
The Critical Role of TRIB2 in Cancer and Therapy Resistance


Abstract
:Simple Summary
Abstract
1. Introduction
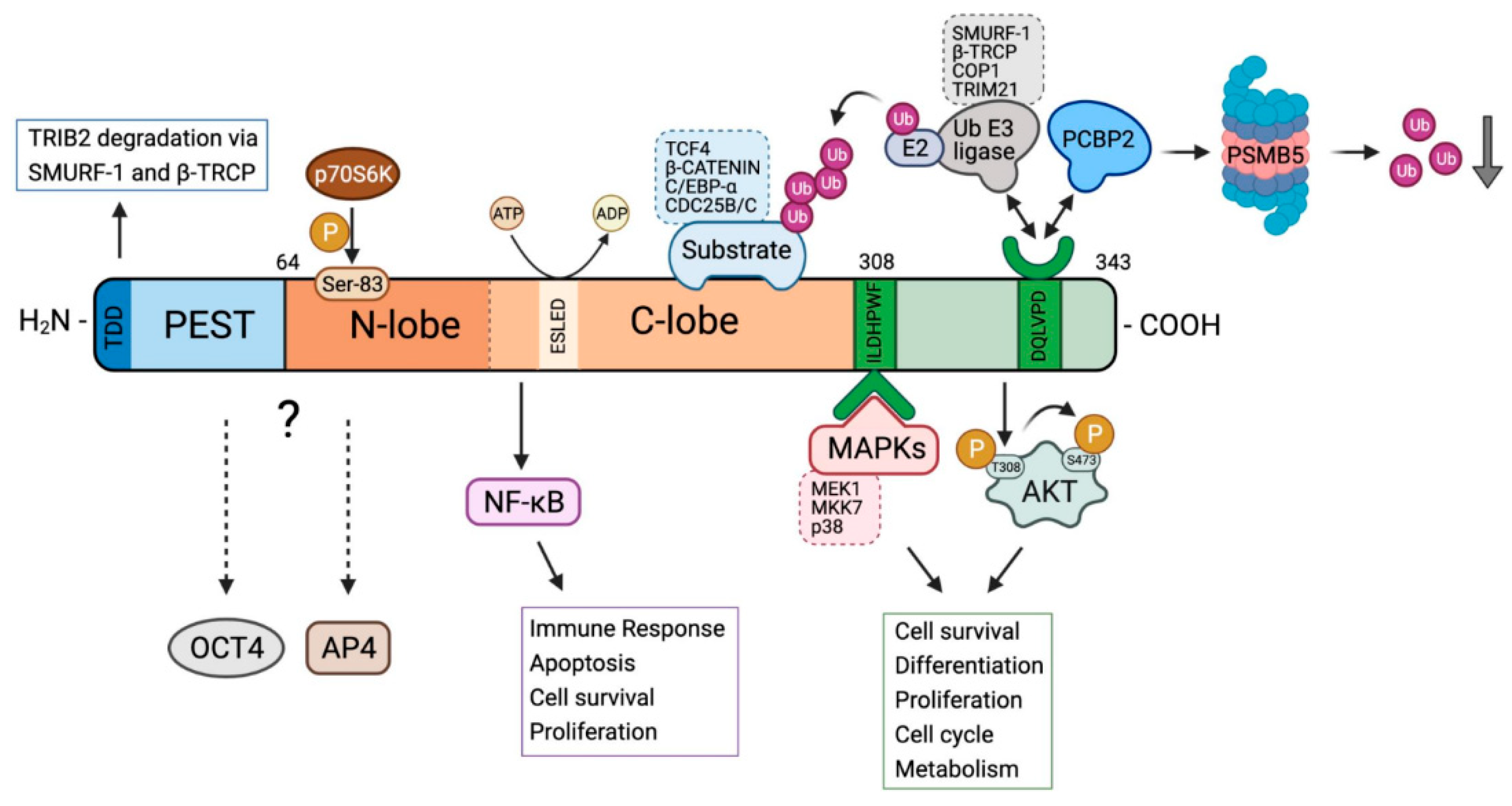
2. Unique Structural Features of TRIB2
3. Regulation of TRIB2 Expression and TRIB2 Signalling
4. TRIB2 in Health and Disease
5. TRIB2 in Cancer
5.1. TRIB2 in Leukemia
5.2. TRIB2 in Liver Cancer
5.3. TRIB2 in Melanoma
6. The Emerging Role of TRIB2 in Therapy Resistance
7. Targeting TRIB2
8. TRIB2 as a Biomarker
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mata, J.; Curado, S.; Ephrussi, A.; Rørth, P. Tribbles Coordinates Mitosis and Morphogenesis in Drosophila by Regulating String/CDC25 Proteolysis. Cell 2000, 101, 511–522. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, S.; Byrne, D.P.; Harris, J.A.; Kannan, N.; Eyers, P.A. Cataloguing the dead: Breathing new life into pseudokinase research. FEBS J. 2020, 287, 4150–4169. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Nakamura, T. Tribbles in disease: Signaling pathways important for cellular function and neoplastic transformation. Cancer Sci. 2011, 102, 1115–1122. [Google Scholar] [CrossRef]
- Eyers, P.A.; Keeshan, K.; Kannan, N. Tribbles in the 21st Century: The Evolving Roles of Tribbles Pseudokinases in Biology and Disease. Trends Cell Biol. 2017, 27, 284–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foulkes, D.M.; Byrne, D.P.; Bailey, F.P.; Eyers, P.A. Tribbles pseudokinases: Novel targets for chemical biology and drug discovery? Biochem. Soc. Trans. 2015, 43, 1095–1103. [Google Scholar] [CrossRef]
- Kiss-Toth, E.; Bagstaff, S.M.; Sung, H.Y.; Jozsa, V.; Dempsey, C.; Caunt, J.C.; Oxley, K.M.; Wyllie, D.H.; Polgar, T.; Harte, M.; et al. Human tribbles, a protein family controlling mitogen-activated protein kinase cascades. J. Biol. Chem. 2004, 279, 42703–42708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soubeyrand, S.; Martinuk, A.; Naing, T.; Lau, P.; McPherson, R. Role of Tribbles Pseudokinase 1 (TRIB1) in human hepatocyte metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 223–232. [Google Scholar] [CrossRef]
- Wennemers, M.; Bussink, J.; van den Beucken, T.; Sweep, F.C.G.J.; Span, P.N. Regulation of TRIB3 mRNA and Protein in Breast Cancer. PLoS ONE 2012, 7, 1–9. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Weng, W.; Qiao, Y.; Ma, L.; Xiao, W.; Yu, Y.; Pan, Q.; Sun, F. Impaired phosphorylation and ubiquitination by p70 S6 kinase (p70S6K) and Smad ubiquitination regulatory factor 1 (Smurf1) promote tribbles homolog 2 (TRIB2) stability and carcinogenic property in liver cancer. J. Biol. Chem. 2013, 288, 33667–33681. [Google Scholar] [CrossRef] [Green Version]
- Hegedus, Z.; Czibula, A.; Kiss-Toth, E. Tribbles: A family of kinase-like proteins with potent signalling regulatory function. Cell. Signal. 2007, 19, 238–250. [Google Scholar] [CrossRef]
- Xu, S.; Tong, M.; Huang, J.; Zhang, Y.; Qiao, Y.; Weng, W.; Liu, W.; Wang, J.; Sun, F. TRIB2 inhibits Wnt/β-Catenin/TCF4 signaling through its associated ubiquitin E3 ligases, β-TrCP, COP1 and Smurf1, in liver cancer cells. FEBS Lett. 2014, 588, 4334–4341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, F.P.; Byrne, D.P.; Oruganty, K.; Eyers, C.E.; Novotny, C.J.; Shokat, K.M.; Kannan, N.; Eyers, P.A. The Tribbles 2 (TRB2) pseudokinase binds to ATP and autophosphorylates in a metal-independent manner. Biochem. J. 2015, 467, 47–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeshan, K.; Bailis, W.; Dedhia, P.H.; Vega, M.E.; Shestova, O.; Xu, L.; Toscano, K.; Uljon, S.N.; Blacklow, S.C.; Pear, W.S. Transformation by Tribbles homolog 2 (Trib2) requires both the Trib2 kinase domain and COP1 binding. Blood 2010, 116, 4948–4957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilby, D.C.; Sung, H.Y.; Winship, P.R.; Goodeve, A.C.; Reilly, J.T.; Kiss-Toth, E. Tribbles-1 and -2 are tumour suppressors, down-regulated in human acute myeloid leukaemia. Immunol. Lett. 2010, 130, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Dobens, L.L.; Bouyain, S. Developmental roles of tribbles protein family members. Dev. Dyn. 2012, 241, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.J.; Mack, E.A.; Rome, K.S.; Pear, W.S. Tribbles in normal and malignant haematopoiesis. Biochem. Soc. Trans. 2015, 43, 1112–1115. [Google Scholar] [CrossRef] [Green Version]
- Richmond, L.; Keeshan, K. Pseudokinases: A tribble-edged sword. FEBS J. 2020, 287, 4170–4182. [Google Scholar] [CrossRef] [Green Version]
- Salomé, M.; Hopcroft, L.; Keeshan, K. Inverse and correlative relationships between TRIBBLES genes indicate non-redundant functions during normal and malignant hemopoiesis. Exp. Hematol. 2018, 66, 63–78.e13. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, S.; Boonvisut, S.; Makishima, S.; Ishizuka, Y.; Watanabe, K.; Nakayama, K. The role of TRIB1 in lipid metabolism; From genetics to pathways. Biochem. Soc. Trans. 2015, 43, 1063–1068. [Google Scholar] [CrossRef]
- Do, E.K.; Park, J.K.; Cheon, H.C.; Kwon, Y.W.; Heo, S.C.; Choi, E.J.; Seo, J.K.; Jang, I.H.; Lee, S.C.; Kim, J.H. Trib2 regulates the pluripotency of embryonic stem cells and enhances reprogramming efficiency. Exp. Mol. Med. 2017, 49, e401. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, K.; Iwamoto, S. An adaptive variant of TRIB2, rs1057001, is associated with higher expression levels of thermogenic genes in human subcutaneous and visceral adipose tissues. J. Physiol. Anthropol. 2017, 36, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Bezy, O.; Vernochet, C.; Gesta, S.; Farmer, S.R.; Kahn, C.R. TRB3 Blocks Adipocyte Differentiation through the Inhibition of C/EBPβ Transcriptional Activity. Mol. Cell. Biol. 2007, 27, 6818–6831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prudente, S.; Sesti, G.; Pandolfi, A.; Andreozzi, F.; Consoli, A.; Trischitta, V. The mammalian tribbles homolog TRIB3, glucose homeostasis, and cardiovascular diseases. Endocr. Rev. 2012, 33, 526–546. [Google Scholar] [CrossRef] [Green Version]
- Soubeyrand, S.; Martinuk, A.; McPherson, R. TRIB1 is a positive regulator of hepatocyte nuclear factor 4-alpha. Sci. Rep. 2017, 7, 5574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, K.; Herzig, S.; Kulkarni, R.N.; Montminy, M. TRB3: A tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 2003, 300, 1574–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, Y.; Zhang, Y.; Wang, J. Ubiquitin E3 ligase SCFβ-TRCP regulates TRIB2 stability in liver cancer cells. Biochem. Biophys. Res. Commun. 2013, 441, 555–559. [Google Scholar] [CrossRef]
- Foulkes, D.M.; Byrne, D.P.; Yeung, W.; Shrestha, S.; Bailey, F.P.; Ferries, S.; Eyers, C.E.; Keeshan, K.; Wells, C.; Drewry, D.H.; et al. Covalent inhibitors of EGFR family protein kinases induce degradation of human Tribbles 2 (TRIB2) pseudokinase in cancer cells. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.-C.; Rosenberg, I.M.; Cao, Z.; Huett, A.S.; Xavier, R.J.; Podolsky, D.K. Tribbles 2 (Trib2) is a novel regulator of toll-like receptor 5 signaling. Inflamm. Bowel Dis. 2012, 18, 877–888. [Google Scholar] [CrossRef] [Green Version]
- Eyers, P.A. TRIBBLES: A Twist in the Pseudokinase Tail. Structure 2015, 23, 1974–1976. [Google Scholar] [CrossRef] [Green Version]
- Liang, K.L.; Paredes, R.; Carmody, R.; Eyers, P.A.; Meyer, S.; McCarthy, T.V.; Keeshan, K. Human TRIB2 oscillates during the cell cycle and promotes ubiquitination and degradation of CDC25C. Int. J. Mol. Sci. 2016, 17, 1378. [Google Scholar] [CrossRef] [Green Version]
- Rishi, L.; Hannon, M.; Salomè, M.; Hasemann, M.; Frank, A.K.; Campos, J.; Timoney, J.; O’Connor, C.; Cahill, M.R.; Porse, B.; et al. Regulation of Trib2 by an E2F1-C/EBPα feedback loop in AML cell proliferation. Blood 2014, 123, 2389–2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanda, T.; Lawton, L.N.; Barrasa, M.I.; Fan, Z.P.; Kohlhammer, H.; Gutierrez, A.; Ma, W.; Tatarek, J.; Ahn, Y.; Kelliher, M.A.; et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell 2012, 22, 209–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.X.; Yan, Y.F.; Liu, Y.M.; Li, Y.J.; Zhang, H.H.; Pang, M.; Hu, J.X.; Zhao, W.; Xie, N.; Zhou, L.; et al. Smad3-related miRNAs regulated oncogenic TRIB2 promoter activity to effectively suppress lung adenocarcinoma growth. Cell Death Dis. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Argiropoulos, B.; Palmqvist, L.; Yung, E.; Kuchenbauer, F.; Heuser, M.; Sly, L.M.; Wan, A.; Krystal, G.; Humphries, R.K. Linkage of Meis1 leukemogenic activity to multiple downstream effectors including Trib2 and Ccl3. Exp. Hematol. 2008, 36, 845–859. [Google Scholar] [CrossRef]
- Nagel, S.; Venturini, L.; Przybylski, G.K.; Grabarczyk, P.; Schneider, B.; Meyer, C.; Kaufmann, M.; Schmidt, C.A.; Scherr, M.; Drexler, H.G.; et al. Activation of Paired-Homeobox Gene PITX1 by Del(5)(q31) in T-Cell Acute Lymphoblastic Leukemia (T-ALL). Blood 2010, 116, 4644. [Google Scholar] [CrossRef]
- Mancini, E.; Sanjuan-Pla, A.; Luciani, L.; Moore, S.; Grover, A.; Zay, A.; Rasmussen, K.D.; Luc, S.; Bilbao, D.; O’Carroll, D.; et al. FOG-1 and GATA-1 act sequentially to specify definitive megakaryocytic and erythroid progenitors. EMBO J. 2012, 31, 351–365. [Google Scholar] [CrossRef]
- Wang, J.; Park, J.S.; Wei, Y.; Rajurkar, M.; Cotton, J.L.; Fan, Q.; Lewis, B.C.; Ji, H.; Mao, J. TRIB2 acts downstream of Wnt/TCF in liver cancer cells to regulate YAP and C/EBPα function. Mol. Cell 2013, 51, 211–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrand, D.; Eberle, M.E.; Wölfle, S.M.; Egler, F.; Sahin, D.; Sähr, A.; Bode, K.A.; Heeg, K. Hsa-miR-99b/let-7e/miR-125a cluster regulates pathogen recognition receptor-stimulated suppressive antigen-presenting cells. Front. Immunol. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Zhang, C.; Chi, Y.L.; Wang, P.Y.; Wang, Y.Q.; Zhang, Y.X.; Deng, J.; Lv, C.J.; Xie, S.Y. miR-511 and miR-1297 Inhibit Human Lung Adenocarcinoma Cell Proliferation by Targeting Oncogene TRIB2. PLoS ONE 2012, 7, e46090. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.Y.; Sun, Y.X.; Zhang, S.; Pang, M.; Zhang, H.H.; Gao, S.Y.; Zhang, C.; Lv, C.J.; Xie, S.Y. Let-7c inhibits A549 cell proliferation through oncogenic TRIB2 related factors. FEBS Lett. 2013, 587, 2675–2681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, G.; Lu, T.; Shen, J.; Wang, J. LncRNA ZEB1-AS1 promotes pancreatic cancer progression by regulating miR-505-3p/TRIB2 axis. Biochem. Biophys. Res. Commun. 2020, 528, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; He, L.; Coppola, M.; Guo, J.; Esposito, N.N.; Coppola, D.; Cheng, J.Q. MicroRNA-155 regulates cell survival, growth, and chemosensitivity by targeting FOXO3a in breast cancer. J. Biol. Chem. 2010, 285, 17869–17879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, H.; Shuaib, A.; Leon, D.D.; Angyal, A.; Salazar, M.; Velasco, G.; Holcombe, M.; Dower, S.K.; Kiss-Toth, E. Competition between members of the tribbles pseudokinase protein family shapes their interactions with mitogen activated protein kinase pathways. Sci. Rep. 2016, 6, 32667. [Google Scholar] [CrossRef] [Green Version]
- Salomé, M.; Magee, A.; Yalla, K.; Chaudhury, S.; Sarrou, E.; Carmody, R.J.; Keeshan, K. A Trib2-p38 axis controls myeloid leukaemia cell cycle and stress response signalling. Cell Death Dis. 2018, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Hou, Z.; Guo, K.; Sun, X.; Hu, F.; Chen, Q.; Luo, X.; Wang, G.; Hu, J.; Sun, L. TRIB2 functions as novel oncogene in colorectal cancer by blocking cellular senescence through AP4/p21 signaling 11 Medical and Health Sciences 1112 Oncology and Carcinogenesis 06 Biological Sciences 0601 Biochemistry and Cell Biology. Mol. Cancer 2018, 17, 1–15. [Google Scholar] [CrossRef]
- Grandinetti, K.B.; Stevens, T.A.; Ha, S.; Salamone, R.J.; Walker, J.R.; Zhang, J.; Agarwalla, S.; Tenen, D.G.; Peters, E.C.; Reddy, V.A. Overexpression of TRIB2 in human lung cancers contributes to tumorigenesis through downregulation of C/EBPα. Oncogene 2011, 30, 3328–3335. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Chen, Y.; Yang, Y.; Zhang, X.; Ma, L.; Xue, X.; Qiao, Y.; Wang, J. TRIB2 modulates proteasome function to reduce ubiquitin stability and protect liver cancer cells against oxidative stress. Cell Death Dis. 2021, 12. [Google Scholar] [CrossRef]
- Hill, R.; Madureira, P.A.; Ferreira, B.; Baptista, I.; Machado, S.; Colaço, L.; Dos Santos, M.; Liu, N.; Dopazo, A.; Ugurel, S.; et al. TRIB2 confers resistance to anti-cancer therapy by activating the serine/threonine protein kinase AKT. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Zanella, F.; Renner, O.; Garcia, B.; Callejas, S.; Dopazo, A.; Peregrina, S.; Carnero, A.; Link, W. Human TRIB2 is a repressor of FOXO that contributes to the malignant phenotype of melanoma cells. Oncogene 2010, 29, 2973–2982. [Google Scholar] [CrossRef] [Green Version]
- Link, W. Tribbles breaking bad: TRIB2 suppresses FOXO and acts as an oncogenic protein in melanoma. Biochem. Soc. Trans. 2015, 43, 1085–1088. [Google Scholar] [CrossRef]
- Takasato, M.; Kobayashi, C.; Okabayashi, K.; Kiyonari, H.; Oshima, N.; Asashima, M.; Nishinakamura, R. Trb2, a mouse homolog of tribbles, is dispensable for kidney and mouse development. Biochem. Biophys. Res. Commun. 2008, 373, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.O.; Nygaard, J.V.; Burns, J.S.; Raarup, M.K.; Nyengaard, J.R.; Bünger, C.; Besenbacher, F.; Howard, K.A.; Kassem, M.; Kjems, J. SiRNA nanoparticle functionalization of nanostructured scaffolds enables controlled multilineage differentiation of stem cells. Mol. Ther. 2010, 18, 2018–2027. [Google Scholar] [CrossRef] [PubMed]
- Naiki, T.; Saijou, E.; Miyaoka, Y.; Sekine, K.; Miyajima, A. TRB2, a mouse tribbles ortholog, suppresses adipocyte differentiation by inhibiting AKT and C/EBP. J. Biol. Chem. 2007, 282, 24075–24082. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, K.; Ogawa, A.; Miyashita, H.; Tabara, Y.; Igase, M.; Kohara, K.; Miki, T.; Kagawa, Y.; Yanagisawa, Y.; Katashima, M.; et al. Positive natural selection of TRIB2, a novel gene that influences visceral fat accumulation, in East Asia. Hum. Genet. 2013, 132, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Basatvat, S.; Carter, D.A.L.; Kiss-Toth, E.; Fazeli, A. Tribbles role in reproduction. Biochem. Soc. Trans. 2015, 43, 1116–1121. [Google Scholar] [CrossRef]
- Warma, A.; Lussier, J.G.; Ndiaye, K. Tribbles pseudokinase 2 (Trib2) regulates expression of binding partners in bovine granulosa cells. Int. J. Mol. Sci. 2021, 22, 1533. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.R.; Yang-Yen, H.F.; Lien, H.W.; Liao, W.H.; Huang, C.J.; Lin, L.I.; Li, C.L.; Yen, J.J.Y. Murine tribbles homolog 2 deficiency affects erythroid progenitor development and confers macrocytic anemia on mice. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Johnston, J.; Basatvat, S.; Ilyas, Z.; Francis, S.; Kiss-Toth, E. Tribbles in inflammation. Biochem. Soc. Trans. 2015, 43, 1069–1074. [Google Scholar] [CrossRef]
- Eder, K.; Guan, H.; Sung, H.Y.; Ward, J.; Angyal, A.; Janas, M.; Sarmay, G.; Duda, E.; Turner, M.; Dower, S.K.; et al. Tribbles-2 is a novel regulator of inflammatory activation of monocytes. Int. Immunol. 2008, 20, 1543–1550. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; James, C.H.; Patel, L.; Smith, A.; Burnand, K.G.; Rahmoune, H.; Lamb, J.R.; Davis, B. Human tribbles homologue 2 is expressed in unstable regions of carotid plaques and regulates macrophage IL-10 in vitro. Clin. Sci. (Lond) 2009, 116, 241–248. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.; Kalathur, R.K.; Colaco, L.; Brandao, R.; Ugurel, S.; Futschik, M.; Link, W. TRIB2 as a biomarker for diagnosis and progression of melanoma. Carcinogenesis 2015, 36, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Hannon, M.M.; Lohan, F.; Erbilgin, Y.; Sayitoglu, M.; O’Hagan, K.; Mills, K.; Ozbek, U.; Keeshan, K. Elevated TRIB2 with NOTCH1 activation in paediatric/adult T-ALL. Br. J. Haematol. 2012, 158, 626–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, K.L.; Rishi, L.; Keeshan, K. Tribbles in acute leukemia. Blood 2013, 121, 4265–4270. [Google Scholar] [CrossRef] [Green Version]
- Kritsch, D.; Hoffmann, F.; Steinbach, D.; Jansen, L.; Mary Photini, S.; Gajda, M.; Mosig, A.S.; Sonnemann, J.; Peters, S.; Melnikova, M.; et al. Tribbles 2 mediates cisplatin sensitivity and DNA damage response in epithelial ovarian cancer. Int. J. Cancer 2017, 141, 1600–1614. [Google Scholar] [CrossRef] [PubMed]
- Miyanaga, A.; Masuda, M.; Motoi, N.; Tsuta, K.; Nakamura, Y.; Nishijima, N.; Watanabe, S.-I.; Asamura, H.; Tsuchida, A.; Seike, M.; et al. Whole-exome and RNA sequencing of pulmonary carcinoid reveals chromosomal rearrangements associated with recurrence. Lung Cancer 2020, 145, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Yu, D.; Perez-Soler, R.; Klostergaard, J.; Zou, Y. TRIB2 contributes to cisplatin resistance in small cell lung cancer. Oncotarget 2017, 8, 109596–109608. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Zhou, X.; Qu, H.; Ma, Y.; Yue, Z.; Shang, W.; Wang, P.; Xie, S.; Li, Y.; Sun, Y. TRIB2 knockdown as a regulator of chemotherapy resistance and proliferation via the ERK/STAT3 signaling pathway in human chronic myelogenous leukemia K562/ADM cells. Oncol. Rep. 2018, 39, 1910–1918. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zuo, J.; Wahafu, A.; Wang, M.-D.; Li, R.-C.; Xie, W.-F. Combined elevation of TRIB2 and MAP3K1 indicates poor prognosis and chemoresistance to temozolomide in glioblastoma. CNS Neurosci. Ther. 2020, 26, 297–308. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, C.; Yalla, K.; Salome, M.; Moka, H.A.; Castaneda, E.G.; Eyers, P.A.; Keeshan, K. Trib2 expression in granulocyte-monocyte progenitors drives a highly drug resistant acute myeloid leukaemia linked to elevated Bcl2. Oncotarget 2018, 9, 14977–14992. [Google Scholar] [CrossRef]
- Machado, S.; Silva, A.; De Sousa-Coelho, A.L.; Duarte, I.; Grenho, I.; Santos, B.; Mayoral-Varo, V.; Megias, D.; Sánchez-Cabo, F.; Dopazo, A.; et al. Harmine and Piperlongumine Revert TRIB2-Mediated Drug Resistance. Cancers 2020, 12, 3689. [Google Scholar] [CrossRef]
- Luan, W.; Shi, Y.; Zhou, Z.; Xia, Y.; Wang, J. circRNA_0084043 promote malignant melanoma progression via miR-153-3p/Snail axis. Biochem. Biophys. Res. Commun. 2018, 502, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chen, J.; Wa, Q.; He, M.; Wang, X.; Zhou, J.; Cen, Y. Knockdown of circ_0084043 suppresses the development of human melanoma cells through miR-429/tribbles homolog 2 axis and Wnt/β-catenin pathway. Life Sci. 2020, 243, 117323. [Google Scholar] [CrossRef] [PubMed]
- Sakai, S.; Miyajima, C.; Uchida, C.; Itoh, Y.; Hayashi, H.; Inoue, Y. Tribbles-Related Protein Family Members as Regulators or Substrates of the Ubiquitin-Proteasome System in Cancer Development. Curr. Cancer Drug Targets 2016, 16, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.; Zhu, X.; Zhang, Y.; Zou, J.; Li, J.; Kong, L.; Zhang, H. Tribbles homolog 2 promotes hepatic fibrosis and hepatocarcinogenesis through phosphatase 1A-Mediated stabilization of yes-associated protein. Liver Int. Off. J. Int. Assoc. Study Liver 2021. [Google Scholar] [CrossRef]
- Liang, K.L.; O’Connor, C.; Veiga, J.P.; McCarthy, T.V.; Keeshan, K. TRIB2 regulates normal and stress-induced thymocyte proliferation. Cell Discov. 2016, 2, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, S.J.; Mack, E.A.; Rome, K.S.; Pajcini, K.V.; Ohtani, T.; Xu, L.; Li, Y.; Meijerink, J.P.P.; Faryabi, R.B.; Pear, W.S. Trib2 suppresses tumor initiation in Notch-driven T-ALL. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeshan, K.; Shestova, O.; Ussin, L.S.; Pear, W.S. Tribbles homolog 2 (Trib2) and HoxA9 cooperate to accelerate acute myelogenous leukemia. Blood Cells Mol. Dis. 2008, 40, 119–121. [Google Scholar] [CrossRef]
- Collins, C.T.; Hess, J.L. Deregulation of the HOXA9/MEIS1 Axis in Acute Leukemia. Curr. Opin. Hematol. 2016, 23, 354–361. [Google Scholar] [CrossRef]
- Pineault, N.; Abramovich, C.; Ohta, H.; Humphries, R.K. Differential and Common Leukemogenic Potentials of Multiple NUP98-Hox Fusion Proteins Alone or with Meis1. Mol. Cell. Biol. 2004, 24, 1907–1917. [Google Scholar] [CrossRef] [Green Version]
- Thorsteinsdottir, U.; Kroon, E.; Jerome, L.; Blasi, F.; Sauvageau, G. Defining roles for HOX and MEIS1 genes in induction of acute myeloid leukemia. Mol. Cell. Biol. 2001, 21, 224–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeshan, K.; He, Y.; Wouters, B.J.; Shestova, O.; Xu, L.; Sai, H.; Rodriguez, C.G.; Maillard, I.; Tobias, J.W.; Valk, P.; et al. Tribbles homolog 2 inactivates C/EBPalpha and causes acute myelogenous leukemia. Cancer Cell 2006, 10, 401–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, C.; Lohan, F.; Campos, J.; Ohlsson, E.; Salomè, M.; Forde, C.; Artschwager, R.; Liskamp, R.M.; Cahill, M.R.; Kiely, P.A.; et al. The presence of C/EBPα and its degradation are both required for TRIB2-mediated leukaemia. Oncogene 2016, 35, 5272–5281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedhia, P.H.; Keeshan, K.; Uljon, S.; Xu, L.; Vega, M.E.; Shestova, O.; Zaks-Zilberman, M.; Romany, C.; Blacklow, S.C.; Pear, W.S. Differential ability of Tribbles family members to promote degradation of C/EBPalpha and induce acute myelogenous leukemia. Blood 2010, 116, 1321–1328. [Google Scholar] [CrossRef]
- Zhang, P.; Iwasaki-Arai, J.; Iwasaki, H.; Fenyus, M.L.; Dayaram, T.; Owens, B.M.; Shigematsu, H.; Levantini, E.; Huettner, C.S.; Lekstrom-Himes, J.A.; et al. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity 2004, 21, 853–863. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, A.; Kato, J.; Nakamae, I.; Yoneda-Kato, N. COP1 targets C/EBPα for degradation and induces acute myeloid leukemia via Trib1. Blood 2013, 122, 1750–1760. [Google Scholar] [CrossRef] [PubMed]
- Kole, C.; Charalampakis, N.; Tsakatikas, S.; Vailas, M.; Moris, D.; Gkotsis, E.; Kykalos, S.; Karamouzis, M.V.; Schizas, D. Immunotherapy for Hepatocellular Carcinoma: A 2021 Update. Cancers 2020, 12, 2859. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Piorońska, W.; Battello, N.; Zimmer, A.D.; Dewidar, B.; Han, M.; Pereira, S.; Blagojevic, B.; Castven, D.; Charlestin, V.; et al. Severe metabolic alterations in liver cancer lead to ERK pathway activation and drug resistance. EBioMedicine 2020, 54, 102699. [Google Scholar] [CrossRef]
- Wang, W.; Smits, R.; Hao, H.; He, C. Wnt/β-Catenin Signaling in Liver Cancers. Cancers 2019, 11, 926. [Google Scholar] [CrossRef] [Green Version]
- Lippert, T.H.; Ruoff, H.-J.; Volm, M. Intrinsic and acquired drug resistance in malignant tumors. The main reason for therapeutic failure. Arzneimittelforschung 2008, 58, 261–264. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Brabec, V.; Kaspárková, J.; Vrána, O.; Nováková, O.; Cox, J.W.; Qu, Y.; Farrell, N. DNA modifications by a novel bifunctional trinuclear platinum phase I anticancer agent. Biochemistry 1999, 38, 6781–6790. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Kang, Y.; Chen, L.; Wang, H.; Liu, J.; Zeng, S.; Yu, L. The Drug-Resistance Mechanisms of Five Platinum-Based Antitumor Agents. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arimoto-Ishida, E.; Ohmichi, M.; Mabuchi, S.; Takahashi, T.; Ohshima, C.; Hayakawa, J.; Kimura, A.; Takahashi, K.; Nishio, Y.; Sakata, M.; et al. Inhibition of phosphorylation of a forkhead transcription factor sensitizes human ovarian cancer cells to cisplatin. Endocrinology 2004, 145, 2014–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcamone, F.; Cassinelli, G.; Fantini, G.; Grein, A.; Orezzi, P.; Pol, C.; Spalla, C. Adriamycin, 14-hydroxydaunomycin, a new antitumor antibiotic from S. peucetius var. caesius. Biotechnol. Bioeng. 1969, 11, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Zunino, F.; Gambetta, R.; Di Marco, A.; Zaccara, A. Interaction of daunomycin and its derivatives with DNA. Biochim. Biophys. Acta Nucleic Acids Protein Synth. 1972, 277, 489–498. [Google Scholar] [CrossRef]
- Baran, Y.; Gür, B.; Kaya, P.; Ural, A.U.; Avcu, F.; Gündüz, U. Upregulation of multi drug resistance genes in doxorubicin resistant human acute myelogeneous leukemia cells and reversal of the resistance. Hematology 2007, 12, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, T.; Kanno, Y.; Yamazaki, Y.; Takahara, T.; Miyata, S.; Nakamura, T. Trib1 links the MEK1/ERK pathway in myeloid leukemogenesis. Blood 2010, 116, 2768–2775. [Google Scholar] [CrossRef]
- Tang, B.; Wu, W.; Zhang, Q.; Sun, Y.; Cui, Y.; Wu, F.; Wei, X.; Qi, G.; Liang, X.; Tang, F.; et al. Inhibition of tribbles protein-1 attenuates radioresistance in human glioma cells. Sci. Rep. 2015, 5, 15961. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Wang, G.; Wang, G.; Zhuang, J.; He, S.; Song, Y.; Ni, J.; Xia, W.; Wang, J. The oncogenic role of Tribbles 1 in hepatocellular carcinoma is mediated by a feedback loop involving microRNA-23a and p53. Front. Physiol. 2017, 8, 789. [Google Scholar] [CrossRef] [Green Version]
- Shahrouzi, P.; Astobiza, I.; Cortazar, A.R.; Torrano, V.; Macchia, A.; Flores, J.M.; Niespolo, C.; Mendizabal, I.; Caloto, R.; Ercilla, A.; et al. Genomic and Functional Regulation of TRIB1 Contributes to Prostate Cancer Pathogenesis. Cancers 2020, 12, 2593. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, N.; Ishii, H.; Mimori, K.; Takatsuno, Y.; Kim, H.; Hirose, H.; Sekimoto, M.; Doki, Y.; Mori, M. Abnormal expression of TRIB3 in colorectal cancer: A novel marker for prognosis. Br. J. Cancer 2009, 101, 1664–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Wu, Q.; Peng, X.; Yu, B. miR-509-5p Inhibits the Proliferation and Invasion of Osteosarcoma by Targeting TRIB2. Biomed. Res. Int. 2019, 2019, 2523032. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020. [Google Scholar] [CrossRef]
- Neklesa, T.K.; Tae, H.S.; Schneekloth, A.R.; Stulberg, M.J.; Corson, T.W.; Sundberg, T.B.; Raina, K.; Holley, S.A.; Crews, C.M. Small-molecule hydrophobic tagging-induced degradation of HaloTag fusion proteins. Nat. Chem. Biol. 2011, 7, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Transcription Factor | Regulation | Cell Type | References |
|---|---|---|---|
| E2F1 | Positive | Acute myeloid leukemia (AML) mice | [32] |
| C/EBPα-p30 | Positive | Acute myeloid leukemia (AML) mice | [32] |
| TAL1 | Positive | Acute lymphoblastic leukemia (ALL) human | [33] |
| NOTCH1 | Positive | Acute lymphoblastic leukemia (ALL) human | [33] |
| Smad3 | Positive | Lung adenocarcinoma human | [34] |
| Mesi1 | Positive | Acute myeloid leukemia (AML) mice | [35] |
| PITX1 | Positive | Acute lymphoblastic leukemia (ALL) human | [36] |
| FOG-1 | Positive | Bone marrow mice | [37] |
| GATA-1 | Positive | Bone marrow mice | [37] |
| TCF | Positive | Liver cancer cells human | [38] |
| FoxA | Positive | Liver cancer cells human | [38] |
| C/EBPα-p42 | Negative | Acute myeloid leukemia (AML) mice | [32] |
| E2A | Negative | Acute lymphoblastic leukemia (ALL) human | [33] |
| Cancer Type | TRIB2 Function | Target | Cell Line/Model | Mode of Action | References |
|---|---|---|---|---|---|
| Lung | Oncogenic | C/EBPα, pSmad3, TRIM21 | A549, LTEP-a-2 cells NOD/SCID mice Patients’ tissue samples | Promotes proliferation, sphere formation and in vivo tumorigenicity | [34,47,66] |
| Colorectal | Oncogenic | AP4/p21, AK | SW48 and LoVo cells Primary tumour samples | Blocks cellular senescence Confers resistance to PI3K inhibitors | [46,49] |
| Pancreatic | Oncogenic | miR505/ ZEB1-AS1 | SW1990 and Capan-1 cells | Promotes viability, migration and invasion | [42] |
| Ovarian | Tumour suppressor | p21 | A2780 cell line Patient tumour samples | Induces of a cisplatin-dependent cell cycle arrest and apoptosis | [65] |
| Melanoma | Oncogenic | FOXO, β-catenin, c-Myc and cyclinD1 | Sk-Mel-28, Sk-Mel-94, Sk-Mel-19, UACC-257, Hs 895T, Sk-Mel-147, A375 and HaCaT cells Melanoma xenograft | Promotes proliferation and drug resistance. Inhibition of apoptosis | [50,72,73,74] |
| Liver | Oncogenic | βTrCP, C/EBPα, SMURF1, PCBP2 | HepG2, Bel-7402, Bel-7404, Hep3B, Huh7, SMMC-7721, Bel-7402 and HL-7702 cells Mouse model of liver fibrosis and HCC | Promotes tumorigenesis, tumour growth and cell survival | [10,12,48,75] |
| Leukemia | Oncogenic | C/EBPα | 32D cells C/EBPαfl/fl mouse model | Cooperates with HOXA9 in AML Confers proliferative advantage and induce AML | [63,64] |
| Tumour suppressor | Notch p38 | Patient samples | Is required for p38 MAPK signalling, induction of cell cycle checkpoint response and apoptosis. Its deficiency accelerates the onset of ALL | [15,45,76] |
| Tumour Type | Anticancer Drug | Cell Line/Model | Role of TRIB2 in Resistance | References |
|---|---|---|---|---|
| Melanoma Osteosarcoma | PI3K inhibitors mTOR inhibitors Dacarbazine Gemcitabine 5-fluorouracil | U2OS osteosarcoma cell line Patient melanoma samples | TRIB2 disrupts AKT/FOXO signalling by increasing AKT and MDM2 phosphorylation | [49] |
| Glioblastoma | Temozolomide | Patient tumour samples | Combined increase in TRIB2 and MAP3K1 | [69] |
| Small Cell Lung Cancer | Cisplatin | NCI-H69 xenograft mouse model, Patient tumour samples | TRIB2 overexpression increases C/EBPα downregulation | [67] |
| Epithelial Ovarian Cancer | Cisplatin | A2780 cell line, Patient tumour samples | There is a correlation with TRIB2 knockdown and resistance | [65] |
| 5′-Aza-Cytidine | A2780 Cisplatin resistant cell line | TRIB2 is overexpressed in resistant cells | ||
| Acute Myeloid Leukemia | Doxorubicin | K562 cell line | TRIB2 promotes the expression of cell membrane transporters MRP1 and MDR1 increasing the efflux of drug | [68] |
| Cytarabine Daunorubicin | K562 cell line | TRIB2 overexpression increases BCL2 expression, promoting antiapoptic signalling |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayoral-Varo, V.; Jiménez, L.; Link, W. The Critical Role of TRIB2 in Cancer and Therapy Resistance. Cancers 2021, 13, 2701. https://doi.org/10.3390/cancers13112701
Mayoral-Varo V, Jiménez L, Link W. The Critical Role of TRIB2 in Cancer and Therapy Resistance. Cancers. 2021; 13(11):2701. https://doi.org/10.3390/cancers13112701
Chicago/Turabian StyleMayoral-Varo, Victor, Lucía Jiménez, and Wolfgang Link. 2021. "The Critical Role of TRIB2 in Cancer and Therapy Resistance" Cancers 13, no. 11: 2701. https://doi.org/10.3390/cancers13112701
APA StyleMayoral-Varo, V., Jiménez, L., & Link, W. (2021). The Critical Role of TRIB2 in Cancer and Therapy Resistance. Cancers, 13(11), 2701. https://doi.org/10.3390/cancers13112701