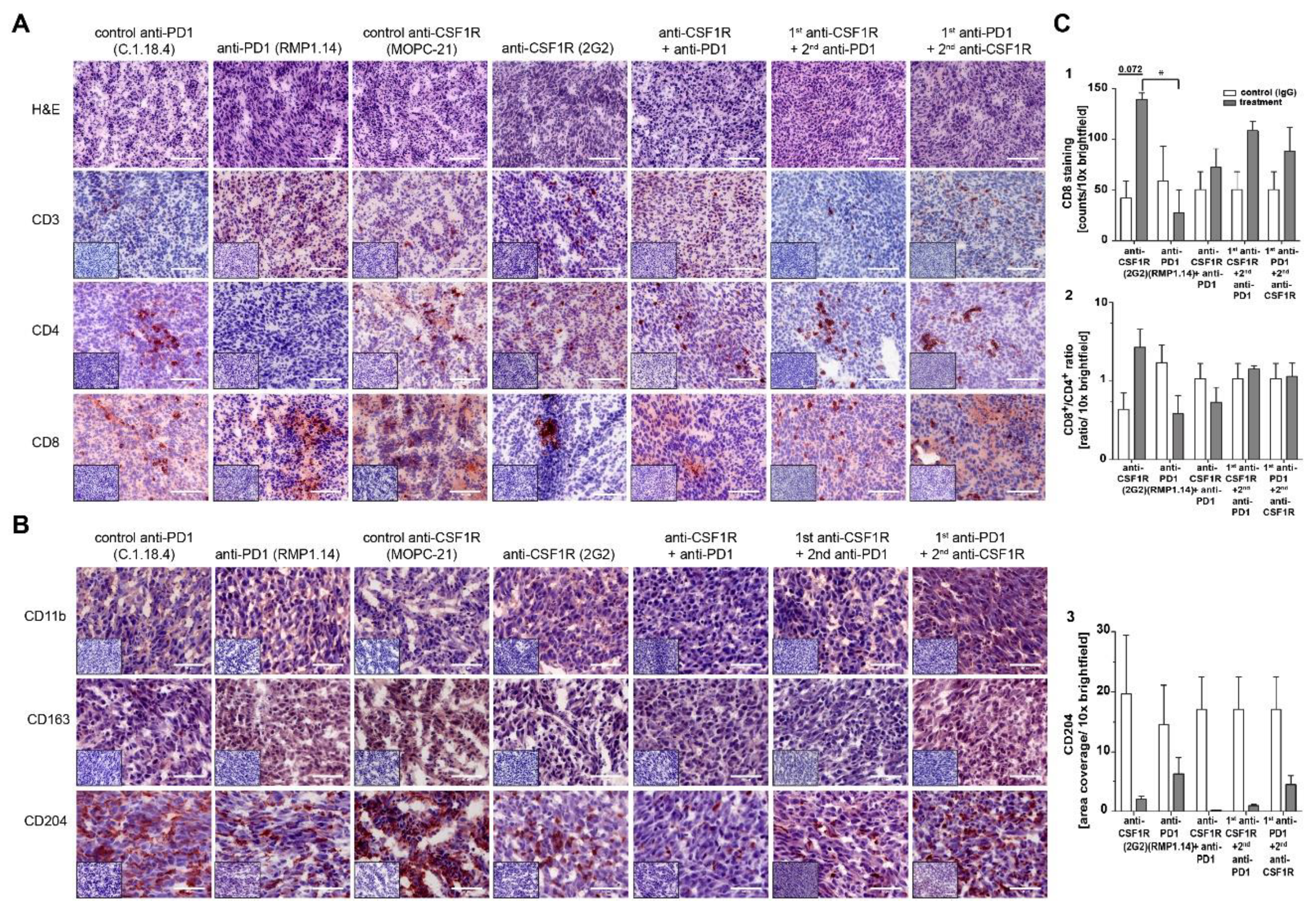
2.3. Combinations of Anti-CSF1R and Anti-PD1 Antibodies Lead to Longer Term Surviving Animals In Vivo If Applied Simultaneously or Sequentially, but Only If PD1 Blockade Follows CSF-1R Blockade
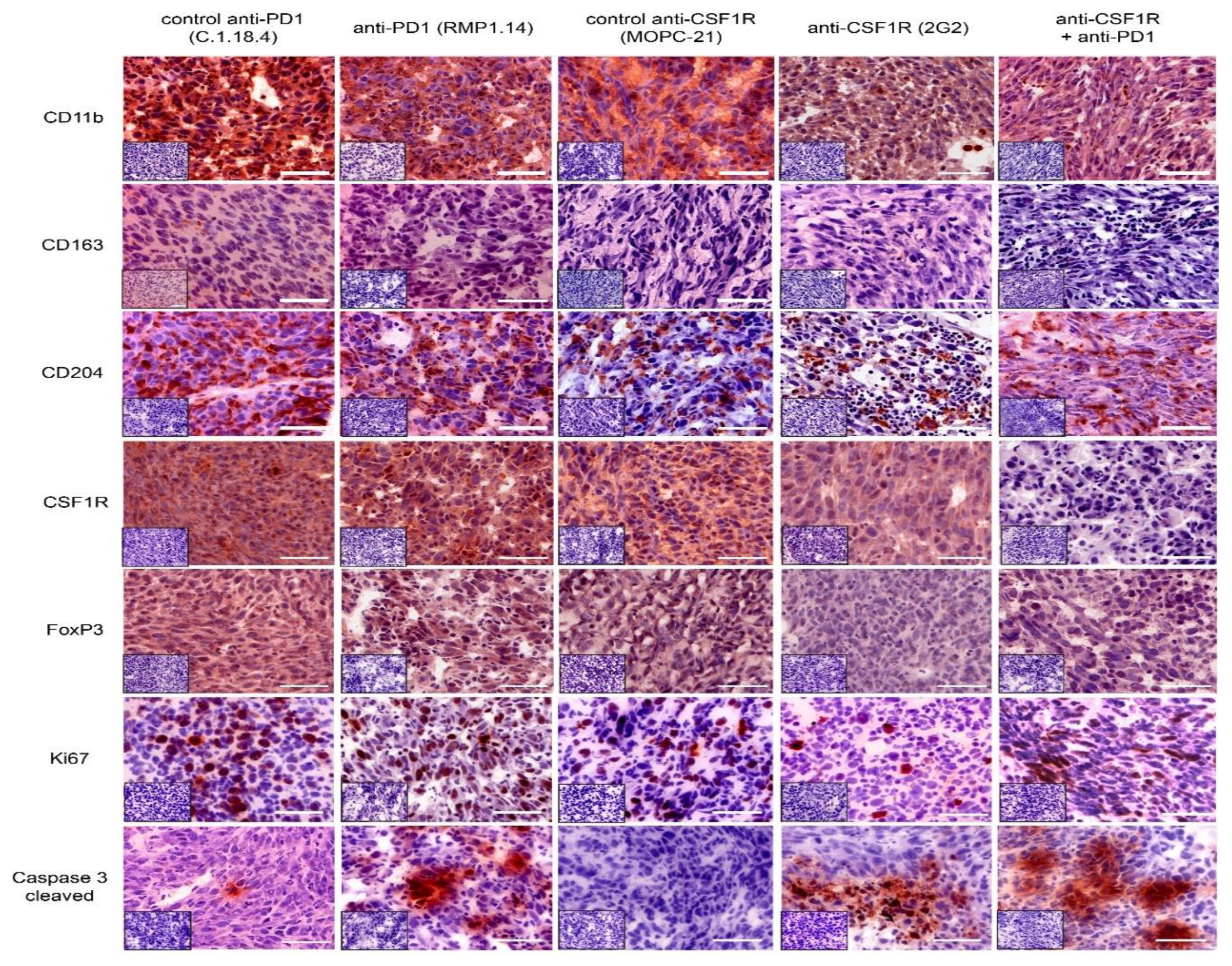
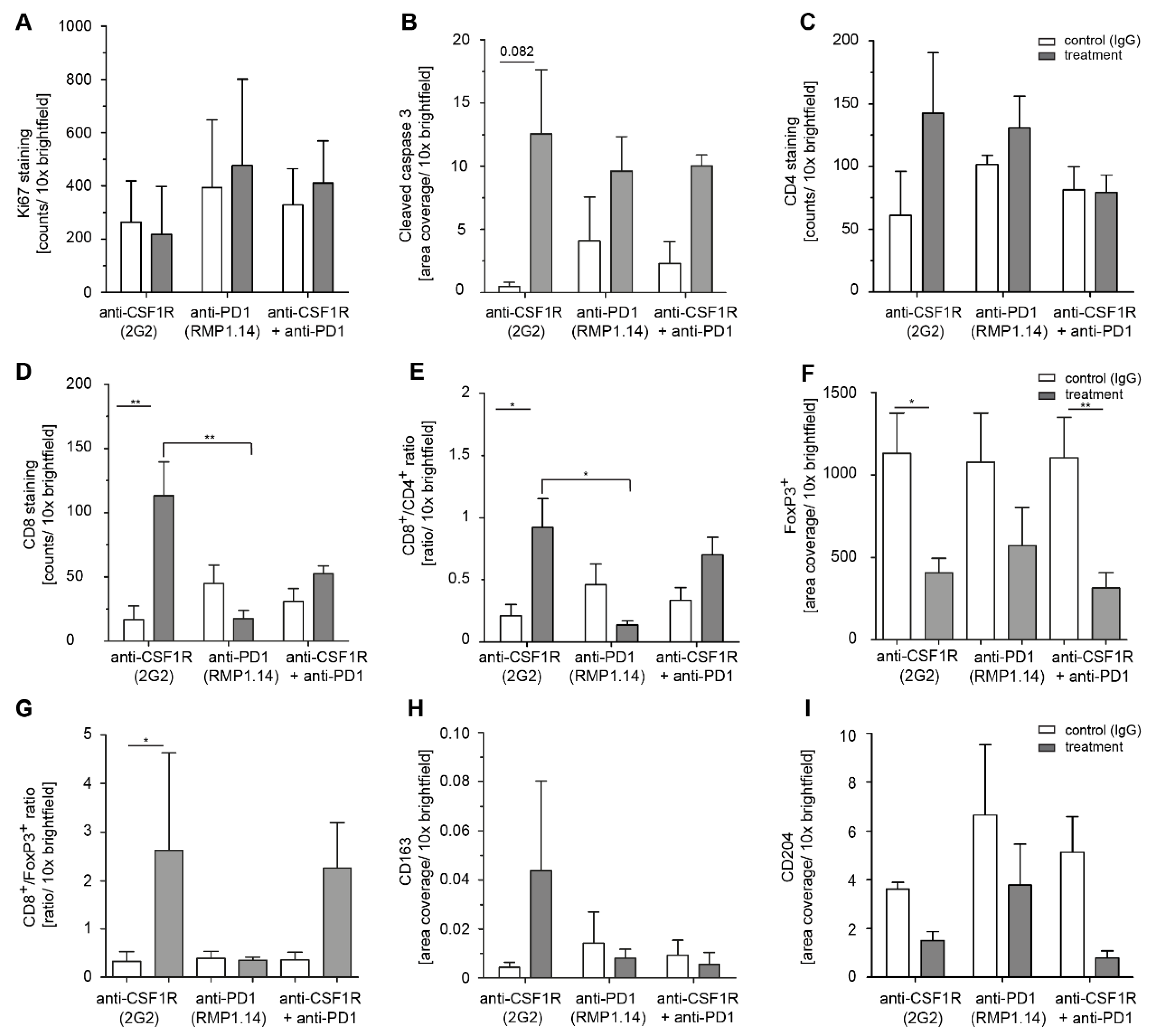
2.4. Combined Anti-CSF1R and PD1 Antibodies Lead to Decreased CSF1R and Increased CD8/CD4 and CD8/FoxP3 Ratios in Post-Treatment Tissues
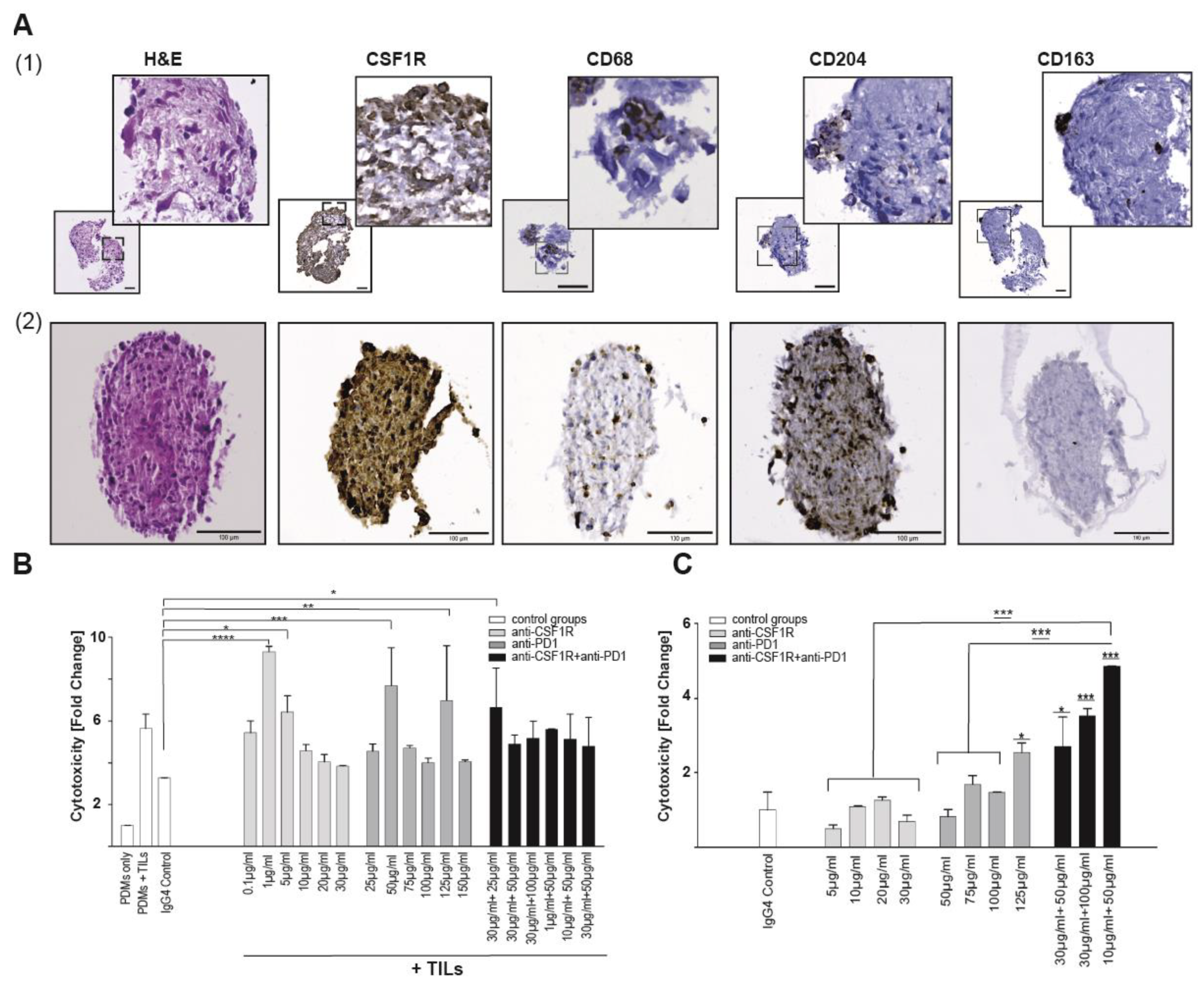
2.5. Coinhibition of CSF1R and PD1 Enhances Cytotoxicity in Glioblastoma PDM/TILs Co-Cultures Ex Vivo
3. Discussion
4. Materials and Methods
4.1. SMA-560 Cell Implantation into Syngeneic VM/Dk Mice
4.2. Treatment Schedules In Vivo
4.3. Scoring of Experimental Animals
4.4. Immunohistochemistry of Murine Tumor Samples
4.5. Immunohistochemistry of Human Glioblastoma Samples
4.6. Patient-Derived Microtumors (PDMs) and Tumor Infiltrating Lymphocytes (TILs)
4.7. Flow Cytometry for the Characterization of PDM-Derived TILs
4.8. Coculture Cytotoxicity Assay
4.9. Immunohistochemistry of PDMs
4.10. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Hegi, M.E.; Mason, W.P.; Bent, M.J.V.D.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-Dense Temozolomide for Newly Diagnosed Glioblastoma: A Randomized Phase III Clinical Trial. J. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef] [Green Version]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy–Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs. Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Vecchione-Koval, T.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro-Oncology 2017, 19 (Suppl. 5), v1–v88. [Google Scholar] [CrossRef] [Green Version]
- Kurz, S.C.; Wen, P.Y. Quo Vadis—Do Immunotherapies Have a Role in Glioblastoma? Curr. Treat. Options Neurol. 2018, 20, 14. [Google Scholar] [CrossRef]
- Wischhusen, J.; Friese, M.A.; Mittelbronn, M.; Meyermann, R.; Weller, M. HLA-E Protects Glioma Cells from NKG2D-Mediated Immune Responses In Vitro: Implications for Immune Escape In Vivo. J. Neuropathol. Exp. Neurol. 2005, 64, 523–528. [Google Scholar] [CrossRef]
- Roth, P.; Mittelbronn, M.; Wick, W.; Meyermann, R.; Tatagiba, M.; Weller, M. Malignant Glioma Cells Counteract Antitumor Immune Responses through Expression of Lectin-Like Transcript-1. Cancer Res. 2007, 67, 3540–3544. [Google Scholar] [CrossRef] [Green Version]
- Kinjyo, I.; Inoue, H.; Hamano, S.; Fukuyama, S.; Yoshimura, T.; Koga, K.; Takaki, H.; Himeno, K.; Takaesu, G.; Kobayashi, T.; et al. Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-β1. J. Exp. Med. 2006, 203, 1021–1031. [Google Scholar] [CrossRef]
- Wei, J.; Barr, J.; Kong, L.-Y.; Wang, Y.; Wu, A.; Sharma, A.K.; Gumin, J.; Henry, V.; Colman, H.; Priebe, W.; et al. Glioblastoma Cancer-Initiating Cells Inhibit T-Cell Proliferation and Effector Responses by the Signal Transducers and Activators of Transcription 3 Pathway. Mol. Cancer Ther. 2010, 9, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti-PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A. Tumor Immunotherapy Directed at PD-1. N. Engl. J. Med. 2012, 366, 2517–2519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neubert, N.J.; Schmittnaegel, M.; Bordry, N.; Nassiri, S.; Wald, N.; Martignier, C.; Tillé, L.; Homicsko, K.; Damsky, W.; Hajjami, H.M.-E.; et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl. Med. 2018, 10, eaan3311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, A.M.; Collier, L.S.; Rodriguez, F.J.; Tieu, C.; Larson, J.D.; Halder, C.; Mahlum, E.; Kollmeyer, T.M.; Akagi, K.; Sarkar, G.; et al. Sleeping Beauty—Mediated Somatic Mutagenesis Implicates CSF1 in the Formation of High-Grade Astrocytomas. Cancer Res. 2010, 70, 3557–3565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; Wei, J.; Kong, L.-Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro-Oncol 2010, 12, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- De, I.; Steffen, M.D.; Clark, P.A.; Patros, C.J.; Sokn, E.; Bishop, S.M.; Litscher, S.; Maklakova, V.I.; Kuo, J.S.; Rodriguez, F.J.; et al. CSF1 Overexpression Promotes High-Grade Glioma Formation without Impacting the Polarization Status of Glioma-Associated Microglia and Macrophages. Cancer Res. 2016, 76, 2552–2560. [Google Scholar] [CrossRef] [Green Version]
- Butowski, N.; Colman, H.; de Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncology 2016, 18, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Kowal, J.; Akkari, L.; Schuhmacher, A.J.; Huse, J.T.; West, B.L.; Joyce, J.A. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene 2017, 36, 6049–6058. [Google Scholar] [CrossRef]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting Tumor-Associated Macrophages with Anti-CSF-1R Antibody Reveals a Strategy for Cancer Therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, CheckMate-143: The game is not over yet. Oncotarget 2017, 8, 91779–91794. [Google Scholar] [CrossRef] [Green Version]
- Szulzewsky, F.; Arora, S.; de Witte, L.; Ulas, T.; Markovic, D.; Schultze, J.L.; Holland, E.C.; Synowitz, M.; Wolf, S.A.; Kettenmann, H. Human glioblastoma-associated microglia/monocytes express a distinct RNA profile compared to human control and murine samples. Glia 2016, 64, 1416–1436. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Bao, J.; Zhao, Q.; Zhou, T.; Sun, X. A Spatio-Temporal Model of Macrophage-Mediated Drug Resistance in Glioma Immunotherapy. Mol. Cancer Ther. 2018, 17, 814–824. [Google Scholar] [CrossRef] [Green Version]
- Mohme, M.; Schliffke, S.; Maire, C.L.; Rünger, A.; Glau, L.; Mende, K.C.; Matschke, J.; Gehbauer, C.; Akyüz, N.; Zapf, S.; et al. Immunophenotyping of Newly Diagnosed and Recurrent Glioblastoma Defines Distinct Immune Exhaustion Profiles in Peripheral and Tumor-infiltrating Lymphocytes. Clin. Cancer Res. 2018, 24, 4187–4200. [Google Scholar] [CrossRef] [Green Version]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wöhrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-Oncology 2015, 17, 1064–1075. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Maxwell, R.; Xia, Y.; Cardarelli, P.; Oyasu, M.; Belcaid, Z.; Kim, E.; Hung, A.; Luksik, A.S.; Garzon-Muvdi, T.; et al. Combination anti-CXCR4 and anti-PD-1 immunotherapy provides survival benefit in glioblastoma through immune cell modulation of tumor microenvironment. J. Neuro-Oncol. 2019, 143, 241–249. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef]
- Antonios, J.P.; Soto, H.; Everson, R.G.; Moughon, D.; Orpilla, J.R.; Shin, N.P.; Sedighim, S.; Treger, J.; Odesa, S.; Tucker, A.; et al. Immunosuppressive tumor-infiltrating myeloid cells mediate adaptive immune resistance via a PD-1/PD-L1 mechanism in glioblastoma. Neuro-Oncology 2017, 19, 796–807. [Google Scholar] [CrossRef]
- Harris-Bookman, S.; Mathios, D.; Martin, A.M.; Xia, Y.; Kim, E.; Xu, H.; Belcaid, Z.; Polanczyk, M.; Barberi, T.; Theodros, D.; et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int. J. Cancer 2018, 143, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Ladomersky, E.; Zhai, L.; Lenzen, A.; Lauing, K.L.; Qian, J.; Scholtens, D.M.; Gritsina, G.; Sun, X.; Liu, Y.; Yu, F.; et al. IDO1 Inhibition Synergizes with Radiation and PD-1 Blockade to Durably Increase Survival Against Advanced Glioblastoma. Clin. Cancer Res. 2018, 24, 2559–2573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; de Andrea, C.; de Cerio, A.L.-D.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Sampson, J.H.; Ashley, D.M.; Archer, G.E.; Fuchs, H.E.; Dranoff, G.; Hale, L.P.; Bigner, D.D. Characterization of a Spontaneous Murine Astrocytoma and Abrogation of Its Tumorigenicity by Cytokine Secretion. Neurosurgery 1997, 41, 1365–1372. [Google Scholar] [CrossRef]
- Serano, R.D.; Pegram, C.N.; Bigner, D.D. Tumorigenic cell culture lines from a spontaneous VM/Dk murine astrocytoma (SMA). Acta Neuropathol. 1980, 51, 53–64. [Google Scholar] [CrossRef]
- Oh, T.; Fakurnejad, S.; Sayegh, E.T.; Clark, A.J.; Ivan, M.E.; Sun, M.Z.; Safaee, M.; Bloch, O.; James, C.D.; Parsa, A.T. Immunocompetent murine models for the study of glioblastoma immunotherapy. J. Transl. Med. 2014, 12, 107. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.; Eisele, G.; Tabatabai, G.; Aulwurm, S.; von Kürthy, G.; Stitz, L.; Roth, P.; Weller, M. Soluble CD70: A novel immunotherapeutic agent for experimental glioblastoma. J. Neurosurg. 2010, 113, 280–285. [Google Scholar] [CrossRef]
- Weller, M.; Tabatabai, G.; Hasenbach, K.; Herrmann, C.; Maurer, G.; Möhle, R.; Marini, P.; Grez, M.; Wick, W. Glioma tropism of lentivirally transduced hematopoietic progenitor cells. Int. J. Oncol. 2010, 36, 1409–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remmele, W.; Stegner, H.E. Vorschlag zur einheitlichen Definition eines Immunreaktiven Score (IRS) für den immunhistochemischen Ostrogenrezeptor-Nachweis (ER-ICA) im Mammakarzinomgewebe (Recommendation for uniform definition of an immunoreactive score (IRS) for immunohistochemical estrogen receptor detection (ER-ICA) in breast cancer tissue). Pathologe 1987, 8, 138–140. [Google Scholar]
- Meyerholz, D.K.; Beck, A.P. Principles and approaches for reproducible scoring of tissue stains in research. Lab. Investig. 2018, 98, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Schönholzer, M.T.; Migliavacca, J.; Alvarez, E.; Kumar, K.S.; Neve, A.; Gries, A.; Ma, M.; Grotzer, M.A.; Baumgartner, M. Real-time sensing of MAPK signaling in medulloblastoma cells reveals cellular evasion mechanism counteracting dasatinib blockade of ERK activation during invasion. Neoplasia 2020, 22, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. A power primer. Psychol. Bull. 1992, 112, 155–159. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Przystal, J.M.; Becker, H.; Canjuga, D.; Tsiami, F.; Anderle, N.; Keller, A.-L.; Pohl, A.; Ries, C.H.; Schmittnaegel, M.; Korinetska, N.; et al. Targeting CSF1R Alone or in Combination with PD1 in Experimental Glioma. Cancers 2021, 13, 2400. https://doi.org/10.3390/cancers13102400
Przystal JM, Becker H, Canjuga D, Tsiami F, Anderle N, Keller A-L, Pohl A, Ries CH, Schmittnaegel M, Korinetska N, et al. Targeting CSF1R Alone or in Combination with PD1 in Experimental Glioma. Cancers. 2021; 13(10):2400. https://doi.org/10.3390/cancers13102400
Chicago/Turabian StylePrzystal, Justyna M., Hannes Becker, Denis Canjuga, Foteini Tsiami, Nicole Anderle, Anna-Lena Keller, Anja Pohl, Carola H. Ries, Martina Schmittnaegel, Nataliya Korinetska, and et al. 2021. "Targeting CSF1R Alone or in Combination with PD1 in Experimental Glioma" Cancers 13, no. 10: 2400. https://doi.org/10.3390/cancers13102400
APA StylePrzystal, J. M., Becker, H., Canjuga, D., Tsiami, F., Anderle, N., Keller, A. -L., Pohl, A., Ries, C. H., Schmittnaegel, M., Korinetska, N., Koch, M., Schittenhelm, J., Tatagiba, M., Schmees, C., Beck, S. C., & Tabatabai, G. (2021). Targeting CSF1R Alone or in Combination with PD1 in Experimental Glioma. Cancers, 13(10), 2400. https://doi.org/10.3390/cancers13102400