
The Carcinogen Cadmium Activates Lysine 63 (K63)-Linked Ubiquitin-Dependent Signaling and Inhibits Selective Autophagy

 ,
,  and
and 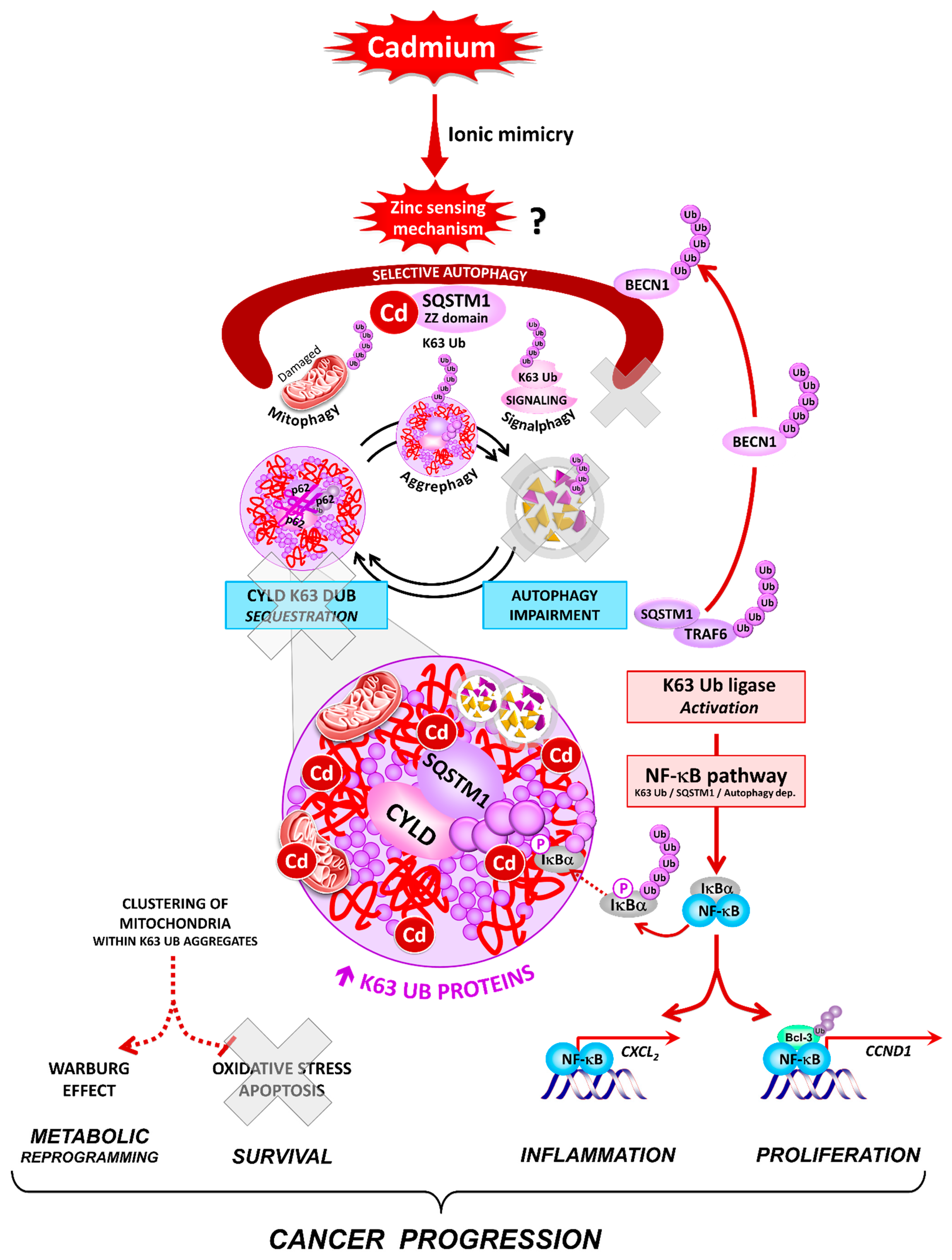{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Detection of Oxidative Stress
2.3. Cell Lysis
2.4. Immunoblotting
2.5. Peptide Preparation for Mass Spectrometry(MS) Analysis
2.6. Mass Spectrometry
2.7. Proteasome Activity
2.8. In Vitro Ubiquitination and Deubiquitination Assays
2.9. Detection of Autophagy
2.10. Electron Microscopy
2.11. Immunofluorescence Staining
2.12. Measurement of Protein Degradation
2.13. Quantitative RT-PCR Analysis
2.14. NF-κB Luciferase Reporter Assay
2.15. Electrophoretic Mobility Shift Assays
2.16. Statistics
3. Results
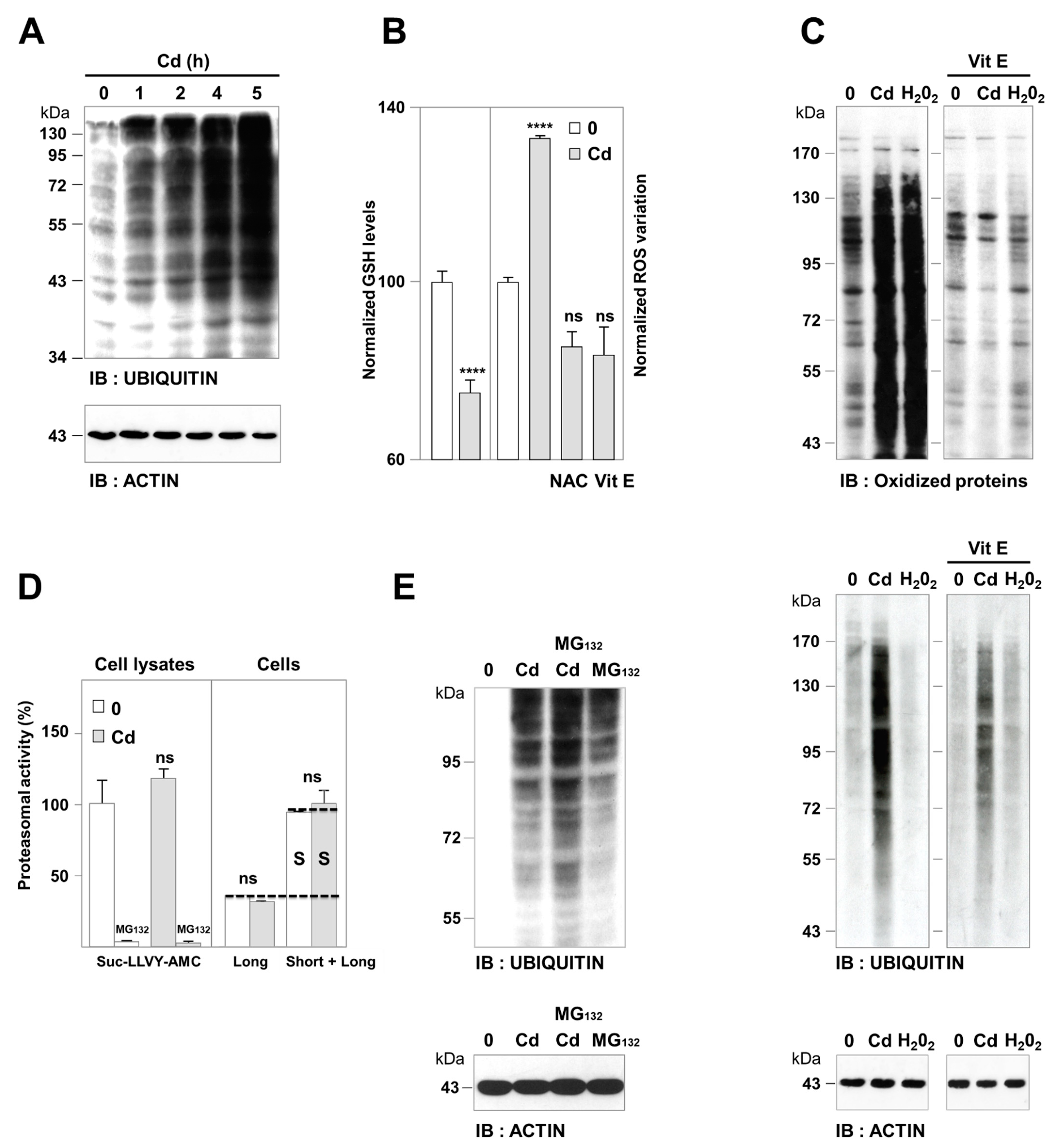
3.1. Cadmium-Induced Protein Ubiquitination was Independent of Oxidative Damage and Proteasome Impairment
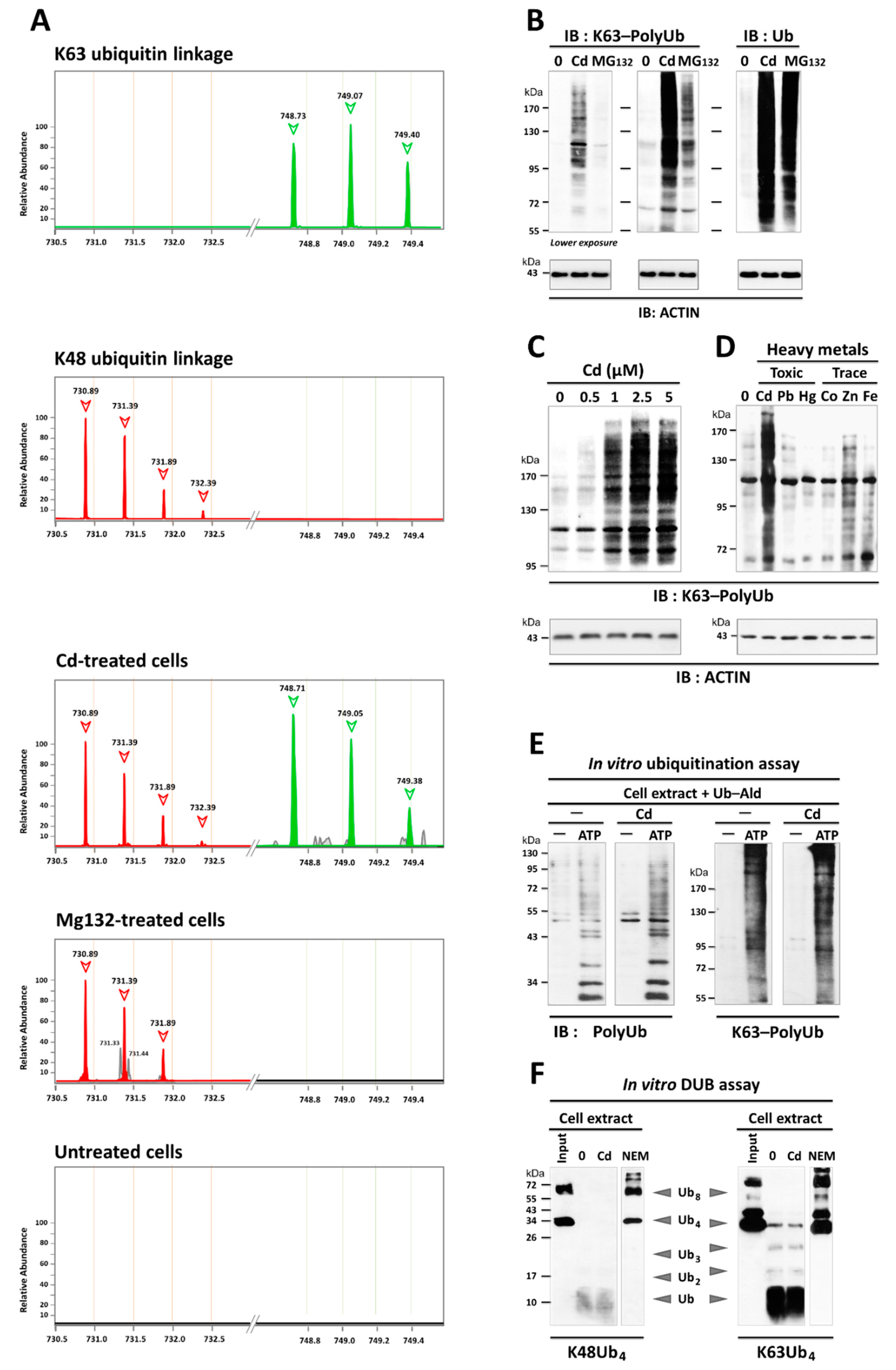
3.2. Cd Is an Activator of K63-Linked Ubiquitination in Lung and Renal Cells
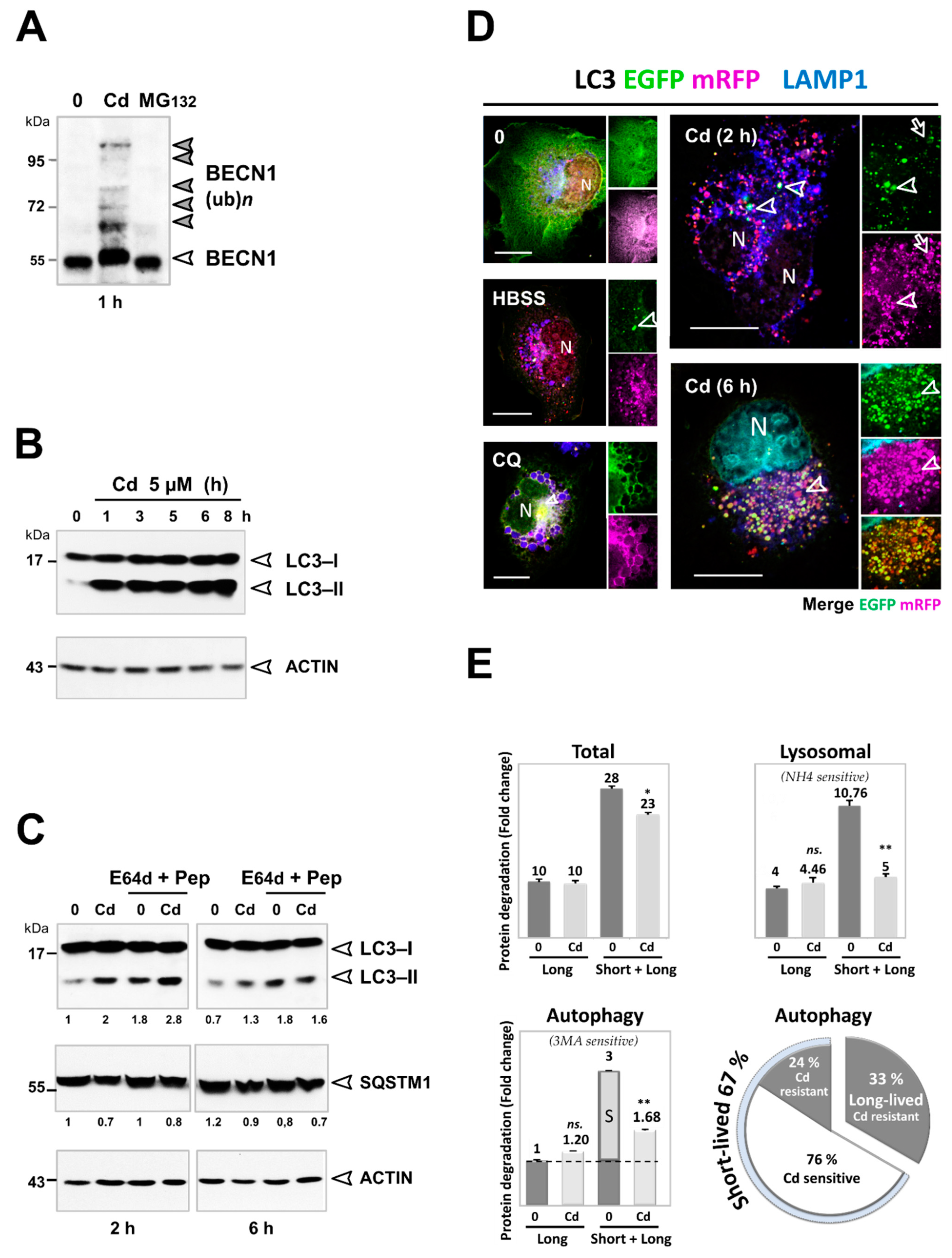
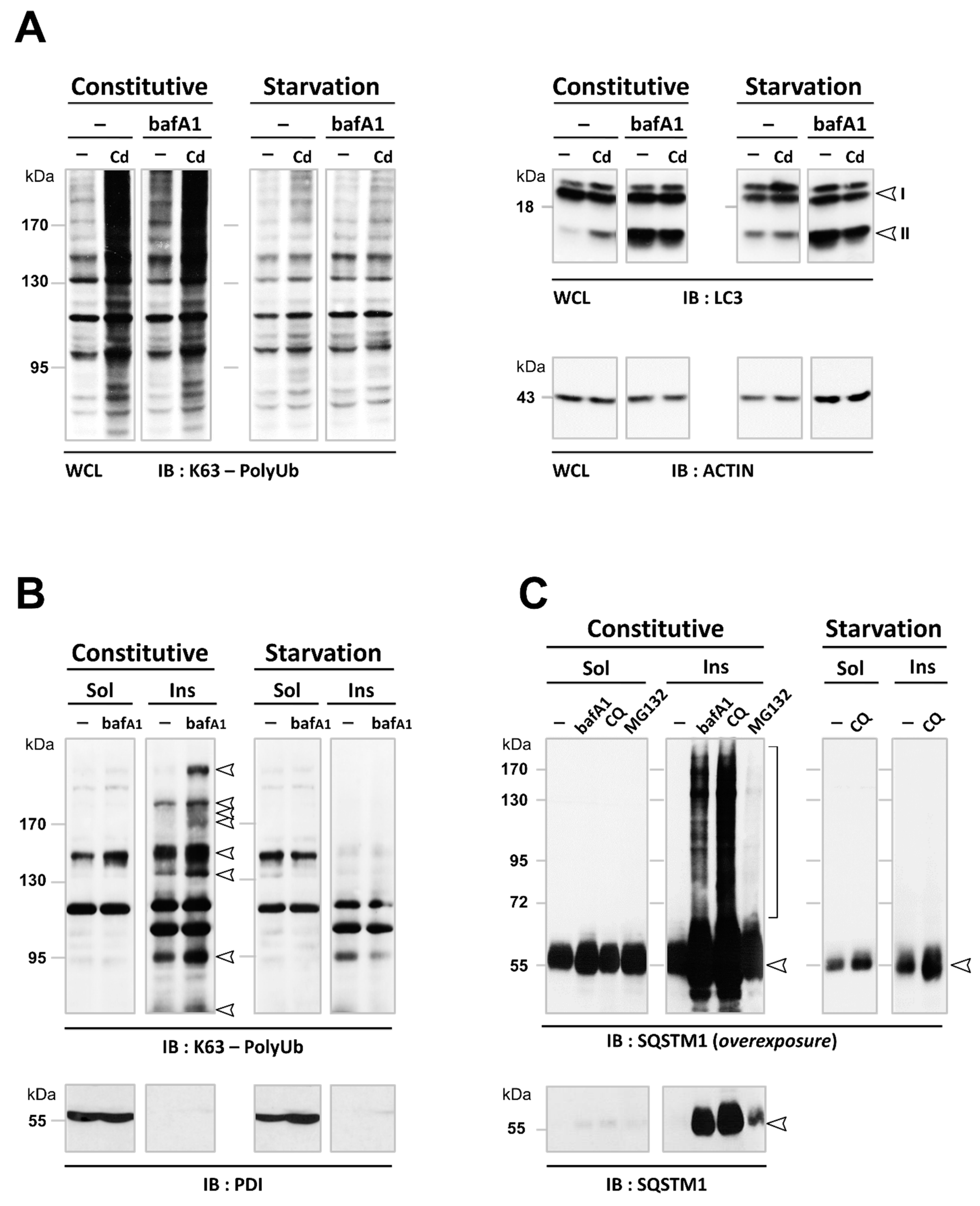
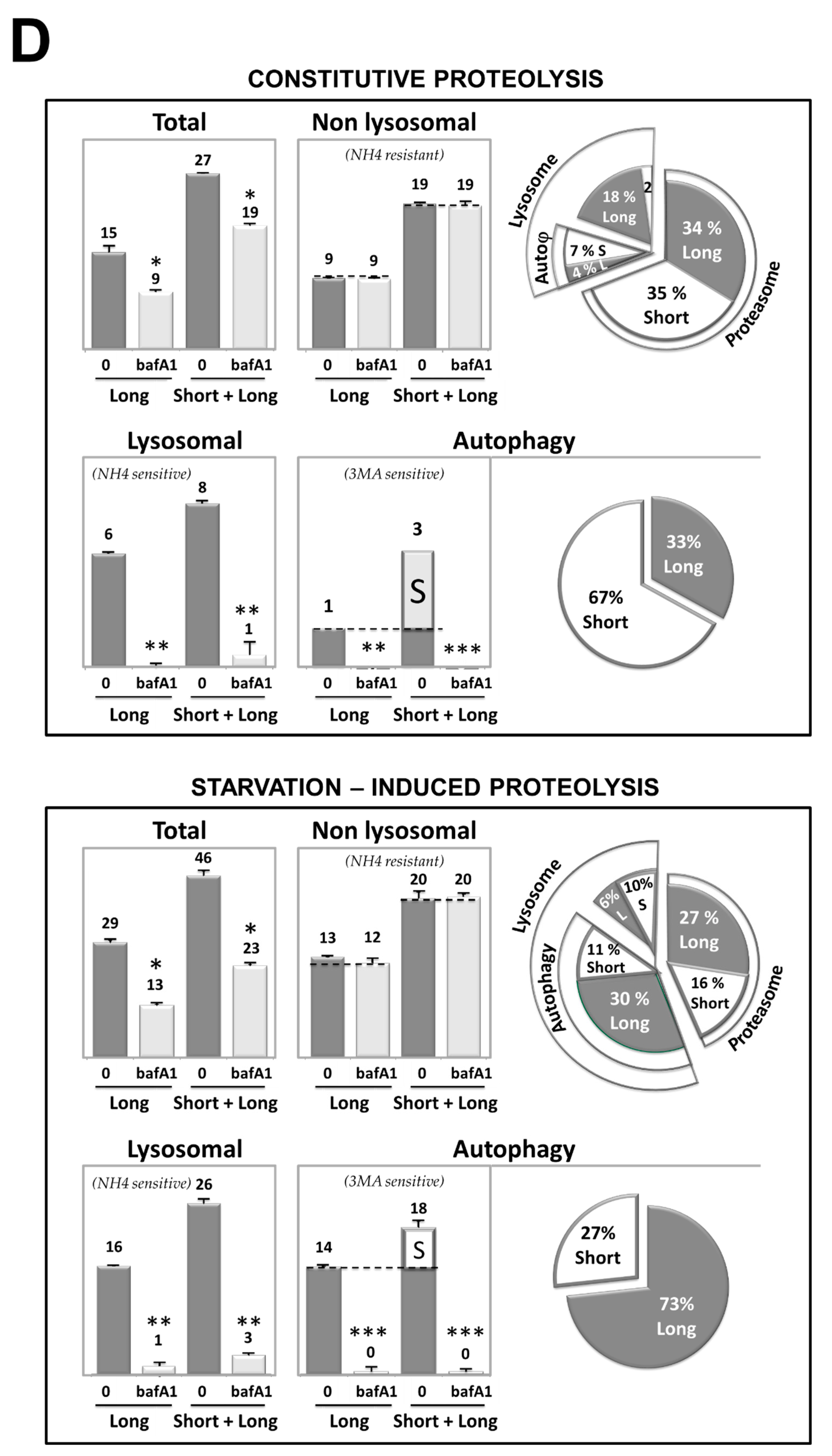
3.3. Cd Selectively Impairs the Autophagy Degradation of Short-Lived Proteins without Interfering with the Autophagy Flux of Long-Lived Proteins
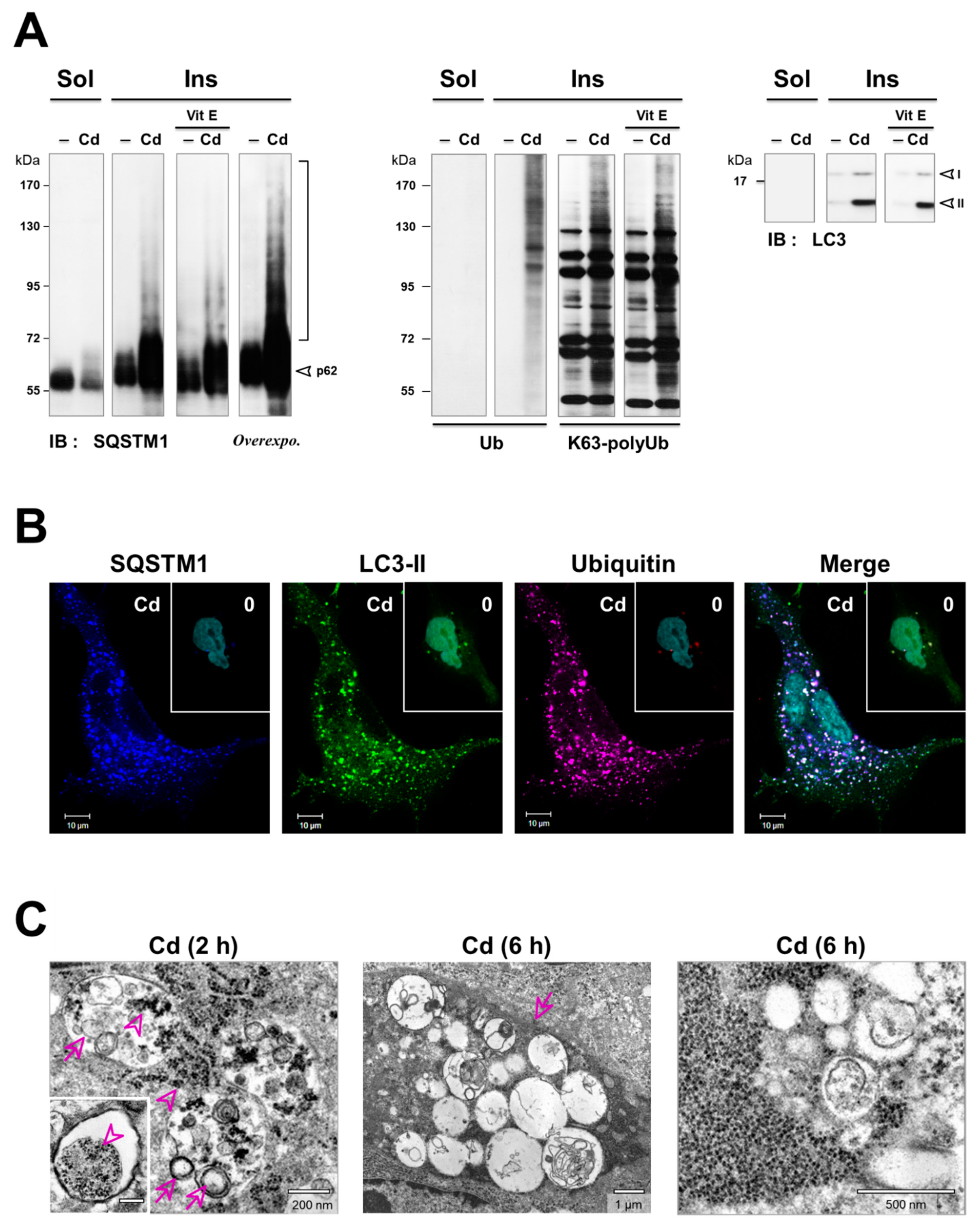
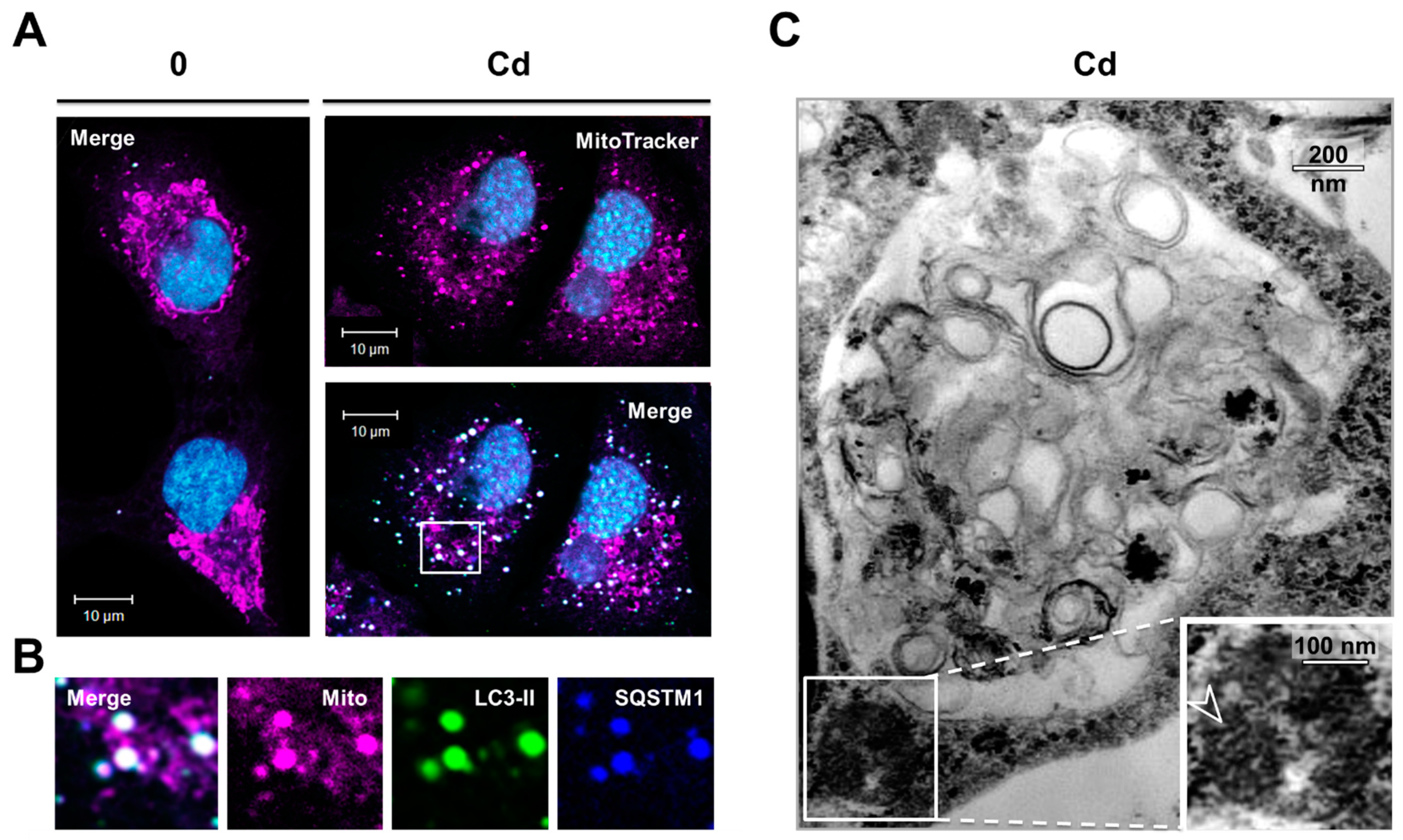
3.4. Inhibition of the Degradation of Three Selective Autophagy Substrates—SQSTM1, Ubiquitinated Proteins, and Mitochondria by Cd
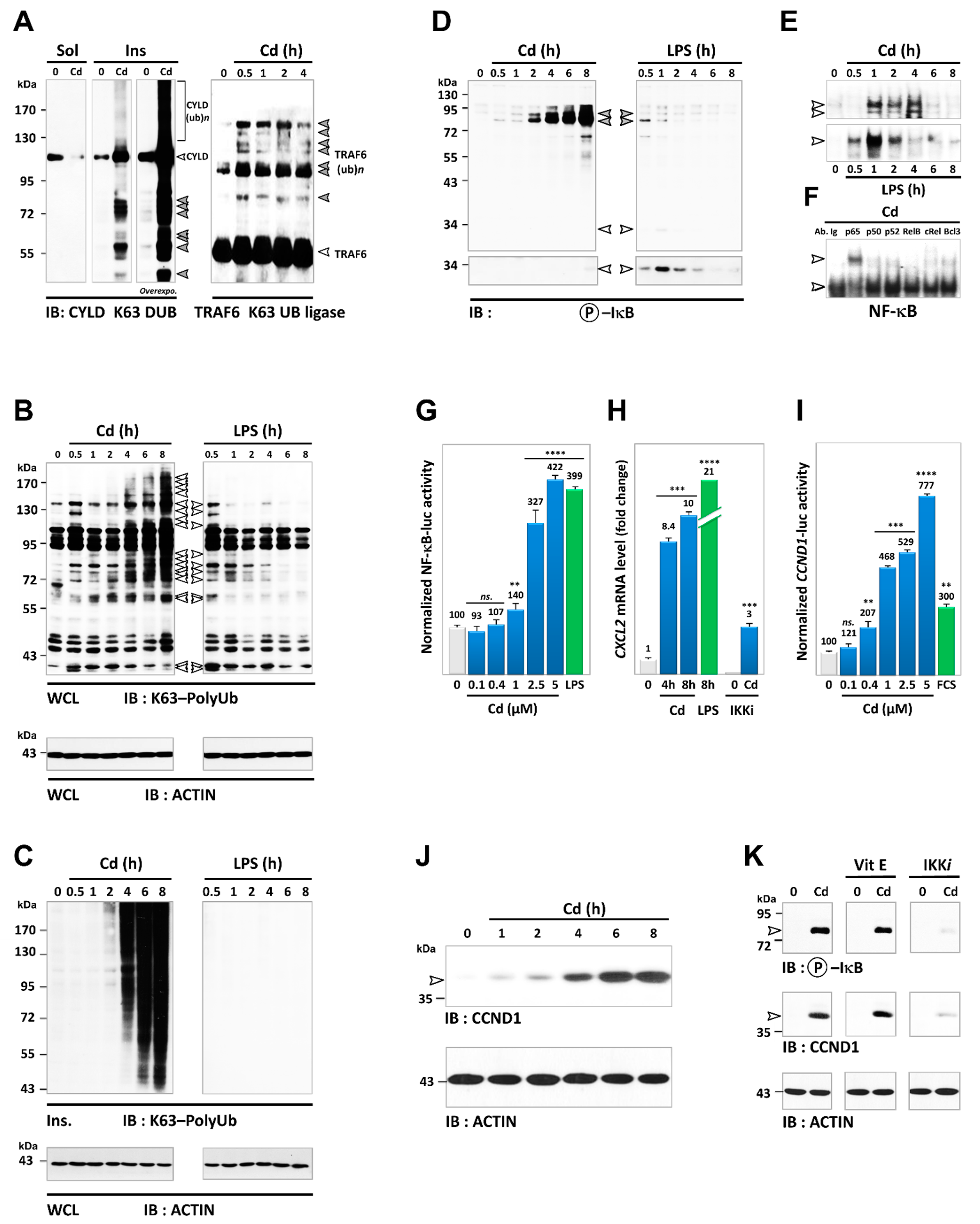
3.5. Cadmium Induces Sustained Activation of the NF-κB Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Qi, J.; Ronai, Z.A. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Rev. Cancer 2018, 18, 69–88. [Google Scholar] [CrossRef]
- French, M.E.; Koehler, C.F.; Hunter, T. Emerging functions of branched ubiquitin chains. Cell Discov. 2021, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, G.; Winklhofer, K.F. Linear Ubiquitin Chains: Cellular Functions and Strategies for Detection and Quantification. Front. Chem. 2020, 7, 915. [Google Scholar] [CrossRef] [PubMed]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Gan, W.; Su, S.; Hauenstein, A.V.; Fu, T.-M.; Brasher, B.; Schwerdtfeger, C.; Liang, A.C.; Xu, M.; Wei, W. K63-linked polyubiquitin chains bind to DNA to facilitate DNA damage repair. Sci. Signal. 2018, 11, eaar8133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, E.L.; Chen, Z.; Van Waes, C. Regulation of NFκB Signalling by Ubiquitination: A Potential Therapeutic Target in Head and Neck Squamous Cell Carcinoma? Cancers 2020, 12, 2877. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef]
- Chen, R.-H.; Chen, Y.-H.; Huang, T.-Y. Ubiquitin-mediated regulation of autophagy. J. Biomed. Sci. 2019, 26, 1–12. [Google Scholar] [CrossRef]
- Lork, M.; Verhelst, K.; Beyaert, R. CYLD, A20 and OTULIN deubiquitinases in NF-κB signaling and cell death: So similar, yet so different. Cell Death Differ. 2017, 24, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)1. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha-Molstad, H.; HyunJoo, C.-M.; Feng, Z.; Lee, S.H.; Kim, J.G.; Yang, P.; Han, B.; Joonsung, H.; Yoo, Y.D.; Hwang, J.; et al. p62/SQSTM1/Sequestosome-1 is an N-recognin of the N-end rule pathway which modulates autophagosome biogenesis. Nat. Commun. 2017, 8, 1–17. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belaid, A.; Ndiaye, P.D.; Filippakis, H.; Roux, J.; Röttinger, É.; Graba, Y.; Brest, P.; Hofman, P.; Mograbi, B. Autophagy: Moving Benchside Promises to Patient Bedsides. Curr. Cancer Drug Targets 2015, 15, 684–702. [Google Scholar] [CrossRef] [PubMed]
- Belaid, A.; Cerezo, M.; Chargui, A.; Corcelle–Termeau, E.; Pedeutour, F.; Giuliano, S.; Ilie, M.; Rubera, I.; Tauc, M.; Barale, S.; et al. Autophagy Plays a Critical Role in the Degradation of Active RHOA, the Control of Cell Cytokinesis, and Genomic Stability. Cancer Res. 2013, 73, 4311–4322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belaid, A.; Ndiaye, P.D.; Cerezo, M.; Cailleteau, L.; Brest, P.; Klionsky, D.J.; Carle, G.F.; Hofman, P.; Mograbi, B. Autophagy and SQSTM1 on the RHOA(d) again. Emerging roles of autophagy in the degradation of signaling proteins. Autophagy 2013, 10, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Belaid, A.; Ndiaye, P.D.; Klionsky, D.J.; Hofman, P.; Mograbi, B. Signalphagy: Scheduled signal termination by macroautophagy. Autophagy 2013, 9, 1629–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [Green Version]
- IARC. Beryllium, cadmium, mercury, and exposures in the glass manufacturing industry, Monographs on the Evaluation of Carcinogenic Risk to Humans. In Proceedings of the IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, Lyon, France, 1993; pp. 119–238. [Google Scholar]
- Jarup, L.; Akesson, A. Current status of cadmium as an environmental health problem. Toxicol. Appl. Pharmacol. 2009, 238, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Moulis, J.-M.; Thévenod, F. New perspectives in cadmium toxicity: An introduction. BioMetals 2010, 23, 763–768. [Google Scholar] [CrossRef] [Green Version]
- NTP (National Toxicology Program). Twelfth Report on Carcinogens, Department of Health and Human Services. In Proceedings of the III-42-III-44, Research Triangle Park. 2011; pp. 80–83. Available online: https://www.ashlandmass.com/DocumentCenter/View/442/National-Toxicology-Program-Report-on-Carcinogens-PDF (accessed on 21 April 2021).
- Barbier, O.; Dauby, A.; Jacquillet, G.; Tauc, M.; Poujeol, P.; Cougnon, M. Zinc and Cadmium Interactions in a Renal Cell Line Derived from Rabbit Proximal Tubule. Nephron Physiol. 2005, 99, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Jacquillet, G.; Barbier, O.; Cougnon, M.; Tauc, M.; Namorado, M.C.; Martin, D.; Reyes, J.L.; Poujeol, P. Zinc protects renal function during cadmium intoxication in the rat. Am. J. Physiol. Ren. Physiol. 2006, 290, F127–F137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, P. Mechanisms of cadmium carcinogenesis. Toxicol. Appl. Pharmacol. 2009, 238, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.C.; Zalups, R.K. Molecular and ionic mimicry and the transport of toxic metals. Toxicol. Appl. Pharmacol. 2005, 204, 274–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chargui, A.; Zekri, S.; Jacquillet, G.; Rubera, I.; Ilie, M.; Belaid, A.; Duranton, C.; Tauc, M.; Hofman, P.; Poujeol, P.; et al. Cadmium-Induced Autophagy in Rat Kidney: An Early Biomarker of Subtoxic Exposure. Toxicol. Sci. 2011, 121, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Wang, T.; Yuan, J.; Sun, J.; Yuan, Y.; Gu, J.; Liu, X.; Bian, J.; Liu, Z. Cadmium-induced cytotoxicity in mouse liver cells is associated with the disruption of autophagic flux via inhibiting the fusion of autophagosomes and lysosomes. Toxicol. Lett. 2020, 321, 32–43. [Google Scholar] [CrossRef]
- Lv, W.; Sui, L.; Yan, X.; Xie, H.; Jiang, L.; Geng, C.; Li, Q.; Yao, X.; Kong, Y.; Cao, J. ROS-dependent Atg4 upregulation mediated autophagy plays an important role in Cd-induced proliferation and invasion in A549 cells. Chem. Biol. Interact. 2018, 279, 136–144. [Google Scholar] [CrossRef]
- Hirano, S.; Kanno, S. Relevance of autophagy markers to cytotoxicity of zinc compounds in macrophages. Toxicol. Vitro 2020, 65, 104816. [Google Scholar] [CrossRef]
- Liuzzi, J.P.; Pazos, R. Interplay Between Autophagy and Zinc. J. Trace Elem. Med. Biol. 2020, 62, 126636. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Fu, Y.; Huang, Z.; Li, M. Transition metals and metal complexes in autophagy and diseases. J. Cell. Physiol. 2021. [Google Scholar] [CrossRef]
- Kawamata, T.; Horie, T.; Matsunami, M.; Sasaki, M.; Ohsumi, Y. Zinc starvation induces autophagy in yeast. J. Biol. Chem. 2017, 292, 8520–8530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Martín, P.; Saito, T.; Komatsu, M. p62/ SQSTM 1: ‘Jack of all trades’ in health and cancer. FEBS J. 2019, 286, 8–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Mi, W.; Xue, Y.; Shi, X.; Kutateladze, T.G. The ZZ domain as a new epigenetic reader and a degradation signal sensor. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.H.; Park, O.H.; Kim, L.; Jung, Y.O.; Park, Y.; Jeong, H.; Hyun, J.; Kim, Y.K.; Song, H.K. Insights into degradation mechanism of N-end rule substrates by p62/SQSTM1 autophagy adapter. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiki, K.; Inamura, H.; Matsuoka, M. PI3K signaling mediates diverse regulation of ATF4 expression for the survival of HK-2 cells exposed to cadmium. Arch. Toxicol. 2014, 88, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Lin, Y.; Jiang, S.; Huang, L.; Yao, H.; Zhuang, Q.; Zhao, R.; Liu, H.; He, C.; Lin, Z. Endoplasmic reticulum stress eIF2α–ATF4 pathway-mediated cyclooxygenase-2 induction regulates cadmium-induced autophagy in kidney. Cell Death Dis. 2016, 7, e2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Witkowska, K.; Afonso Guerra-Assunção, J.; Ren, M.; Ng, F.L.; Mauro, C.; Tucker, A.T.; Caulfield, M.J.; Ye, S. A blood pressure-associated variant of the SLC39A8 gene influences cellular cadmium accumulation and toxicity. Hum. Mol. Genet. 2016, 25, 4117–4126. [Google Scholar] [CrossRef] [Green Version]
- Jenkinson, S.E.; Chung, G.W.; Van Loon, E.; Bakar, N.S.; Dalzell, A.M.; Brown, C.D.A. The limitations of renal epithelial cell line HK-2 as a model of drug transporter expression and function in the proximal tubule. Pflüg. Arch. Eur. J. Physiol. 2012, 464, 601–611. [Google Scholar] [CrossRef]
- L’Hoste, S.; Chargui, A.; Belfodil, R.; Duranton, C.; Rubera, I.; Mograbi, B.; Poujeol, C.; Tauc, M.; Poujeol, P. CFTR mediates cadmium-induced apoptosis through modulation of ROS level in mouse proximal tubule cells. Free Radic. Biol. Med. 2009, 46, 1017–1031. [Google Scholar] [CrossRef] [PubMed]
- Corcelle, E.; Nebout, M.; Bekri, S.; Gauthier, N.; Hofman, P.; Poujeol, P.; Fénichel, P.; Mograbi, B. Disruption of autophagy at the maturation step by the carcinogen lindane is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Cancer Res. 2006, 66, 6861–6870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuertes, G.; Villarroya, A.; Knecht, E. Role of proteasomes in the degradation of short-lived proteins in human fibroblasts under various growth conditions. Int. J. Biochem. Cell Biol. 2003, 35, 651–664. [Google Scholar] [CrossRef]
- Branca, J.J.V.; Fiorillo, C.; Carrino, D.; Paternostro, F.; Taddei, N.; Gulisano, M.; Pacini, A.; Becatti, M. Cadmium-Induced Oxidative Stress: Focus on the Central Nervous System. Antioxidants 2020, 9, 492. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, T.; Priya, S.; Sharma, S.K.; Andersson, S.; Jakobsson, S.; Tanghe, R.; Ashouri, A.; Rauch, S.; Goloubinoff, P.; Christen, P.; et al. Cadmium Causes Misfolding and Aggregation of Cytosolic Proteins in Yeast. Mol. Cell Biol. 2017, 37, e00490-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanthasamy, A.; Choi, C.; Jin, H.; Harischandra, D.; Anantharam, V. Effect of divalent metals on the neuronal proteasomal system, prion protein ubiquitination and aggregation. Toxicol. Lett. 2012, 214, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo-Pereira, M.E.; Li, Z.; Jansen, M.; Rockwell, P. N-Acetylcysteine and Celecoxib Lessen Cadmium Cytotoxicity Which Is Associated with Cyclooxygenase-2 Up-regulation in Mouse Neuronal Cells. J. Biol. Chem. 2002, 277, 25283–25289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-Y.; Xia, M.-Z.; Wang, H.; Liu, X.-J.; Hu, Y.-F.; Chen, Y.-H.; Zhang, C.; Xu, D.-X. Cadmium Selectively Induces MIP-2 and COX-2 Through PTEN-Mediated Akt Activation in RAW264.7 Cells. Toxicol. Sci. 2014, 138, 310–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Sidhu, J.S.; Hong, S.; Robinson, J.F.; Ponce, R.A.; Faustman, E.M. Cadmium Induced p53-Dependent Activation of Stress Signaling, Accumulation of Ubiquitinated Proteins, and Apoptosis in Mouse Embryonic Fibroblast Cells. Toxicol. Sci. 2011, 120, 403–412. [Google Scholar] [CrossRef] [Green Version]
- Othumpangat, S.; Kashon, M.; Joseph, P. Eukaryotic Translation Initiation Factor 4E Is a Cellular Target for Toxicity and Death Due to Exposure to Cadmium Chloride. J. Biol. Chem. 2005, 280, 25162–25169. [Google Scholar] [CrossRef] [Green Version]
- Thévenod, F.; Friedmann, J.M. Cadmium-mediated oxidative stress in kidney proximal tubule cells induces degradation of Na+/K+-ATPase through proteasomal and endo-/lysosomal proteolytic pathways. FASEB J. 1999, 13, 1751–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Arnaud, L.; Rockwell, P.; Figueiredo-Pereira, M.E. A single amino acid substitution in a proteasome subunit triggers aggregation of ubiquitinated proteins in stressed neuronal cells. J. Neurochem. 2004, 90, 19–28. [Google Scholar] [CrossRef]
- WHO. Ten Chemicals of Major Public Health Concern; World Health Organization: Geneva, Switzerland, 2010; Available online: https://www.who.int/ipcs/assessment/public_health/chemicals_phc/en/ (accessed on 5 February 2013).
- Chmielowska-Bąk, J.; Izbiańska, K.; Deckert, J. The toxic Doppelganger: On the ionic and molecular mimicry of cadmium. Acta Biochim. Pol. 2013, 60, 369–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnesano, F.; Belviso, B.D.; Caliandro, R.; Falini, G.; Fermani, S.; Natile, G.; Siliqi, D. Crystallographic Analysis of Metal-Ion Binding to Human Ubiquitin. Chem. Eur. J. 2010, 17, 1569–1578. [Google Scholar] [CrossRef]
- Camara-Artigas, A.; Plaza-Garrido, M.; Martinez-Rodriguez, S.; Bacarizo, J. New crystal form of human ubiquitin in the presence of magnesium. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 29–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Hao, S.; Qiu, Z.; Wang, Y.; Zhao, Y.; Li, Y.; Gao, W.; Wu, Y.; Liu, C.; Xu, X.; et al. Cadmium disrupts the DNA damage response by destabilizing RNF168. Food Chem. Toxicol. 2019, 133, 110745. [Google Scholar] [CrossRef]
- Shi, C.-S.; Kehrl, J.H. TRAF6 and A20 Regulate Lysine 63-Linked Ubiquitination of Beclin-1 to Control TLR4-Induced Autophagy. Sci. Signal. 2010, 3, ra42. [Google Scholar] [CrossRef]
- Herak-Kramberger, C.M.; Brown, D.; Sabolic, I. Cadmium inhibits vacuolar H+-ATPase and endocytosis in rat kidney cortex. Kidney Int. 1998, 53, 1713–1726. [Google Scholar] [CrossRef] [Green Version]
- Messner, B.; Ploner, C.; Laufer, G.; Bernhard, D. Cadmium activates a programmed, lysosomal membrane permeabilization-dependent necrosis pathway. Toxicol. Lett. 2012, 212, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Medicherla, B.; Goldberg, A.L. Heat shock and oxygen radicals stimulate ubiquitin-dependent degradation mainly of newly synthesized proteins. J. Cell Biol. 2008, 182, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Johnston, H.E.; Samant, R.S. Alternative systems for misfolded protein clearance: Life beyond the proteasome. FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F. Cadmium and cellular signaling cascades: To be or not to be? Toxicol. Appl. Pharmacol. 2009, 238, 221–239. [Google Scholar] [CrossRef] [PubMed]
- Staal, J.; Driege, Y.; Bekaert, T.; Demeyer, A.; Muyllaert, D.; Van Damme, P.; Gevaert, K.; Beyaert, R. T-cell receptor-induced JNK activation requires proteolytic inactivation of CYLD by MALT1. EMBO J. 2011, 30, 1742–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, M.; Scheidereit, C. The IκB kinase complex in NF -κB regulation and beyond. EMBO Rep. 2014, 15, 46–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.; Gopinathan, G.; Sukumar, J.T.; Gribben, J.G. Blocking Autophagy Prevents Bortezomib-Induced NF-κB Activation by Reducing I-κBα Degradation in Lymphoma Cells. PLoS ONE 2012, 7, e32584. [Google Scholar] [CrossRef] [PubMed]
- Coogan, T.P.; Bare, R.M.; Waalkes, M.P. Cadmium-induced DNA strand damage in cultured liver cells: Reduction in cadmium genotoxicity following zinc pretreatment. Toxicol. Appl. Pharmacol. 1992, 113, 227–233. [Google Scholar] [CrossRef]
- Satarug, S. Dietary Cadmium Intake and Its Effects on Kidneys. Toxics 2018, 6, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.B.; Hwang, E.S. High Levels of ROS Impair Lysosomal Acidity and Autophagy Flux in Glucose-Deprived Fibroblasts by Activating ATM and Erk Pathways. Biomolecules 2020, 10, 761. [Google Scholar] [CrossRef] [PubMed]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Zinc through the Three Domains of Life. J. Proteom. Res. 2006, 5, 3173–3178. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shen, C.; Nakamura, E.; Ando, K.; Signoretti, S.; Beroukhim, R.; Cowley, G.S.; Lizotte, P.; Liberzon, E.; Bair, S.; et al. SQSTM1 Is a Pathogenic Target of 5q Copy Number Gains in Kidney Cancer. Cancer Cell 2013, 24, 738–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Wang, C.; Croce, C.M.; Guan, J.-L. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev. 2014, 28, 1204–1216. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; McBride, S.J.; Boyd, W.A.; Alper, S.; Freedman, J.H. Toxicogenomic analysis of Caenorhabditis elegans reveals novel genes and pathways involved in the resistance to cadmium toxicity. Genome Biol. 2007, 8, R122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, K.S.; Zdraljevic, S.; Stevens, L.; Collins, K.; Tanny, R.E.; Andersen, E.C. Natural variation in the sequestosome-related gene, sqst-5, underlies zinc homeostasis in Caenorhabditis elegans. PLoS Genet. 2020, 16, e1008986. [Google Scholar] [CrossRef]
- Verzella, D.; Pescatore, A.; Capece, D.; Vecchiotti, D.; Ursini, M.V.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Life, death, and autophagy in cancer: NF-κB turns up everywhere. Cell Death Dis. 2020, 11, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef]
- Hossein-Khannazer, N.; Azizi, G.; Eslami, S.; Mohammed, H.A.; Fayyaz, F.; Hosseinzadeh, R.; Usman, A.B.; Kamali, A.N.; Mohammadi, H.; Jadidi-Niaragh, F.; et al. The effects of cadmium exposure in the induction of inflammation. Immunopharmacol. Immunotoxicol. 2019, 42, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F.; Lee, W.-K.; Garrick, M.D. Iron and Cadmium Entry Into Renal Mitochondria: Physiological and Toxicological Implications. Front. Cell Dev. Biol. 2020, 8, 848. [Google Scholar] [CrossRef]
- Xiao, B.; Deng, X.; Lim, G.G.; Zhou, W.; Saw, W.-T.; Zhou, Z.D.; Lim, K.-L.; Tan, E.-K. p62-Mediated mitochondrial clustering attenuates apoptosis induced by mitochondrial depolarization. Biochim. Biophys. Acta BBA Mol. Cell Res. 2017, 1864, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.R.; Spandidos, D.A.; Tsatsakis, A.; Schweitzer, A.; Djordjevic, V.; Djordjevic, A.B. Potential interaction of cadmium chloride with pancreatic mitochondria: Implications for pancreatic cancer. Int. J. Mol. Med. 2019, 44, 145–156. [Google Scholar] [CrossRef] [PubMed]
- De Smet, F.; Rubio, M.S.; Hompes, D.; Naus, E.; De Baets, G.; Langenberg, T.; Hipp, M.S.; Houben, B.; Claes, F.; Charbonneau, S.; et al. Nuclear inclusion bodies of mutant and wild-type p53 in cancer: A hallmark of p53 inactivation and proteostasis remodelling by p53 aggregation. J. Pathol. 2016, 242, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.-C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Trnka, M.J.; Guan, S.; Kwon, D.; Kim, D.-H.; Chen, J.-J.; Greer, P.A.; Burlingame, A.L.; Correia, M.A. A Novel Mechanism for NF-κB-activation via IκB-aggregation: Implications for Hepatic Mallory-Denk-Body Induced Inflammation. Mol. Cell. Proteom. 2020, 19, 1968–1986. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chargui, A.; Belaid, A.; Ndiaye, P.D.; Imbert, V.; Samson, M.; Guigonis, J.-M.; Tauc, M.; Peyron, J.-F.; Poujeol, P.; Brest, P.; et al. The Carcinogen Cadmium Activates Lysine 63 (K63)-Linked Ubiquitin-Dependent Signaling and Inhibits Selective Autophagy. Cancers 2021, 13, 2490. https://doi.org/10.3390/cancers13102490
Chargui A, Belaid A, Ndiaye PD, Imbert V, Samson M, Guigonis J-M, Tauc M, Peyron J-F, Poujeol P, Brest P, et al. The Carcinogen Cadmium Activates Lysine 63 (K63)-Linked Ubiquitin-Dependent Signaling and Inhibits Selective Autophagy. Cancers. 2021; 13(10):2490. https://doi.org/10.3390/cancers13102490
Chicago/Turabian StyleChargui, Abderrahman, Amine Belaid, Papa Diogop Ndiaye, Véronique Imbert, Michel Samson, Jean-Marie Guigonis, Michel Tauc, Jean-François Peyron, Philippe Poujeol, Patrick Brest, and et al. 2021. "The Carcinogen Cadmium Activates Lysine 63 (K63)-Linked Ubiquitin-Dependent Signaling and Inhibits Selective Autophagy" Cancers 13, no. 10: 2490. https://doi.org/10.3390/cancers13102490
APA StyleChargui, A., Belaid, A., Ndiaye, P. D., Imbert, V., Samson, M., Guigonis, J. -M., Tauc, M., Peyron, J. -F., Poujeol, P., Brest, P., Hofman, P., & Mograbi, B. (2021). The Carcinogen Cadmium Activates Lysine 63 (K63)-Linked Ubiquitin-Dependent Signaling and Inhibits Selective Autophagy. Cancers, 13(10), 2490. https://doi.org/10.3390/cancers13102490