
Recent Advances in Glioma Therapy: Combining Vascular Normalization and Immune Checkpoint Blockade


Abstract
:Simple Summary
Abstract
1. Introduction
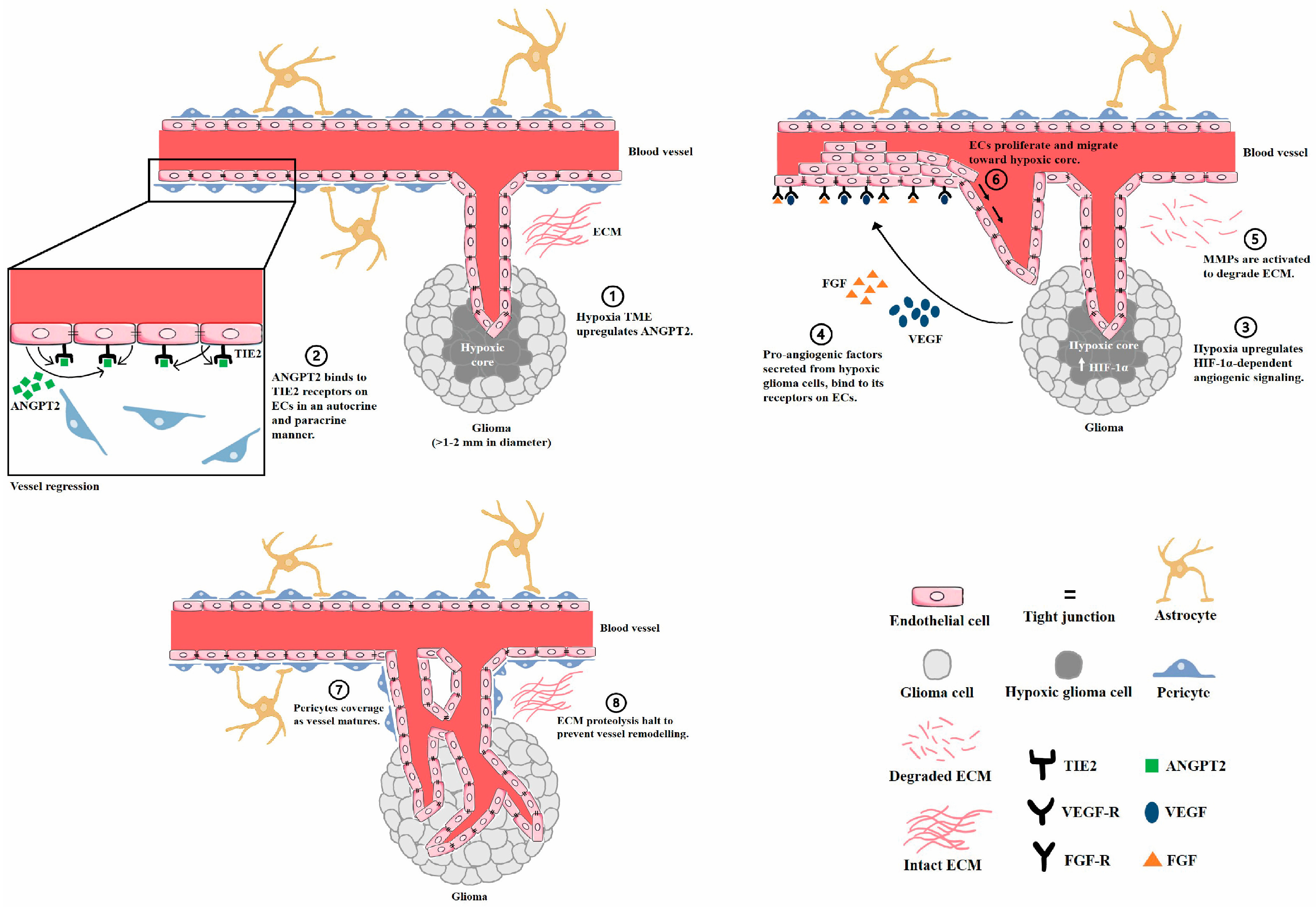
2. Glioma Angiogenesis
Perivascular Niche and Vascular Minicry in the Glioma Microenvironment
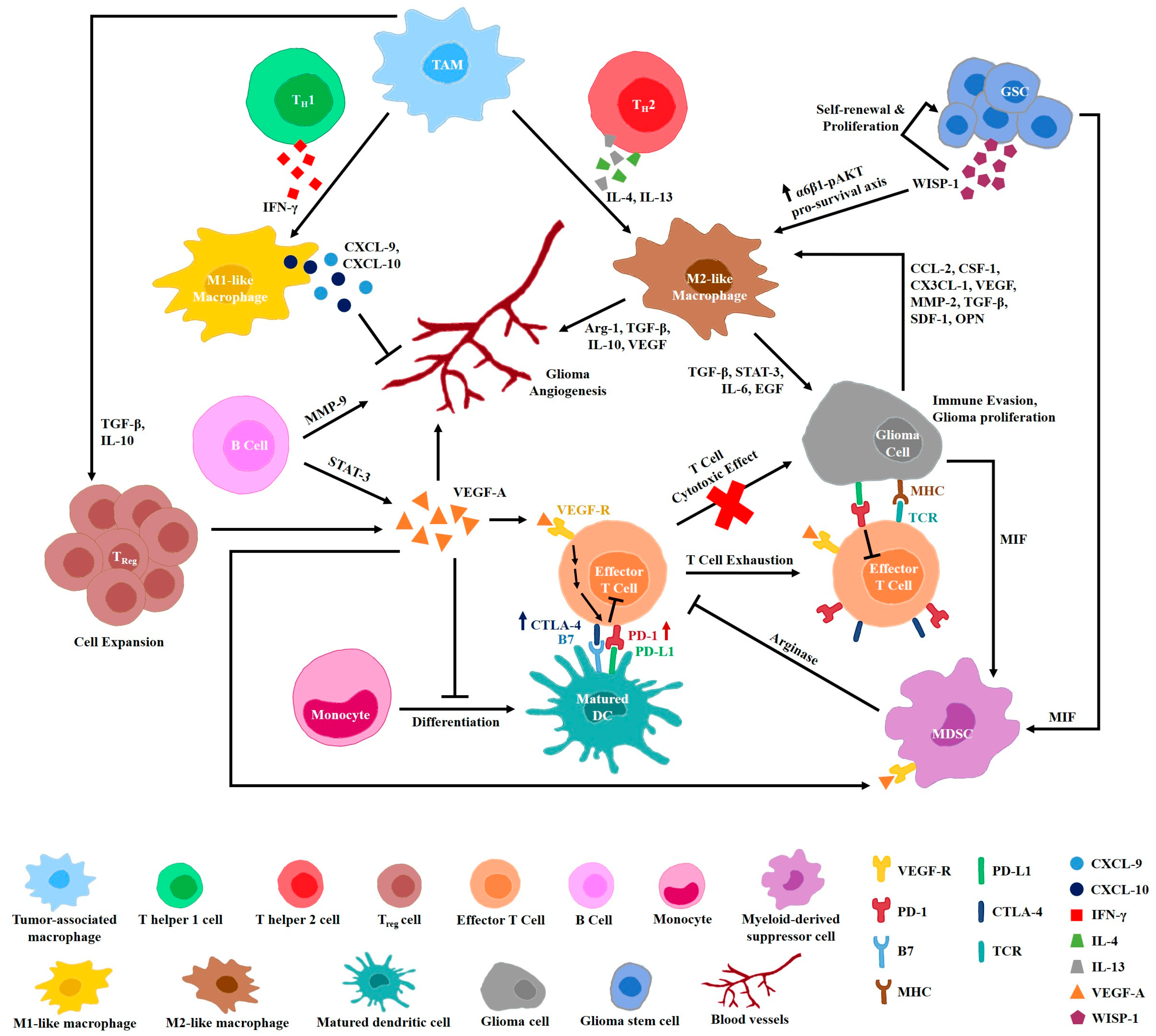
3. Glioma Immune Microenvironment
Crosstalk between Immune Cells and Angiogenesis
4. Therapeutic Intervention
4.1. Antiangiogenic Therapy
4.2. Immune Checkpoint Blockade
4.2.1. PD-1/PD-L1 Axis
4.2.2. CTLA-4/CD28/B7 Axis
4.2.3. LAG-3
4.2.4. TIGIT
4.2.5. TIM-3
4.3. Strategies to Increase Treatment Efficacy
4.3.1. Vascular Normalization Increases T-Cell Infiltration
4.3.2. Combining ICB with Antiangiogenic Molecules to Increase Treatment Efficacy
4.4. Challenges to ICB in GBM
4.4.1. Discordance between Preclinical and Clinical Study Settings
4.4.2. “Windows” of Opportunity
4.4.3. Biomarkers
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Kesari, S.; Steinberg, D.M.; Toms, S.A.; Taylor, L.P.; Lieberman, F.; Silvani, A.; Fink, K.L.; et al. Maintenance Therapy with Tumor-Treating Fields Plus Temozolomide vs. Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. JAMA 2015, 314, 2535–2543. [Google Scholar] [CrossRef]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015, 25, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Teo, W.Y.; Sekar, K.; Seshachalam, P.; Shen, J.; Chow, W.Y.; Lau, C.C.; Yang, H.; Park, J.; Kang, S.G.; Li, X.; et al. Relevance of a TCGA-derived Glioblastoma Subtype Gene-Classifier among Patient Populations. Sci. Rep. 2019, 9, 7442. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Fonseca, A.C.; Badie, B. Microglia and macrophages in malignant gliomas: Recent discoveries and implications for promising therapies. Clin. Dev. Immunol. 2013, 2013, 264124. [Google Scholar] [CrossRef]
- Montana, V.; Sontheimer, H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J. Neurosci. 2011, 31, 4858–4867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellis-Davies, G.; Sontheimer, H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 2014, 5, 4196. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Xu, S.L.; Duan, J.J.; Yi, L.; Guo, Y.F.; Shi, Y.; Li, L.; Yang, Z.Y.; Liao, X.M.; Cai, J.; et al. Invasion of white matter tracts by glioma stem cells is regulated by a NOTCH1-SOX2 positive-feedback loop. Nat. Neurosci. 2019, 22, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; ElAli, A.; Virgintino, D.; Gilbert, M.R. Blood-brain barrier pericyte importance in malignant gliomas: What we can learn from stroke and Alzheimer’s disease. Neuro-Oncology 2017, 19, 1173–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Tonini, T.; Rossi, F.; Claudio, P.P. Molecular basis of angiogenesis and cancer. Oncogene 2003, 22, 6549–6556. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [Green Version]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef]
- Genoud, V.; Marinari, E.; Nikolaev, S.I.; Castle, J.C.; Bukur, V.; Dietrich, P.Y.; Okada, H.; Walker, P.R. Responsiveness to anti-PD-1 and anti-CTLA-4 immune checkpoint blockade in SB28 and GL261 mouse glioma models. Oncoimmunology 2018, 7, e1501137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol. Res. 2016, 4, 124–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, L.; Xiao, H.; Xu, M.; Ye, X.; Hu, J.; Li, F.; Li, M.; Luo, C.; Yu, S.; Bian, X.; et al. Glioma-initiating cells: A predominant role in microglia/macrophages tropism to glioma. J. Neuroimmunol. 2011, 232, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Aslan, K.; Turco, V.; Blobner, J.; Sonner, J.K.; Liuzzi, A.R.; Nunez, N.G.; De Feo, D.; Kickingereder, P.; Fischer, M.; Green, E.; et al. Heterogeneity of response to immune checkpoint blockade in hypermutated experimental gliomas. Nat. Commun. 2020, 11, 931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habimana-Griffin, L.; Ye, D.; Carpenter, J.; Prior, J.; Sudlow, G.; Marsala, L.; Mixdorf, M.; Rubin, J.; Chen, H.; Achilefu, S. Intracranial glioma xenograft model rapidly reestablishes blood-brain barrier integrity for longitudinal imaging of tumor progression using fluorescence molecular tomography and contrast agents. J. Biomed. Opt. 2020, 25. [Google Scholar] [CrossRef] [PubMed]
- Scharpfenecker, M.; Fiedler, U.; Reiss, Y.; Augustin, H.G. The Tie-2 ligand angiopoietin-2 destabilizes quiescent endothelium through an internal autocrine loop mechanism. J. Cell Sci. 2005, 118, 771–780. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Guo, P.; Fang, Q.; Tao, H.Q.; Wang, D.; Nagane, M.; Huang, H.J.; Gunji, Y.; Nishikawa, R.; Alitalo, K.; et al. Angiopoietin-2 induces human glioma invasion through the activation of matrix metalloprotease-2. Proc. Natl. Acad. Sci. USA 2003, 100, 8904–8909. [Google Scholar] [CrossRef] [Green Version]
- Brat, D.J.; Castellano-Sanchez, A.A.; Hunter, S.B.; Pecot, M.; Cohen, C.; Hammond, E.H.; Devi, S.N.; Kaur, B.; Van Meir, E.G. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 2004, 64, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Winkler, F.; Kozin, S.V.; Tong, R.T.; Chae, S.S.; Booth, M.F.; Garkavtsev, I.; Xu, L.; Hicklin, D.J.; Fukumura, D.; di Tomaso, E.; et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: Role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell 2004, 6, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Bullitt, E.; Zeng, D.; Gerig, G.; Aylward, S.; Joshi, S.; Smith, J.K.; Lin, W.; Ewend, M.G. Vessel tortuosity and brain tumor malignancy: A blinded study. Acad. Radiol. 2005, 12, 1232–1240. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Mao, S.; Li, H.; Zheng, M.; Yi, L.; Lin, J.M.; Lin, Z.X. The pathological structure of the perivascular niche in different microvascular patterns of glioblastoma. PLoS ONE 2017, 12, e0182183. [Google Scholar] [CrossRef] [Green Version]
- Eyler, C.E.; Wu, Q.; Yan, K.; MacSwords, J.M.; Chandler-Militello, D.; Misuraca, K.L.; Lathia, J.D.; Forrester, M.T.; Lee, J.; Stamler, J.S.; et al. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell 2011, 146, 53–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krusche, B.; Ottone, C.; Clements, M.P.; Johnstone, E.R.; Goetsch, K.; Lieven, H.; Mota, S.G.; Singh, P.; Khadayate, S.; Ashraf, A.; et al. EphrinB2 drives perivascular invasion and proliferation of glioblastoma stem-like cells. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, M.G.; Nyanyo, D.; Hung, C.K.; Goerger, J.P.; Warren, R.Z.; Williams, R.M.; Nishimura, N.; Fischbach, C. Endothelial cells promote 3D invasion of GBM by IL-8-dependent induction of cancer stem cell properties. Sci. Rep. 2019, 9, 9069. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef] [Green Version]
- Ho, I.A.W.; Shim, W.S.N. Contribution of the Microenvironmental Niche to Glioblastoma Heterogeneity. Biomed. Res. Int. 2017, 2017, 9634172. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and Niche Concept. Cancers 2018, 11, 5. [Google Scholar] [CrossRef] [Green Version]
- Shiraki, Y.; Mii, S.; Enomoto, A.; Momota, H.; Han, Y.P.; Kato, T.; Ushida, K.; Kato, A.; Asai, N.; Murakumo, Y.; et al. Significance of perivascular tumour cells defined by CD109 expression in progression of glioma. J. Pathol. 2017, 243, 468–480. [Google Scholar] [CrossRef]
- Minata, M.; Audia, A.; Shi, J.; Lu, S.; Bernstock, J.; Pavlyukov, M.S.; Das, A.; Kim, S.H.; Shin, Y.J.; Lee, Y.; et al. Phenotypic Plasticity of Invasive Edge Glioma Stem-like Cells in Response to Ionizing Radiation. Cell Rep. 2019, 26, 1893–1905. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhang, Q.; Zhou, L.; Guan, Y.; Chen, S.; Zhang, Y.; Han, X. Inhibitory effects of compound DMBT on hypoxia-induced vasculogenic mimicry in human breast cancer. Biomed. Pharmacother. 2017, 96, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, Y.; Zhang, Q.; Guo, C.; Chen, F.; Xi, S.; Zeng, J.; Ke, C.; Sharma, H.S.; Chen, Z. Expression of Twist associated to microcirculation patterns of human glioma correlated with progression and survival of the patient. Int. Rev. Neurobiol. 2020, 151, 201–217. [Google Scholar] [CrossRef]
- Zhang, D.; Sun, B.; Zhao, X.; Sun, H.; An, J.; Lin, X.; Zhu, D.; Zhao, X.; Wang, X.; Liu, F.; et al. Twist1 accelerates tumour vasculogenic mimicry by inhibiting Claudin15 expression in triple-negative breast cancer. J. Cell. Mol. Med. 2020, 24, 7163–7174. [Google Scholar] [CrossRef]
- Cao, W.; Xu, C.; Li, X.; Yang, X. Twist1 promotes astrocytoma development by stimulating vasculogenic mimicry. Oncol. Lett. 2019, 18, 846–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Xu, Y.; Sun, J.; Chen, W.; Zhao, L.; Ma, C.; Wang, Q.; Sun, J.; Huang, B.; Zhang, Y.; et al. M2-like tumor-associated macrophages drive vasculogenic mimicry through amplification of IL-6 expression in glioma cells. Oncotarget 2017, 8, 819–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, S.; Francescone, R.; Faibish, M.; Bentley, B.; Taylor, S.L.; Oh, D.; Schapiro, R.; Moral, L.; Yan, W.; Shao, R. Transdifferentiation of glioblastoma stem-like cells into mural cells drives vasculogenic mimicry in glioblastomas. J. Neurosci. 2012, 32, 12950–12960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef]
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S.; et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4274–4280. [Google Scholar] [CrossRef] [Green Version]
- Fessler, E.; Borovski, T.; Medema, J.P. Endothelial cells induce cancer stem cell features in differentiated glioblastoma cells via bFGF. Mol. Cancer 2015, 14, 157. [Google Scholar] [CrossRef] [Green Version]
- Baisiwala, S.; Auffinger, B.; Caragher, S.P.; Shireman, J.M.; Ahsan, R.; Lee, G.; Hasan, T.; Park, C.; Saathoff, M.R.; Christensen, A.C.; et al. Chemotherapeutic Stress Induces Transdifferentiation of Glioblastoma Cells to Endothelial Cells and Promotes Vascular Mimicry. Stem Cells Int. 2019, 2019, 6107456. [Google Scholar] [CrossRef]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, F.J.; Orr, B.A.; Ligon, K.L.; Eberhart, C.G. Neoplastic cells are a rare component in human glioblastoma microvasculature. Oncotarget 2012, 3, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Wang, Q.; Wang, Y.A.; Hua, S.; Sauve, C.G.; Ong, D.; Lan, Z.D.; Chang, Q.; Ho, Y.W.; Monasterio, M.M.; et al. Epigenetic Activation of WNT5A Drives Glioblastoma Stem Cell Differentiation and Invasive Growth. Cell 2016, 167, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, S.; Baluk, P.; Kaidoh, T.; Haskell, A.; Jain, R.K.; McDonald, D.M. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2002, 160, 985–1000. [Google Scholar] [CrossRef] [Green Version]
- Caspani, E.M.; Crossley, P.H.; Redondo-Garcia, C.; Martinez, S. Glioblastoma: A pathogenic crosstalk between tumor cells and pericytes. PLoS ONE 2014, 9, e101402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinton, L.; Masetto, E.; Vettore, M.; Solito, S.; Magri, S.; D’Andolfi, M.; Del Bianco, P.; Lollo, G.; Benoit, J.P.; Okada, H.; et al. The immune suppressive microenvironment of human gliomas depends on the accumulation of bone marrow-derived macrophages in the center of the lesion. J. Immunother. Cancer 2019, 7, 58. [Google Scholar] [CrossRef]
- Martinez-Lage, M.; Lynch, T.M.; Bi, Y.; Cocito, C.; Way, G.P.; Pal, S.; Haller, J.; Yan, R.E.; Ziober, A.; Nguyen, A.; et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol. Commun. 2019, 7, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2012, 60, 502–514. [Google Scholar] [CrossRef]
- Kushchayev, S.V.; Kushchayeva, Y.S.; Wiener, P.C.; Scheck, A.C.; Badie, B.; Preul, M.C. Monocyte-derived cells of the brain and malignant gliomas: The double face of Janus. World Neurosurg. 2014, 82, 1171–1186. [Google Scholar] [CrossRef]
- Rutledge, W.C.; Kong, J.; Gao, J.; Gutman, D.A.; Cooper, L.A.; Appin, C.; Park, Y.; Scarpace, L.; Mikkelsen, T.; Cohen, M.L.; et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin. Cancer Res. 2013, 19, 4951–4960. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, B.C.; Showers, C.R.; Anderson, D.E.; Anderson, L.; Canoll, P.; Bruce, J.N.; Anderson, R.C. Tumor-associated macrophages in glioma: Friend or foe? J. Oncol. 2013, 2013, 486912. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Graeber, M.B. The molecular profile of microglia under the influence of glioma. Neuro-Oncology 2012, 14, 958–978. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.Y.; Ling, X.; Caruso, H.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Investig. 2019, 129, 137–149. [Google Scholar] [CrossRef]
- Zhang, J.; Sarkar, S.; Cua, R.; Zhou, Y.; Hader, W.; Yong, V.W. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis 2012, 33, 312–319. [Google Scholar] [CrossRef]
- Wang, S.C.; Hong, J.H.; Hsueh, C.; Chiang, C.S. Tumor-secreted SDF-1 promotes glioma invasiveness and TAM tropism toward hypoxia in a murine astrocytoma model. Lab Investig. 2012, 92, 151–162. [Google Scholar] [CrossRef]
- Atai, N.A.; Bansal, M.; Lo, C.; Bosman, J.; Tigchelaar, W.; Bosch, K.S.; Jonker, A.; De Witt Hamer, P.C.; Troost, D.; McCulloch, C.A.; et al. Osteopontin is up-regulated and associated with neutrophil and macrophage infiltration in glioblastoma. Immunology 2011, 132, 39–48. [Google Scholar] [CrossRef]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro-Oncology 2010, 12, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.T.; Lee, H.W.; Lee, H.O.; Song, H.J.; Jeong da, E.; Shin, S.; Kim, H.; Shin, Y.; Nam, D.H.; Jeong, B.C.; et al. Application of single-cell RNA sequencing in optimizing a combinatorial therapeutic strategy in metastatic renal cell carcinoma. Genome Biol. 2016, 17, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [Green Version]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [Green Version]
- Song, L.P.; Zhang, J.; Wu, S.F.; Huang, Y.; Zhao, Q.; Cao, J.P.; Wu, Y.L.; Wang, L.S.; Chen, G.Q. Hypoxia-inducible factor-1alpha-induced differentiation of myeloid leukemic cells is its transcriptional activity independent. Oncogene 2008, 27, 519–527. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P.C. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef]
- Matsubara, T.; Kanto, T.; Kuroda, S.; Yoshio, S.; Higashitani, K.; Kakita, N.; Miyazaki, M.; Sakakibara, M.; Hiramatsu, N.; Kasahara, A.; et al. TIE2-expressing monocytes as a diagnostic marker for hepatocellular carcinoma correlates with angiogenesis. Hepatology 2013, 57, 1416–1425. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Liu, D.; Cortes-Santiago, N.; Hossain, M.B.; Conrad, C.A.; Aldape, K.D.; Fuller, G.N.; Marini, F.C.; Alonso, M.M.; Idoate, M.A.; et al. Anti-vascular endothelial growth factor therapy-induced glioma invasion is associated with accumulation of Tie2-expressing monocytes. Oncotarget 2014, 5, 2208–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Palma, M.; Venneri, M.A.; Galli, R.; Sergi Sergi, L.; Politi, L.S.; Sampaolesi, M.; Naldini, L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 2005, 8, 211–226. [Google Scholar] [CrossRef] [Green Version]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4+ T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baer, C.; Squadrito, M.L.; Laoui, D.; Thompson, D.; Hansen, S.K.; Kiialainen, A.; Hoves, S.; Ries, C.H.; Ooi, C.H.; De Palma, M. Suppression of microRNA activity amplifies IFN-gamma-induced macrophage activation and promotes anti-tumour immunity. Nat. Cell Biol. 2016, 18, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Lee, H.; Pal, S.; Jove, V.; Deng, J.; Zhang, W.; Hoon, D.S.; Wakabayashi, M.; Forman, S.; Yu, H. B cells promote tumor progression via STAT3 regulated-angiogenesis. PLoS ONE 2013, 8, e64159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreu, P.; Johansson, M.; Affara, N.I.; Pucci, F.; Tan, T.; Junankar, S.; Korets, L.; Lam, J.; Tawfik, D.; DeNardo, D.G.; et al. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell 2010, 17, 121–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffelt, S.B.; Chen, Y.Y.; Muthana, M.; Welford, A.F.; Tal, A.O.; Scholz, A.; Plate, K.H.; Reiss, Y.; Murdoch, C.; De Palma, M.; et al. Angiopoietin 2 stimulates TIE2-expressing monocytes to suppress T cell activation and to promote regulatory T cell expansion. J. Immunol. 2011, 186, 4183–4190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, L.O.; Poirier, M.B.; Fortin, D. Transforming growth factor-beta and its implication in the malignancy of gliomas. Target Oncol. 2015, 10, 1–14. [Google Scholar] [CrossRef]
- Razavi, S.M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune Evasion Strategies of Glioblastoma. Front. Surg. 2016, 3, 11. [Google Scholar] [CrossRef]
- Tao, W.; Chu, C.; Zhou, W.; Huang, Z.; Zhai, K.; Fang, X.; Huang, Q.; Zhang, A.; Wang, X.; Yu, X.; et al. Dual Role of WISP1 in maintaining glioma stem cells and tumor-supportive macrophages in glioblastoma. Nat. Commun. 2020, 11, 3015. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.B.; Gao, K.D.; Jiang, T.; Cheng, S.J.; Li, W.B. Bevacizumab combined with chemotherapy for glioblastoma: A meta-analysis of randomized controlled trials. Oncotarget 2017, 8, 57337–57344. [Google Scholar] [CrossRef] [Green Version]
- Vredenburgh, J.J.; Desjardins, A.; Herndon, J.E., 2nd; Marcello, J.; Reardon, D.A.; Quinn, J.A.; Rich, J.N.; Sathornsumetee, S.; Gururangan, S.; Sampson, J.; et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J. Clin. Oncol. 2007, 25, 4722–4729. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Yan, H.; Zhao, P.; Yang, Y.; Cao, B. Efficacy and Safety of Bevacizumab Combined with Chemotherapy for Managing Metastatic Breast Cancer: A Meta-Analysis of Randomized Controlled Trials. Sci. Rep. 2015, 5, 15746. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.; Zheng, Y.; Xu, H.; Ge, W.; Xu, X. Bevacizumab improves survival in metastatic colorectal cancer patients with primary tumor resection: A meta-analysis. Sci. Rep. 2019, 9, 20326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, N.; Hillan, K.J.; Gerber, H.P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef]
- Diaz, R.J.; Ali, S.; Qadir, M.G.; De La Fuente, M.I.; Ivan, M.E.; Komotar, R.J. The role of bevacizumab in the treatment of glioblastoma. J. Neurooncol. 2017, 133, 455–467. [Google Scholar] [CrossRef]
- Helfrich, I.; Scheffrahn, I.; Bartling, S.; Weis, J.; von Felbert, V.; Middleton, M.; Kato, M.; Ergun, S.; Augustin, H.G.; Schadendorf, D. Resistance to antiangiogenic therapy is directed by vascular phenotype, vessel stabilization, and maturation in malignant melanoma. J. Exp. Med. 2010, 207, 491–503. [Google Scholar] [CrossRef] [Green Version]
- De Groot, J.F.; Fuller, G.; Kumar, A.J.; Piao, Y.; Eterovic, K.; Ji, Y.; Conrad, C.A. Tumor invasion after treatment of glioblastoma with bevacizumab: Radiographic and pathologic correlation in humans and mice. Neuro-Oncology 2010, 12, 233–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, P.Y.; Yung, W.K.; Lamborn, K.R.; Dahia, P.L.; Wang, Y.; Peng, B.; Abrey, L.E.; Raizer, J.; Cloughesy, T.F.; Fink, K.; et al. Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium Study 99-08. Clin. Cancer Res. 2006, 12, 4899–4907. [Google Scholar] [CrossRef] [Green Version]
- Hainsworth, J.D.; Ervin, T.; Friedman, E.; Priego, V.; Murphy, P.B.; Clark, B.L.; Lamar, R.E. Concurrent radiotherapy and temozolomide followed by temozolomide and sorafenib in the first-line treatment of patients with glioblastoma multiforme. Cancer 2010, 116, 3663–3669. [Google Scholar] [CrossRef] [PubMed]
- Neyns, B.; Sadones, J.; Chaskis, C.; Dujardin, M.; Everaert, H.; Lv, S.; Duerinck, J.; Tynninen, O.; Nupponen, N.; Michotte, A.; et al. Phase II study of sunitinib malate in patients with recurrent high-grade glioma. J. Neurooncol. 2011, 103, 491–501. [Google Scholar] [CrossRef]
- Brandes, A.A.; Carpentier, A.F.; Kesari, S.; Sepulveda-Sanchez, J.M.; Wheeler, H.R.; Chinot, O.; Cher, L.; Steinbach, J.P.; Capper, D.; Specenier, P.; et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro-Oncology 2016, 18, 1146–1156. [Google Scholar] [CrossRef] [Green Version]
- Wick, A.; Desjardins, A.; Suarez, C.; Forsyth, P.; Gueorguieva, I.; Burkholder, T.; Cleverly, A.L.; Estrem, S.T.; Wang, S.; Lahn, M.M.; et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor I, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Investig. New Drugs 2020, 38, 1570–1579. [Google Scholar] [CrossRef] [Green Version]
- Anderson, A.R. A hybrid mathematical model of solid tumour invasion: The importance of cell adhesion. Math. Med. Biol. 2005, 22, 163–186. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solimando, A.G.; Da Via, M.C.; Leone, P.; Borrelli, P.; Croci, G.A.; Tabares, P.; Brandl, A.; Di Lernia, G.; Bianchi, F.P.; Tafuri, S.; et al. Halting the vicious cycle within the multiple myeloma ecosystem: Blocking JAM-A on bone marrow endothelial cells restores the angiogenic homeostasis and suppresses tumor progression. Haematologica 2020. [Google Scholar] [CrossRef]
- Harjunpää, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Cirkel, G.A.; Kerklaan, B.M.; Vanhoutte, F.; Van der Aa, A.; Lorenzon, G.; Namour, F.; Pujuguet, P.; Darquenne, S.; de Vos, F.Y.; Snijders, T.J.; et al. A dose escalating phase I study of GLPG0187, a broad spectrum integrin receptor antagonist, in adult patients with progressive high-grade glioma and other advanced solid malignancies. Investig. New Drugs 2016, 34, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.Y.; Zhan, Y.P.; Zong, W.J.; Yu, C.J.; Li, J.F.; Qu, Y.M.; Han, S. The PD-1/B7-H1 pathway modulates the natural killer cells versus mouse glioma stem cells. PLoS ONE 2015, 10, e0134715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris-Bookman, S.; Mathios, D.; Martin, A.M.; Xia, Y.; Kim, E.; Xu, H.; Belcaid, Z.; Polanczyk, M.; Barberi, T.; Theodros, D.; et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int. J. Cancer 2018, 143, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T.; et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology 2018, 7, e1466769. [Google Scholar] [CrossRef]
- Kim, J.E.; Patel, M.A.; Mangraviti, A.; Kim, E.S.; Theodros, D.; Velarde, E.; Liu, A.; Sankey, E.W.; Tam, A.; Xu, H.; et al. Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin. Cancer Res. 2017, 23, 124–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairfax, B.P.; Taylor, C.A.; Watson, R.A.; Nassiri, I.; Danielli, S.; Fang, H.; Mahe, E.A.; Cooper, R.; Woodcock, V.; Traill, Z.; et al. Peripheral CD8+ T cell characteristics associated with durable responses to immune checkpoint blockade in patients with metastatic melanoma. Nat. Med. 2020, 26, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Karachaliou, N.; Gonzalez-Cao, M.; Crespo, G.; Drozdowskyj, A.; Aldeguer, E.; Gimenez-Capitan, A.; Teixido, C.; Molina-Vila, M.A.; Viteri, S.; De Los Llanos Gil, M.; et al. Interferon gamma, an important marker of response to immune checkpoint blockade in non-small cell lung cancer and melanoma patients. Ther. Adv. Med. Oncol. 2018, 10, 1758834017749748. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Gettinger, S.; Rizvi, N.A.; Chow, L.Q.; Borghaei, H.; Brahmer, J.; Ready, N.; Gerber, D.E.; Shepherd, F.A.; Antonia, S.; Goldman, J.W.; et al. Nivolumab Monotherapy for First-Line Treatment of Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 2980–2987. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.B.; Gettinger, S.N.; Mahajan, A.; Chiang, A.C.; Herbst, R.S.; Sznol, M.; Tsiouris, A.J.; Cohen, J.; Vortmeyer, A.; Jilaveanu, L.; et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: Early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 976–983. [Google Scholar] [CrossRef] [Green Version]
- Tawbi, H.A.; Forsyth, P.A.; Algazi, A.; Hamid, O.; Hodi, F.S.; Moschos, S.J.; Khushalani, N.I.; Lewis, K.; Lao, C.D.; Postow, M.A.; et al. Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. N. Engl. J. Med. 2018, 379, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Ma, L.; Lv, J.; Dong, Y.; Zhang, X.; Li, X.; Zhang, H.; Nong, J.; Zhang, Q.; Qin, N.; Yang, X.; et al. PD-L1 Expression and Its Regulation in Lung Adenocarcinoma with ALK Translocation. Interdiscip. Sci. 2019, 11, 266–272. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, CheckMate-143: The game is not over yet. Oncotarget 2017, 8, 91779–91794. [Google Scholar] [CrossRef] [Green Version]
- Collins, A.V.; Brodie, D.W.; Gilbert, R.J.; Iaboni, A.; Manso-Sancho, R.; Walse, B.; Stuart, D.I.; van der Merwe, P.A.; Davis, S.J. The interaction properties of costimulatory molecules revisited. Immunity 2002, 17, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 1996, 183, 2533–2540. [Google Scholar] [CrossRef]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Fecci, P.E.; Ochiai, H.; Mitchell, D.A.; Grossi, P.M.; Sweeney, A.E.; Archer, G.E.; Cummings, T.; Allison, J.P.; Bigner, D.D.; Sampson, J.H. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin. Cancer Res. 2007, 13, 2158–2167. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res. 2014, 20, 5290–5301. [Google Scholar] [CrossRef] [Green Version]
- Duerinck, J.; Schwarze, J.K.; Awada, G.; Tijtgat, J.; Vaeyens, F.; Bertels, C.; Geens, W.; Klein, S.; Seynaeve, L.; Cras, L.; et al. Intracerebral administration of CTLA-4 and PD-1 immune checkpoint blocking monoclonal antibodies in patients with recurrent glioblastoma: A phase I clinical trial. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef]
- Kisielow, M.; Kisielow, J.; Capoferri-Sollami, G.; Karjalainen, K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur. J. Immunol. 2005, 35, 2081–2088. [Google Scholar] [CrossRef]
- Grosso, J.F.; Kelleher, C.C.; Harris, T.J.; Maris, C.H.; Hipkiss, E.L.; De Marzo, A.; Anders, R.; Netto, G.; Getnet, D.; Bruno, T.C.; et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J. Clin. Investig. 2007, 117, 3383–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreae, S.; Piras, F.; Burdin, N.; Triebel, F. Maturation and activation of dendritic cells induced by lymphocyte activation gene-3 (CD223). J. Immunol. 2002, 168, 3874–3880. [Google Scholar] [CrossRef] [Green Version]
- Maruhashi, T.; Okazaki, I.M.; Sugiura, D.; Takahashi, S.; Maeda, T.K.; Shimizu, K.; Okazaki, T. LAG-3 inhibits the activation of CD4+ T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat. Immunol. 2018, 19, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Camisaschi, C.; Casati, C.; Rini, F.; Perego, M.; De Filippo, A.; Triebel, F.; Parmiani, G.; Belli, F.; Rivoltini, L.; Castelli, C. LAG-3 expression defines a subset of CD4+CD25highFoxp3+ regulatory T cells that are expanded at tumor sites. J. Immunol. 2010, 184, 6545–6551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Meng, Q.; Bartek, J., Jr.; Poiret, T.; Persson, O.; Rane, L.; Rangelova, E.; Illies, C.; Peredo, I.H.; Luo, X.; et al. Tumor-infiltrating lymphocytes (TILs) from patients with glioma. Oncoimmunology 2017, 6, e1252894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef] [Green Version]
- Durham, N.M.; Nirschl, C.J.; Jackson, C.M.; Elias, J.; Kochel, C.M.; Anders, R.A.; Drake, C.G. Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS ONE 2014, 9, e109080. [Google Scholar] [CrossRef] [Green Version]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.; Tan, T.G.; Sefik, E.; Yajnik, V.; et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 2014, 40, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boles, K.S.; Vermi, W.; Facchetti, F.; Fuchs, A.; Wilson, T.J.; Diacovo, T.G.; Cella, M.; Colonna, M. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur. J. Immunol. 2009, 39, 695–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano, E.; Dominguez-Villar, M.; Kuchroo, V.; Hafler, D.A. The TIGIT/CD226 axis regulates human T cell function. J. Immunol. 2012, 188, 3869–3875. [Google Scholar] [CrossRef]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Merrill, M.K.; Bernhardt, G.; Sampson, J.H.; Wikstrand, C.J.; Bigner, D.D.; Gromeier, M. Poliovirus receptor CD155-targeted oncolysis of glioma. Neuro-Oncology 2004, 6, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Freeman, G.J.; Casasnovas, J.M.; Umetsu, D.T.; DeKruyff, R.H. TIM genes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol. Rev. 2010, 235, 172–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, Y.; Li, G.; Huang, H.; Zhang, G.; Wang, F.; Sun, J.; Yang, Q.; Zhang, X.; Lu, B. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS ONE 2012, 7, e30676. [Google Scholar] [CrossRef] [Green Version]
- Komohara, Y.; Morita, T.; Annan, D.A.; Horlad, H.; Ohnishi, K.; Yamada, S.; Nakayama, T.; Kitada, S.; Suzu, S.; Kinoshita, I.; et al. The Coordinated Actions of TIM-3 on Cancer and Myeloid Cells in the Regulation of Tumorigenicity and Clinical Prognosis in Clear Cell Renal Cell Carcinomas. Cancer Immunol. Res. 2015, 3, 999–1007. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Liu, X.; Ma, H.; Zhang, H.; Song, X.; Gao, L.; Liang, X.; Ma, C. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut 2015, 64, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.W.; Dutta, A.; Chang, L.Y.; Mahalingam, J.; Lin, Y.C.; Chiang, J.M.; Hsu, C.Y.; Huang, C.T.; Su, W.T.; Chu, Y.Y.; et al. Apoptosis of tumor infiltrating effector TIM-3+CD8+ T cells in colon cancer. Sci. Rep. 2015, 5, 15659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Wang, Z.; Zhang, C.; Liu, X.; Cai, J.; Wang, Z.; Hu, H.; Wu, F.; Bao, Z.; Liu, Y.; et al. Molecular and clinical characterization of TIM-3 in glioma through 1,024 samples. Oncoimmunology 2017, 6, e1328339. [Google Scholar] [CrossRef]
- Zhang, J.; Sai, K.; Wang, X.L.; Ye, S.Q.; Liang, L.J.; Zhou, Y.; Chen, Z.J.; Hu, W.M.; Liu, J.M. Tim-3 Expression and MGMT Methylation Status Association With Survival in Glioblastoma. Front. Pharmacol. 2020, 11, 584652. [Google Scholar] [CrossRef]
- Liu, Z.; Han, H.; He, X.; Li, S.; Wu, C.; Yu, C.; Wang, S. Expression of the galectin-9-Tim-3 pathway in glioma tissues is associated with the clinical manifestations of glioma. Oncol. Lett. 2016, 11, 1829–1834. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Chang, C.Y.; Yoon, H.J.; Kim, K.S.; Koh, H.S.; Kim, S.S.; Lee, S.J.; Kane, L.P.; Park, E.J. Glial TIM-3 Modulates Immune Responses in the Brain Tumor Microenvironment. Cancer Res. 2020, 80, 1833–1845. [Google Scholar] [CrossRef] [Green Version]
- Bristol-Myers Squibb. Bristol-Myers Squibb Announces Phase 3 CheckMate-498 Study Did Not Meet Primary Endpoint of Overall Survival with Opdivo (nivolumab) Plus Radiation in Patients with Newly Diagnosed MGMT-Unmethylated Glioblastoma Multiforme; Bristol Myers Squibb: New York, NY, USA, 9 May 2019. [Google Scholar]
- Bristol-Myers Squibb. Bristol Myers Squibb Announces Update on Phase 3 CheckMate-548 Trial Evaluating Patients with Newly Diagnosed MGMT-Methylated Glioblastoma Multiforme; Bristol Myers Squibb: New York, NY, USA, 23 December 2020. [Google Scholar]
- Lim, M.; Ye, X.; Piotrowski, A.F.; Desai, A.S.; Ahluwalia, M.S.; Walbert, T.; Fisher, J.D.; Desideri, S.; Nabors, L.B.; Wen, P.Y.; et al. Updated safety phase I trial of anti-LAG-3 alone and in combination with anti-PD-1 in patients with recurrent GBM. J. Clin. Oncol. 2020, 38. [Google Scholar] [CrossRef]
- Nayak, L.; Molinaro, A.M.; Peters, K.; Clarke, J.L.; Jordan, J.T.; de Groot, J.; Nghiemphu, L.; Kaley, T.; Colman, H.; McCluskey, C.; et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2021, 27, 1048–1057. [Google Scholar] [CrossRef]
- Awada, G.; Ben Salama, L.; De Cremer, J.; Schwarze, J.K.; Fischbuch, L.; Seynaeve, L.; Du Four, S.; Vanbinst, A.M.; Michotte, A.; Everaert, H.; et al. Axitinib plus avelumab in the treatment of recurrent glioblastoma: A stratified, open-label, single-center phase 2 clinical trial (GliAvAx). J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; Lopez-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inoges, S.; de Andrea, C.; Lopez-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef]
- Reardon, D.A.; Kaley, T.J.; Dietrich, J.; Clarke, J.L.; Dunn, G.; Lim, M.; Cloughesy, T.F.; Gan, H.K.; Park, A.J.; Schwarzenberger, P.; et al. Phase II study to evaluate safety and efficacy of MEDI4736 (durvalumab) + radiotherapy in patients with newly diagnosed unmethylated MGMT glioblastoma (new unmeth GBM). J. Clin. Oncol. 2019, 37. [Google Scholar] [CrossRef]
- Reardon, D.; Kaley, T.; Dietrich, J.; Clarke, J.; Dunn, G.; Lim, M.; Cloughesy, T.; Gan, H.; Park, A.; Schwarzenberger, P.; et al. ATIM-38. Phase 2 study to evaluate the clinical efficacy and safety of medi4736 (durvalumab, durva) + bevacizumab (bev) in bev-naïve patients with recurrent glioblastoma (gbm). Neuro-Oncology 2018, 20. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.; Kaley, T.; Dietrich, J.; Clarke, J.L.; Dunn, G.P.; Lim, M.; Cloughesy, T.; Gan, H.K.; Park, A.; Schwarzenberger, P.; et al. ATIM-12. Phase 2 study to evaluate the clinical efficacy and safety of medi4736 (durvalumab [dur]) in patients with bevacizumab (bev)-refractory recurrent glioblastoma (gbm). Neuro-Oncology 2017, 19. [Google Scholar] [CrossRef]
- Ahluwalia, M.; Peereboom, D.; Schilero, C.; Forst, D.; Wong, E.; Wen, P.; Reardon, D. RBTT-01. Randomized phase 2 open label study of nivolumab plus standard dose bevacizumab versus nivolumab plus low dose bevacizumab in recurrent glioblastoma. Neuro-Oncology 2018, 20, vi234. [Google Scholar] [CrossRef]
- Haslam, A.; Prasad, V. Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw. Open 2019, 2, e192535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missiaen, R.; Mazzone, M.; Bergers, G. The reciprocal function and regulation of tumor vessels and immune cells offers new therapeutic opportunities in cancer. Semin. Cancer Biol. 2018, 52, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Goldstein, A.; Wang, H.; Ching Lo, H.; Sun Kim, I.; Welte, T.; Sheng, K.; Dobrolecki, L.E.; Zhang, X.; Putluri, N.; et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 2017, 544, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozao-Choy, J.; Ma, G.; Kao, J.; Wang, G.X.; Meseck, M.; Sung, M.; Schwartz, M.; Divino, C.M.; Pan, P.Y.; Chen, S.H. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009, 69, 2514–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Ting, K.K.; Li, J.; Cogger, V.C.; Chen, J.; Johansson-Percival, A.; Ngiow, S.F.; Holst, J.; Grau, G.; Goel, S.; et al. Targeting Vascular Endothelial-Cadherin in Tumor-Associated Blood Vessels Promotes T-cell-Mediated Immunotherapy. Cancer Res. 2017, 77, 4434–4447. [Google Scholar] [CrossRef] [Green Version]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Bonacchi, A.; Romagnani, P.; Romanelli, R.G.; Efsen, E.; Annunziato, F.; Lasagni, L.; Francalanci, M.; Serio, M.; Laffi, G.; Pinzani, M.; et al. Signal transduction by the chemokine receptor CXCR3: Activation of Ras/ERK, Src, and phosphatidylinositol 3-kinase/Akt controls cell migration and proliferation in human vascular pericytes. J. Biol. Chem. 2001, 276, 9945–9954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef] [Green Version]
- Hamzah, J.; Jugold, M.; Kiessling, F.; Rigby, P.; Manzur, M.; Marti, H.H.; Rabie, T.; Kaden, S.; Grone, H.J.; Hammerling, G.J.; et al. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature 2008, 453, 410–414. [Google Scholar] [CrossRef]
- Mazzone, M.; Dettori, D.; de Oliveira, R.L.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.M.; Lanahan, A.A.; Pollard, P.; de Almodovar, C.R.; et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 2009, 136, 839–851. [Google Scholar] [CrossRef] [Green Version]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulieres, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Wu, X.; Giobbie-Hurder, A.; Liao, X.; Connelly, C.; Connolly, E.M.; Li, J.; Manos, M.P.; Lawrence, D.; McDermott, D.; Severgnini, M.; et al. Angiopoietin-2 as a Biomarker and Target for Immune Checkpoint Therapy. Cancer Immunol. Res. 2017, 5, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Di Tacchio, M.; Macas, J.; Weissenberger, J.; Sommer, K.; Bahr, O.; Steinbach, J.P.; Senft, C.; Seifert, V.; Glas, M.; Herrlinger, U.; et al. Tumor Vessel Normalization, Immunostimulatory Reprogramming, and Improved Survival in Glioblastoma with Combined Inhibition of PD-1, Angiopoietin-2, and VEGF. Cancer Immunol. Res. 2019, 7, 1910–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauri, D.N.; Ebner, R.; Montgomery, R.I.; Kochel, K.D.; Cheung, T.C.; Yu, G.L.; Ruben, S.; Murphy, M.; Eisenberg, R.J.; Cohen, G.H.; et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 1998, 8, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Lo, J.C.; Foster, A.; Yu, P.; Chen, H.M.; Wang, Y.; Tamada, K.; Chen, L.; Fu, Y.X. The regulation of T cell homeostasis and autoimmunity by T cell-derived LIGHT. J. Clin. Investig. 2001, 108, 1771–1780. [Google Scholar] [CrossRef]
- Giles, D.A.; Zahner, S.; Krause, P.; Van Der Gracht, E.; Riffelmacher, T.; Morris, V.; Tumanov, A.; Kronenberg, M. The Tumor Necrosis Factor Superfamily Members TNFSF14 (LIGHT), Lymphotoxin beta and Lymphotoxin beta Receptor Interact to Regulate Intestinal Inflammation. Front. Immunol. 2018, 9, 2585. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Jabouille, A.; Steri, V.; Johansson-Percival, A.; Michael, I.P.; Kotamraju, V.R.; Junckerstorff, R.; Nowak, A.K.; Hamzah, J.; Lee, G.; et al. Vascular targeting of LIGHT normalizes blood vessels in primary brain cancer and induces intratumoural high endothelial venules. J. Pathol. 2018, 245, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Ager, A.; May, M.J. Understanding high endothelial venules: Lessons for cancer immunology. Oncoimmunology 2015, 4, e1008791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinet, L.; Garrido, I.; Filleron, T.; Le Guellec, S.; Bellard, E.; Fournie, J.J.; Rochaix, P.; Girard, J.P. Human solid tumors contain high endothelial venules: Association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res. 2011, 71, 5678–5687. [Google Scholar] [CrossRef] [Green Version]
- Martinet, L.; Le Guellec, S.; Filleron, T.; Lamant, L.; Meyer, N.; Rochaix, P.; Garrido, I.; Girard, J.P. High endothelial venules (HEVs) in human melanoma lesions: Major gateways for tumor-infiltrating lymphocytes. Oncoimmunology 2012, 1, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Low, S.; Sakai, Y.; Hoshino, H.; Hirokawa, M.; Kawashima, H.; Higuchi, K.; Imamura, Y.; Kobayashi, M. High endothelial venule-like vessels and lymphocyte recruitment in diffuse sclerosing variant of papillary thyroid carcinoma. Pathology 2016, 48, 666–674. [Google Scholar] [CrossRef] [Green Version]
- Sakai, Y.; Hoshino, H.; Kitazawa, R.; Kobayashi, M. High endothelial venule-like vessels and lymphocyte recruitment in testicular seminoma. Andrology 2014, 2, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.A.; Hwang, H.W.; Kim, M.K.; Lee, T.J.; Yim, K.; Won, H.S.; Sun, S.; Kim, E.Y.; Ko, Y.H. High Endothelial Venule with Concomitant High CD8+ Tumor-Infiltrating Lymphocytes Is Associated with a Favorable Prognosis in Resected Gastric Cancer. J. Clin. Med. 2020, 9, 2628. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Nduom, E.K.; Wei, J.; Yaghi, N.K.; Huang, N.; Kong, L.Y.; Gabrusiewicz, K.; Ling, X.; Zhou, S.; Ivan, C.; Chen, J.Q.; et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro-Oncology 2016, 18, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Zhai, L.; Ladomersky, E.; Lauing, K.L.; Wu, M.; Genet, M.; Gritsina, G.; Gyorffy, B.; Brastianos, P.K.; Binder, D.C.; Sosman, J.A.; et al. Infiltrating T Cells Increase IDO1 Expression in Glioblastoma and Contribute to Decreased Patient Survival. Clin. Cancer Res. 2017, 23, 6650–6660. [Google Scholar] [CrossRef] [Green Version]
- Lu-Emerson, C.; Snuderl, M.; Kirkpatrick, N.D.; Goveia, J.; Davidson, C.; Huang, Y.; Riedemann, L.; Taylor, J.; Ivy, P.; Duda, D.G.; et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro-Oncology 2013, 15, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.R.; Chen, Z.Q.; Yang, X.Y.; Ding, Z.N.; Liu, K.X.; Yan, L.J.; Meng, G.X.; Yang, Y.F.; Yan, Y.C.; Yao, S.Y.; et al. RECK expression is associated with angiogenesis and immunogenic Tumor Microenvironment in Hepatocellular Carcinoma, and is a prognostic factor for better survival. J. Cancer 2021, 12, 3827–3840. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wohrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-Oncology 2015, 17, 1064–1075. [Google Scholar] [CrossRef] [Green Version]
- Pratt, D.; Dominah, G.; Lobel, G.; Obungu, A.; Lynes, J.; Sanchez, V.; Adamstein, N.; Wang, X.; Edwards, N.A.; Wu, T.; et al. Programmed Death Ligand 1 Is a Negative Prognostic Marker in Recurrent Isocitrate Dehydrogenase-Wildtype Glioblastoma. Neurosurgery 2019, 85, 280–289. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. Cancer Immunology. The “cancer immunogram”. Science 2016, 352, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Sun, H.; Zhang, H.; Wang, X. An Immunogram for an Individualized Assessment of the Antitumor Immune Response in Patients with Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 1189. [Google Scholar] [CrossRef] [PubMed]
- Karasaki, T.; Nagayama, K.; Kuwano, H.; Nitadori, J.I.; Sato, M.; Anraku, M.; Hosoi, A.; Matsushita, H.; Morishita, Y.; Kashiwabara, K.; et al. An Immunogram for the Cancer-Immunity Cycle: Towards Personalized Immunotherapy of Lung Cancer. J. Thorac. Oncol. 2017, 12, 791–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, Y.; Kushihara, Y.; Saito, N.; Yamaguchi, S.; Kakimi, K. A novel scoring method based on RNA-Seq immunograms describing individual cancer-immunity interactions. Cancer Sci. 2020, 111, 4031–4040. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Sorensen, A.G.; di Tomaso, E.; Zhang, W.T.; Duda, D.G.; Cohen, K.S.; Kozak, K.R.; Cahill, D.P.; Chen, P.J.; Zhu, M.; et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 2007, 11, 83–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528. [Google Scholar] [CrossRef]
- Khalsa, J.K.; Cheng, N.; Keegan, J.; Chaudry, A.; Driver, J.; Bi, W.L.; Lederer, J.; Shah, K. Immune phenotyping of diverse syngeneic murine brain tumors identifies immunologically distinct types. Nat. Commun. 2020, 11, 3912. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Target | Treatment | Setting | Study Design | No. of Patients (n) | Primary Endpoint(s) | Primary Outcome | Identifier | Reference |
|---|---|---|---|---|---|---|---|---|
| PD-1 | Nivolumab vs. Bevacizumab | R | Open-label phase III | 369 | OS | Primary endpoint not met | CheckMate-143 NCT02017717 | [123] |
| PD-1 | Nivolumab + RT vs. TMZ + RT | ND, unmethylated MGMT | Open-label phase III | 553 | OS | Primary endpoint not met | CheckMate-498 NCT02617589 | [158] |
| PD-1 | Nivolumab + TMZ + RT vs. Placebo + TMZ + RT | ND, methylated MGMT | Triple-blinded phase III | 693 (targeted) | PFS, OS | Primary endpoints not met | CheckMate-548 NCT02667587 | [159] |
| PD-1 | Pembrolizumab + TMZ + RT | ND | Open-label phase II | 56 (targeted) | OS | Recruiting participants | NCT03899857 | |
| CTLA-4 | Ipilimumab + Nivolumab | R | Open-label phase I | 27 | PFS, OS | mPFS 2.7 mo mOS 8.7 mo | NCT03233152 | [131] |
| CTLA-4 | Ipilimumab + Nivolumab + RT vs. TMZ + RT | ND, unmethylated MGMT | Open-label phase II/III | 485 (targeted) | PFS, OS | Recruiting participants | NCT04396860 | |
| LAG-3 | Anti-LAG-3 mAb (BMS-986016) +/− Nivolumab | R | Open-label phase I | 33 | MTD | Late-onset DLT, DLT rate <33% | NCT02658981 | [160] |
| TIGIT | Anti-TIGIT mAb (ASP8374) +/− Pembrolizumab | Advanced solid tumors | Open-label phase Ib | 169 | Safety and tolerability: DLT and AE | Result pending | NCT03260322 | |
| TIGIT | Anti-TIGIT mAb (BMS-986207) +/− Nivolumab | Advanced solid tumors | Open-label phase I/IIa | 130 (targeted) | AE, SAE, ORR, mDOR, PSF rate | Recruiting participants | NCT02913313 | |
| TIM-3 | Anti-TIM-3 mAb (MBG453) + spartalizumab + SRS | R | Open-label phase I | 15 (targeted) | SAE | Recruiting participants | NCT03961971 | |
| PD-1 + VEGF | Pembrolizumab +/− Bevacizumab | R | Open-label, randomized phase II | 80 | MTD, DLT, PFS6 | MTD 200 mg/3 weeks No DLT PFS6 26% vs. 6.7% | NCT02337491 | [161] |
| PD-L1 + VEGFR | Avelumab + Axitinib | R | Open-label phase II (2 cohorts) | 54 | PSF6 | PSF6 22.2% vs. 18.5% | NCT03291314 | [162] |
| PD-1 | Neoadjuvant +/− adjuvant Pembrolizumab | R | Open-label, randomized phase II | 35 | PSF | mPFS 3.3 mo vs. 2.4 mo | NCT02337686 | [163] |
| PD-1 | Neoadjuvant Nivolumab | ND & R | Open-label phase II | 30 | Changes in PD-L1 percentage and expression levels by lymphocytes | Nivolumab vs. control: no significant changes in immune cells’ PD-L1 expression | NCT02550249 | [164] |
| PD-L1 | Durvalumab + RT (Cohort A) | ND, unmethylated MGMT (Cohort A) | Open-label, multi-cohort phase II | 40 | OS-12 | mOS 15.1 mo OS-12 60% | NCT02336165 | [165] |
| PD-L1 + VEGF | Durvalumab +/− Bevacizumab (Cohort B) | R, bevacizumab- naïve | Open-label, multi-cohort phase II | 97 | PFS-6 | PFS-6 B1: 20%; B2: 15.2%; B3: 21.1% | NCT02336165 | [166] |
| Durvalumab + Bevacizumab (Cohort C) | R, bevacizumab- refractory | Open-label, multi-cohort phase II | 22 | OS-6 | OS-6 36.4% ≥22 weeks | NCT02336165 | [167] | |
| PD-1 + VEGF | Neoadjuvant Nivolumab + low/standard Bevacizumab dosage | R | Open-label, randomized phase II | 90 | OS-12 | OS-12 58% | NCT03452579 | [168] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, R.L.Y.; Ho, I.A.W. Recent Advances in Glioma Therapy: Combining Vascular Normalization and Immune Checkpoint Blockade. Cancers 2021, 13, 3686. https://doi.org/10.3390/cancers13153686
Ho RLY, Ho IAW. Recent Advances in Glioma Therapy: Combining Vascular Normalization and Immune Checkpoint Blockade. Cancers. 2021; 13(15):3686. https://doi.org/10.3390/cancers13153686
Chicago/Turabian StyleHo, Rachel L. Y., and Ivy A. W. Ho. 2021. "Recent Advances in Glioma Therapy: Combining Vascular Normalization and Immune Checkpoint Blockade" Cancers 13, no. 15: 3686. https://doi.org/10.3390/cancers13153686
APA StyleHo, R. L. Y., & Ho, I. A. W. (2021). Recent Advances in Glioma Therapy: Combining Vascular Normalization and Immune Checkpoint Blockade. Cancers, 13(15), 3686. https://doi.org/10.3390/cancers13153686