
Evaluating Targeted Therapies in Ovarian Cancer Metabolism: Novel Role for PCSK9 and Second Generation mTOR Inhibitors

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfections
2.2. Cell Viability and Inhibitor Treatment
2.3. Western Blot
2.4. Drug Testing
2.5. Gene Expression Analysis
3. Results
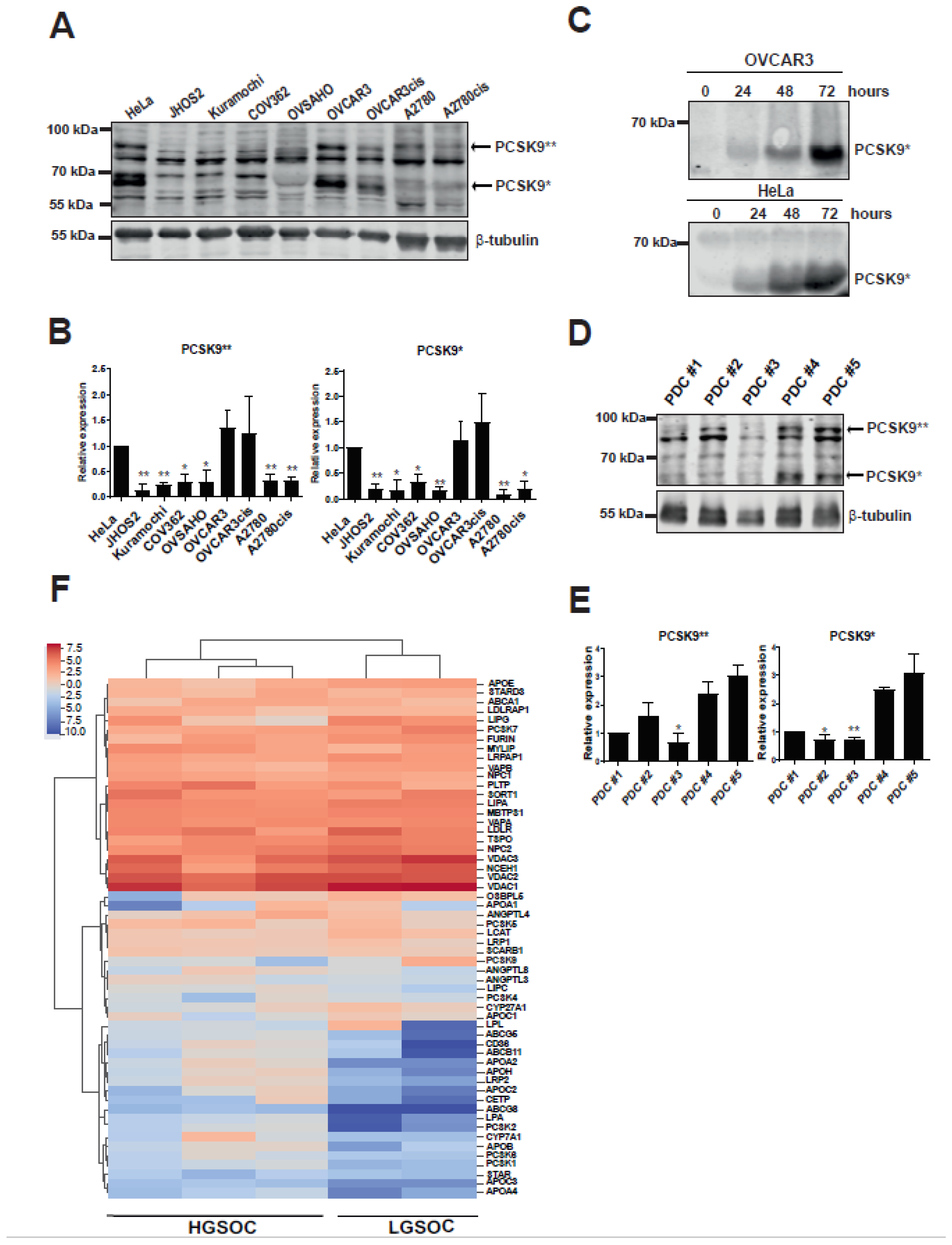
3.1. Cholesterol Modulating pCSK9 Proprotein Is Expressed in OC Cell Lines and Patient Samples
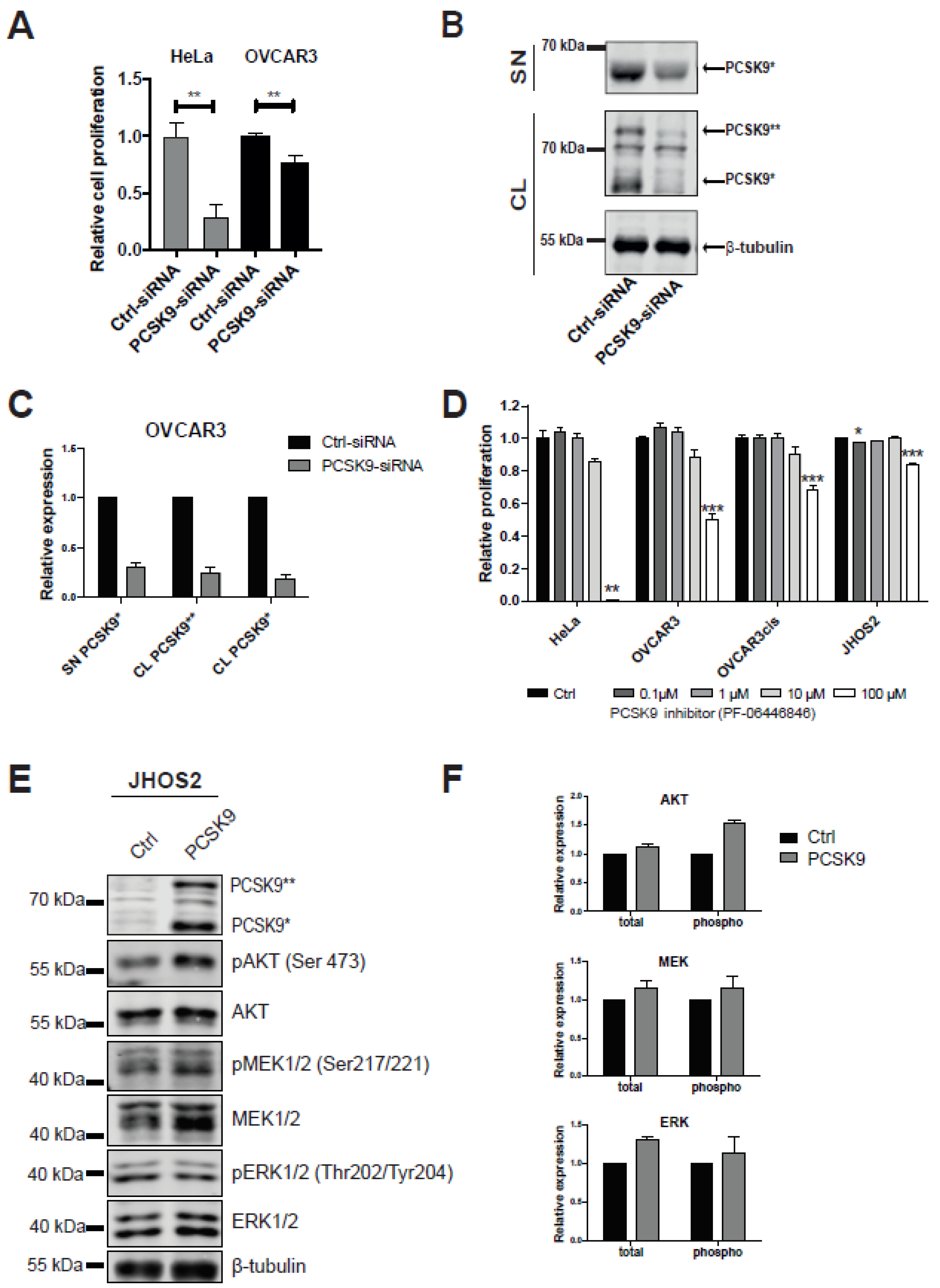
3.2. Targeting PCSK9 Expression Impairs OC Cell Viability
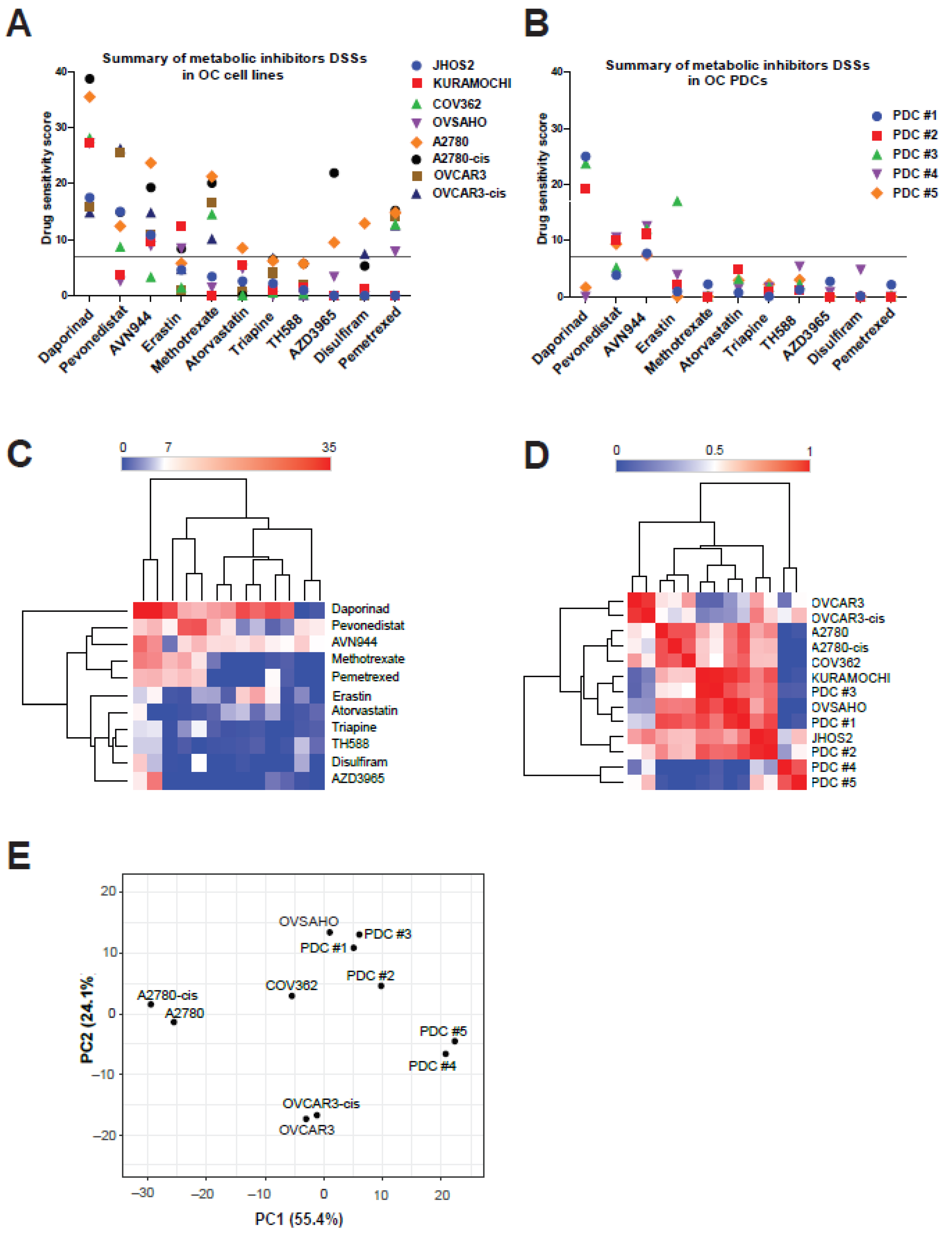
3.3. Anti-Metabolic Drugs Show OC Subtype-Specific Efficacies
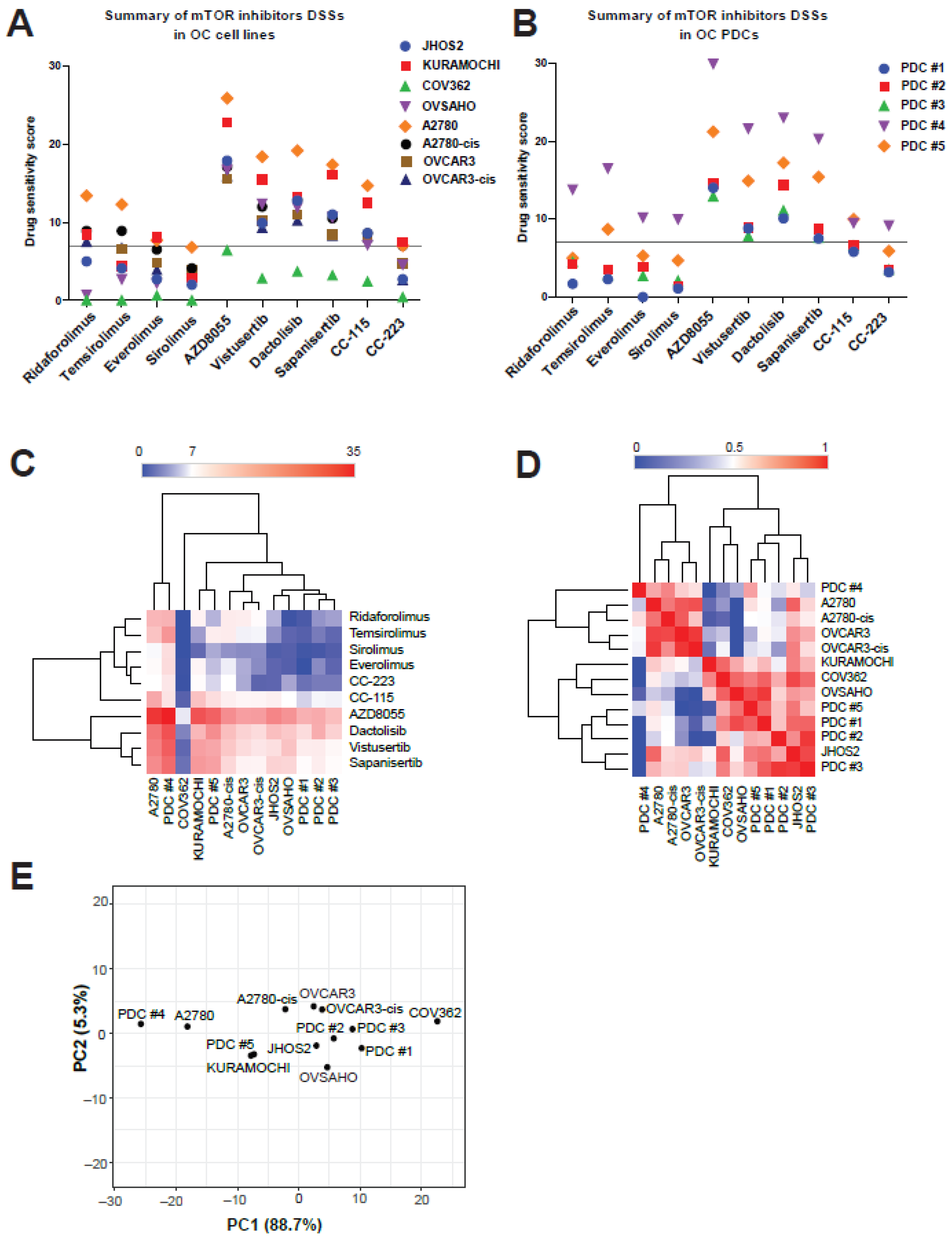
3.4. mTOR Inhibitors Elicit Diverse Efficacies in OC Cell Lines and PDCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| A2780cis | Cis-platin resistant A2780 |
| APO | Apolipoprotein |
| ARID1A | AT-Rich Interaction Domain 1A |
| BRAF | v-raf murine sarcoma viral oncogene homolog B1 |
| CRLs | Cullin-RING E3 ligases |
| DEPTOR | DEP domain containing mTOR interacting protein |
| DSS | Drug sensitivity score |
| EOC | Epithelial ovarian cancer |
| FRBP-12 | 12 kDa FK506-binding protein |
| GTP | guanosine triphosphate |
| HDL | High-density lipoprotein |
| HeLa | Henrietta Lacks’ cervical carcinoma cell line |
| HepG2 | Hepatoma G2 |
| HER2 | Human epidermal growth factor receptor 2 |
| HGSOC | High-grade serous ovarian cancer |
| HR | Homologous recombination |
| IMPDH | Inosine monosphosphate dehydrogenase |
| IMPDH2 | Inosine monosphosphate dehydrogenase 2 |
| KRAS | Kirsten Rat Sarcoma Viral Proto-Oncogene |
| LDLR | Low-density lipoprotein receptor |
| LGSOC | Low-grade serous ovarian cancer |
| MAPK | Mitogen-Activated Protein Kinase |
| MCT1 | Monocarboxylate transporter 1 |
| mLST8 | Mammalian lethal with Sec13 protein 8 |
| mSin | Regulatory subunit |
| mTOR | Mammalian target of Rapamycin |
| mTORC | Mammalian target of Rapamycin Complex |
| NAE | Nedd8 activating enzyme |
| NAMPT | Nicotinamide phosphoribosyltransferase |
| NARC-1 | Neural Apoptosis-Regulated Convertase 1 |
| Nedd8 | Neural Precursor Cell Expressed, Developmentally Downregulated 8 |
| OC | Ovarian cancer |
| OVCAR3cis | Cisplatin resistant OVCAR3 |
| PACE4 | Paired Basic Amino Acid Cleaving Enzyme 4 |
| PC | Previously known as Proprotein Convertase Subtilisin/Kexin Type 5 or Proprotein Convertase Subtilisin/Kexin |
| PCA | Principal components analysis |
| PCs | Proprotein convertases |
| PCSK | Proprotein Convertase Subtilisin/Kexin |
| PDCs | Patient-derived cell cultures |
| PI3K | Phosphoinositide 3-kinase |
| PIKK | Phosphoinositide 3-kinase-related kinase family |
| PRAS40 | Proline-Rich Akt Substrate |
| Protor1/2 | Regulatory subunit |
| Rictor | Rapamycin insensitive companion of mTOR |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| RT-qPCR | Real time quantitative polymerase chain reaction |
| siRNA | Small interference RNA |
| SREBP | sterol regulatory element-binding protein |
| SKI-1/S1P | Subtilisin kexin isozyme 1 |
| TAK 117 | Serabelisib |
| TAK 228 | Sapanisertib |
| TH588 | Selective Human MutT Homolog 1 (NUDT1) inhibitor |
| TP53 | Cellular Tumor Antigen P53 |
References
- Narod, S. Can advanced-stage ovarian cancer be cured? Nat. Rev. Clin. Oncol. 2016, 13, 255–261. [Google Scholar] [CrossRef]
- Lheureux, S.; Msc, M.B.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef] [Green Version]
- Bowtell, D.D.; Böhm, S.; Ahmed, A.A.; Aspuria, P.-J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef]
- Rojas, V.; Hirshfield, K.M.; Ganesan, S.; Rodriguez-Rodriguez, L. Molecular Characterization of Epithelial Ovarian Cancer: Implications for Diagnosis and Treatment. Int. J. Mol. Sci. 2016, 17, 2113. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Phan, L.M.; Yeung, S.J.; Lee, M.-H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [PubMed]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackerman, D.; Simon, M.C. Hypoxia, lipids, and cancer: Surviving the harsh tumor microenvironment. Trends Cell Biol. 2014, 24, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Geng, X.; Li, Y. Milky spots: Omental functional units and hotbeds for peritoneal cancer metastasis. Tumor Biol. 2016, 37, 5715–5726. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.; Krishnan, V.; Schoof, M.; Rodriguez, I.; Theriault, B.; Chekmareva, M.; Rinker-Schaeffer, C. Milky Spots Promote Ovarian Cancer Metastatic Colonization of Peritoneal Adipose in Experimental Models. Am. J. Pathol. 2013, 183, 576–591. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Stenvers, K.L. Getting to Know Ovarian Cancer Ascites: Opportunities for Targeted Therapy-Based Translational Research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam, R.A.; Adam, Y.G. Malignant ascites: Past, present, and future. J. Am. Coll. Surg. 2004, 198, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Yadav, B.; Pemovska, T.; Szwajda, A.; Kulesskiy, E.; Kontro, M.; Karjalainen, R.; Majumder, M.M.; Malani, D.; Murumägi, A.; Knowles, J.; et al. Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci. Rep. 2015, 4, 5193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morpheus. Available online: https://software.broadinstitute.org/morpheus (accessed on 2 May 2021).
- Kumar, A.; Kankainen, M.; Parsons, A.; Kallioniemi, O.; Matti, K.; Heckman, C.A. The impact of RNA sequence library construction protocols on transcriptomic profiling of leukemia. BMC Genom. 2017, 18, 629. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, D355–D360. [Google Scholar] [CrossRef] [Green Version]
- Oliphant, T.E. Python for Scientific Computing. Comput. Sci. Eng. 2007, 9, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Waskom, M.; Botvinnik, O.; Hobson, P.; Cole, J.B.; Halchenko, Y.; Hoyer, S.; Miles, A.; Augspurger, T.; Yarkoni, T.; Megies, T.; et al. Seaborn: V0.5.0 (November 2014) 2014. Zenodo 2014. [Google Scholar] [CrossRef]
- Page, R.E.; Klein-Szanto, A.J.P.; Litwin, S.; Nicolas, E.; Al-Jumaily, R.; Alexander, P.; Godwin, A.K.; Ross, E.A.; Schilder, R.J.; Bassi, D.E. Increased Expression of the Pro-Protein Convertase Furin Predicts Decreased Survival in Ovarian Cancer. Cell. Oncol. 2007, 29, 289–299. [Google Scholar] [CrossRef]
- Seidah, N.G.; Sadr, M.S.; Chrétien, M.; Mbikay, M. The Multifaceted Proprotein Convertases: Their Unique, Redundant, Complementary, and Opposite Functions. J. Biol. Chem. 2013, 288, 21473–21481. [Google Scholar] [CrossRef] [Green Version]
- Kwok, S.C.; Chakraborty, D.; Soares, M.J.; Dai, G. Relative expression of proprotein convertases in rat ovaries during pregnancy. J. Ovarian Res. 2013, 6, 91. [Google Scholar] [CrossRef] [Green Version]
- Fan, D.; Yancey, P.G.; Qiu, S.; Ding, L.; Weeber, E.J.; Linton, M.F.; Fazio, S. Self-Association of Human PCSK9 Correlates with Its LDLR-Degrading Activity. Biochemistry 2008, 47, 1631–1639. [Google Scholar] [CrossRef] [Green Version]
- Nassoury, N.; Blasiole, D.A.; Oler, A.T.; Benjannet, S.; Hamelin, J.; Poupon, V.; McPherson, P.S.; Attie, A.D.; Prat, A.; Seidah, N.G. The Cellular Trafficking of the Secretory Proprotein Convertase PCSK9 and Its Dependence on the LDLR. Traffic 2007, 8, 718–732. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.; Wiviott, S.D.; Raal, F.J.; Blom, D.; Robinson, J.; Ballantyne, C.M.; Somaratne, R.; Legg, J.; Wasserman, S.M.; et al. Efficacy and Safety of Evolocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1500–1509. [Google Scholar] [CrossRef] [Green Version]
- Fasano, T.; Sun, X.-M.; Patel, D.D.; Soutar, A.K. Degradation of LDLR protein mediated by ‘gain of function’ PCSK9 mutants in normal and ARH cells. Atherosclerosis 2009, 203, 166–171. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Hernandez, L.; Kim, M.K.; Lyle, L.T.; Bunch, K.P.; House, C.D.; Ning, F.; Noonan, A.; Annunziata, C.M. Characterization of ovarian cancer cell lines as in vivo models for preclinical studies. Gynecol. Oncol. 2016, 142, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, A.; O’Donnell, R.; Drew, Y.; Curtin, N.J.; Saha, S.S. Characterisation of Ovarian Cancer Cell Line NIH-OVCAR3 and Implications of Genomic, Transcriptomic, Proteomic and Functional DNA Damage Response Biomarkers for Therapeutic Targeting. Cancers 2020, 12, 1939. [Google Scholar] [CrossRef] [PubMed]
- Karvonen, H.; Arjama, M.; Kaleva, L.; Niininen, W.; Barker, H.; Koivisto-Korander, R.; Tapper, J.; Pakarinen, P.; Lassus, H.; Loukovaara, M.; et al. Glucocorticoids induce differentiation and chemoresistance in ovarian cancer by promoting ROR1-mediated stemness. Cell Death Dis. 2020, 11, 790. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, Y.; Hobbs, H.H.; Cohen, J.C. Molecular characterization of proprotein convertase subtilisin/kexin type 9-mediated degradation of the LDLR. J. Lipid Res. 2012, 53, 1932–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodack, D.P.; Farago, A.F.; Dastur, A.; Held, M.A.; Dardaei, L.; Friboulet, L.; von Flotow, F.; Damon, L.J.; Lee, D.; Parks, M.; et al. Primary Patient-Derived Cancer Cells and Their Potential for Personalized Cancer Patient Care. Cell Rep. 2017, 21, 3298–3309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germain, N.; Dhayer, M.; Boileau, M.; Fovez, Q.; Kluza, J.; Marchetti, P. Lipid Metabolism and Resistance to Anticancer Treatment. Biol. 2020, 9, 474. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A Comprehensive Resource for in Silico Drug Discovery and Exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nat. Cell Biol. 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Naffouje, R.; Grover, P.; Yu, H.; Sendilnathan, A.; Wolfe, K.; Majd, N.; Smith, E.P.; Takeuchi, K.; Senda, T.; Kofuji, S.; et al. Anti-Tumor Potential of IMP Dehydrogenase Inhibitors: A Century-Long Story. Cancers 2019, 11, 1346. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Qiu, C.; Hou, M.; Wang, X.; Huang, C.; Zou, J.; Liu, T.; Qu, J. Ferroptosis in Ovarian Cancer: A Novel Therapeutic Strategy. Front. Oncol. 2021, 11, 1364. [Google Scholar] [CrossRef]
- Li, P.; Zheng, Y.; Chen, X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front. Pharmacol. 2017, 8, 460. [Google Scholar] [CrossRef]
- Malhotra, H.S.; Goa, K.L. Atorvastatin. Drugs 2001, 61, 1835–1881. [Google Scholar] [CrossRef]
- Finch, R.A.; Liu, M.-C.; Grill, S.P.; Rose, W.C.; Loomis, R.; Vasquez, K.M.; Cheng, Y.-C.; Sartorelli, A.C. Triapine (3-aminopyridine-2-carboxaldehyde- thiosemicarbazone): A potent inhibitor of ribonucleotide reductase activity with broad spectrum antitumor activity. Biochem. Pharmacol. 2000, 59, 983–991. [Google Scholar] [CrossRef]
- Gad, H.; Koolmeister, T.; Jemth, A.-S.; Eshtad, S.; Jacques, S.A.; Ström, C.E.; Svensson, L.M.; Schultz, N.; Lundbäck, T.; Einarsdottir, B.; et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nat. Cell Biol. 2014, 508, 215–221. [Google Scholar] [CrossRef]
- Wang, N.; Jiang, X.; Zhang, S.; Zhu, A.; Yuan, Y.; Xu, H.; Lei, J.; Yan, C. Structural basis of human monocarboxylate transporter 1 inhibition by anti-cancer drug candidates. Cell 2021, 184, 370–383.e13. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, M.; Mylonas, I.; Kast, R.E.; Bruning, A. Disulfiram/copper causes redox-related proteotoxicity and concomitant heat shock response in ovarian cancer cells that is augmented by auranofin-mediated thioredoxin inhibition. Oncoscience 2013, 1, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Egloff, H.; Jatoi, A. Pemetrexed for Ovarian Cancer: A Systematic Review of the Published Literature and a Consecutive Series of Patients Treated in a Nonclinical Trial Setting. Case Rep. Oncol. 2014, 7, 541–549. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [Green Version]
- Sehgal, S. Sirolimus: Its discovery, biological properties, and mechanism of action. Transplant. Proc. 2003, 35, S7–S14. [Google Scholar] [CrossRef]
- Pike, K.G.; Malagu, K.; Hummersone, M.G.; Menear, K.A.; Duggan, H.M.; Gomez, S.; Martin, N.M.; Ruston, L.; Pass, S.L.; Pass, M. Optimization of potent and selective dual mTORC1 and mTORC2 inhibitors: The discovery of AZD8055 and AZD2014. Bioorganic Med. Chem. Lett. 2013, 23, 1212–1216. [Google Scholar] [CrossRef] [PubMed]
- Oishi, T.; Itamochi, H.; Kudoh, A.; Nonaka, M.; Kato, M.; Nishimura, M.; Oumi, N.; Sato, S.; Naniwa, J.; Sato, S.; et al. The PI3K/mTOR dual inhibitor NVP-BEZ235 reduces the growth of ovarian clear cell carcinoma. Oncol. Rep. 2014, 32, 553–558. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.B.; Williams, K.A.; Krie, A.K.; De, P.; Dey, N.; Leyland-Jones, B.; Starks, D.; Rojas-Espaillat, L.A. Results of a phase Ib trial evaluating the safety and clinical activity of sapanisertib (TAK 228) in combination with serabelisib (TAK 117) and paclitaxel in patients with advanced ovarian, endometrial, or breast cancer. J. Clin. Oncol. 2020, 38, 3604. [Google Scholar] [CrossRef]
- Mortensen, D.S.; Perrin-Ninkovic, S.M.; Shevlin, G.; Elsner, J.; Zhao, J.; Whitefield, B.; Tehrani, L.; Sapienza, J.; Riggs, J.R.; Parnes, J.S.; et al. Optimization of a Series of Triazole Containing Mammalian Target of Rapamycin (mTOR) Kinase Inhibitors and the Discovery of CC-115. J. Med. Chem. 2015, 58, 5599–5608. [Google Scholar] [CrossRef]
- Mortensen, D.S.; Perrin-Ninkovic, S.M.; Shevlin, G.; Zhao, J.; Packard, G.; Bahmanyar, S.; Correa, M.; Elsner, J.; Harris, R.; Lee, B.G.S.; et al. Discovery of Mammalian Target of Rapamycin (mTOR) Kinase Inhibitor CC-223. J. Med. Chem. 2015, 58, 5323–5333. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Baretić, D.; Williams, R.L. The structural basis for mTOR function. Semin. Cell Dev. Biol. 2014, 36, 91–101. [Google Scholar] [CrossRef]
- Zhang, Y.; Nicholatos, J.; Dreier, J.R.; Ricoult, S.J.H.; Widenmaier, S.; Hotamisligil, G.S.; Kwiatkowski, D.J.; Manning, B.D. Coordinated regulation of protein synthesis and degradation by mTORC1. Nat. Cell Biol. 2014, 513, 440–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Abraham, R.T.; Wiederrecht, G.J. Immunopharmacology of rapamycin. Annu. Rev. Immunol. 1996, 14, 483–510. [Google Scholar] [CrossRef]
- Liu, Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006, 9, 230–234. [Google Scholar] [CrossRef] [Green Version]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernlund, E.; Ihrlund, L.S.; Khan, O.; Ates, Y.O.; Linder, S.; Panaretakis, T.; Shoshan, M.C. Potentiation of chemotherapeutic drugs by energy metabolism inhibitors 2-deoxyglucose and etomoxir. Int. J. Cancer 2008, 123, 476–483. [Google Scholar] [CrossRef] [Green Version]
- Ji, Z.; Shen, Y.; Feng, X.; Kong, Y.; Shao, Y.; Meng, J.; Zhang, X.; Yang, G. Deregulation of Lipid Metabolism: The Critical Factors in Ovarian Cancer. Front. Oncol. 2020, 10, 593017. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, W.; Li, S.; Yang, H. The role of cholesterol metabolism in cancer. Am. J. Cancer Res. 2019, 9, 219–227. [Google Scholar] [PubMed]
- Helzlsouer, K.J.; Alberg, A.J.; Norkus, E.P.; Morris, J.S.; Hoffman, S.C.; Comstock, G.W. Prospective Study of Serum Micronutrients and Ovarian Cancer. J. Natl. Cancer Inst. 1996, 88, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Essalmani, R.; Day, R.; Khatib, A.M.; Seidah, N.; Prat, A. Proprotein Convertase Subtilisin/Kexin Type 9 Deficiency Reduces Melanoma Metastasis in Liver. Neoplasia 2012, 14, 1122–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Li, S.; Fang, Y.; Zou, Y.; Song, D.; Zhang, S.; Cai, Y. Proprotein Convertase Subtilisin/Kexin Type 9 Promotes Gastric Cancer Metastasis and Suppresses Apoptosis by Facilitating MAPK Signaling Pathway Through HSP70 Up-Regulation. Front. Oncol. 2021, 10, 609663. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. ApoA1 and ApoA1-specific self-antibodies in cardiovascular disease. Lab. Investig. 2016, 96, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Ko, C.-W.; Tso, P.; Bhargava, A. Apolipoprotein A-IV: A Multifunctional Protein Involved in Protection against Atherosclerosis and Diabetes. Cells 2019, 8, 319. [Google Scholar] [CrossRef] [Green Version]
- Mather, K.A.; Thalamuthu, A.; Oldmeadow, C.; Song, F.; Armstrong, N.J.; Poljak, A.; Holliday, E.G.; McEvoy, M.; Kwok, J.B.; Assareh, A.A.; et al. Genome-wide significant results identified for plasma apolipoprotein H levels in middle-aged and older adults. Sci. Rep. 2016, 6, 23675. [Google Scholar] [CrossRef] [Green Version]
- Wolska, A.; Reimund, M.; Remaley, A.T. Apolipoprotein C-II. Curr. Opin. Lipidol. 2020, 31, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Yi, J.; Li, W.; Zheng, X.; Liu, J.; Wang, J.; Du, G. Apolipoproteins and cancer. Cancer Med. 2019, 8, 7032–7043. [Google Scholar] [CrossRef] [Green Version]
- Su, F.; Lang, J.; Kumar, A.; Ng, C.; Hsieh, B.; Suchard, M.A.; Reddy, S.T.; Farias-Eisner, R. Validation of Candidate Serum Ovarian Cancer Biomarkers for Early Detection. Biomark. Insights 2007, 2, 369–375. [Google Scholar] [CrossRef]
- Dieplinger, H.; Ankerst, D.; Burges, A.; Lenhard, M.; Lingenhel, A.; Fineder, L.; Buchner, H.; Stieber, P. Afamin and Apolipoprotein A-IV: Novel Protein Markers for Ovarian Cancer. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1127–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, G.; Yilmaz, A.; Kumar, P.; Turkoglu, O.; Mutch, D.G.; Powell, M.A.; Rosen, B.; Bahado-Singh, R.O.; Graham, S.F. Targeted metabolomic profiling of low and high grade serous epithelial ovarian cancer tissues: A pilot study. Metabolomics 2018, 14, 154. [Google Scholar] [CrossRef]
- Elia, I.; Doglioni, G.; Fendt, S.-M. Metabolic Hallmarks of Metastasis Formation. Trends Cell Biol. 2018, 28, 673–684. [Google Scholar] [CrossRef]
- Olesen, U.H.; Petersen, J.G.; Garten, A.; Kiess, W.; Yoshino, J.; Imai, S.-I.; Christensen, M.K.; Fristrup, P.; Thougaard, A.V.; Björkling, F.; et al. Target enzyme mutations are the molecular basis for resistance towards pharmacological inhibition of nicotinamide phosphoribosyltransferase. BMC Cancer 2010, 10, 677. [Google Scholar] [CrossRef] [Green Version]
- Floryk, D.; Thompson, T.C. Antiproliferative effects of AVN944, a novel inosine 5-monophosphate dehydrogenase inhibitor, in prostate cancer cells. Int. J. Cancer 2008, 123, 2294–2302. [Google Scholar] [CrossRef] [Green Version]
- Weaver, Z.; Strovel, J.; Chakiath, M.; Natarajan, P.; Lawrence, T.; Bol, D. Antiangiogenic properties of the IMPDH inhibitor AVN944. Cancer Res. 2007, 67 (Suppl. 9), 3983. Available online: http://cancerres.aacrjournals.org/content/67/9_Supplement/3983.abstract (accessed on 14 June 2021).
- Tian, Y.; Zhang, J.; Chen, L.; Zhang, X. The expression and prognostic role of IMPDH2 in ovarian cancer. Ann. Diagn. Pathol. 2020, 46, 151511. [Google Scholar] [CrossRef]
- Xu, B.; Deng, Y.; Bi, R.; Guo, H.; Shu, C.; Shah, N.K.; Chang, J.; Liu, G.; Du, Y.; Wei, W.; et al. A first-in-class inhibitor, MLN4924 (pevonedistat), induces cell-cycle arrest, senescence, and apoptosis in human renal cell carcinoma by suppressing UBE2M-dependent neddylation modification. Cancer Chemother. Pharmacol. 2018, 81, 1083–1093. [Google Scholar] [CrossRef]
- Nawrocki, S.T.; Kelly, K.R.; Smith, P.G.; Espitia, C.M.; Possemato, A.; Beausoleil, S.A.; Milhollen, M.; Blakemore, S.; Thomas, M.; Berger, A.; et al. Disrupting Protein NEDDylation with MLN4924 Is a Novel Strategy to Target Cisplatin Resistance in Ovarian Cancer. Clin. Cancer Res. 2013, 19, 3577–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lockhart, A.C.; Bauer, T.M.; Aggarwal, C.; Lee, C.B.; Harvey, R.D.; Cohen, R.B.; Sedarati, F.; Nip, T.K.; Faessel, H.; Dash, A.B.; et al. Phase Ib study of pevonedistat, a NEDD8-activating enzyme inhibitor, in combination with docetaxel, carboplatin and paclitaxel, or gemcitabine, in patients with advanced solid tumors. Investig. New Drugs 2019, 37, 87–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, J.; Van Leeuwen, J.E.; Elbaz, M.; Branchard, E.; Penn, L.Z. Statins as Anticancer Agents in the Era of Precision Medicine. Clin. Cancer Res. 2020, 26, 5791–5800. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin Use and Reduced Cancer-Related Mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Emons, G.; Kurzeder, C.; Schmalfeldt, B.; Neuser, P.; de Gregorio, N.; Pfisterer, J.; Park-Simon, T.-W.; Mahner, S.; Schröder, W.; Lück, H.-J.; et al. Temsirolimus in women with platinum-refractory/resistant ovarian cancer or advanced/recurrent endometrial carcinoma. A phase II study of the AGO-study group (AGO-GYN8). Gynecol. Oncol. 2016, 140, 450–456. [Google Scholar] [CrossRef]
- Hisamatsu, T.; Mabuchi, S.; Matsumoto, Y.; Kawano, M.; Sasano, T.; Takahashi, R.; Sawada, K.; Ito, K.; Kurachi, H.; Schilder, R.J.; et al. Potential Role of mTORC2 as a Therapeutic Target in Clear Cell Carcinoma of the Ovary. Mol. Cancer Ther. 2013, 12, 1367–1377. [Google Scholar] [CrossRef] [Green Version]
- Caumanns, J.J.; Berns, K.; Wisman, G.B.A.; Fehrmann, R.; Tomar, T.; Klip, H.; Meersma, G.J.; Hijmans, E.M.; Gennissen, A.M.; Duiker, E.; et al. Integrative Kinome Profiling Identifies mTORC1/2 Inhibition as Treatment Strategy in Ovarian Clear Cell Carcinoma. Clin. Cancer Res. 2018, 24, 3928–3940. [Google Scholar] [CrossRef] [Green Version]
- Pétigny-Lechartier, C.; Duboc, C.; Jebahi, A.; Louis, M.-H.; Abeilard, E.; Denoyelle, C.; Gauduchon, P.; Poulain, L.; Villedieu, M. The mTORC1/2 Inhibitor AZD8055 Strengthens the Efficiency of the MEK Inhibitor Trametinib to Reduce the Mcl-1/[Bim and Puma] ratio and to Sensitize Ovarian Carcinoma Cells to ABT-737. Mol. Cancer Ther. 2016, 16, 102–115. [Google Scholar] [CrossRef] [Green Version]
- Basu, B.; Krebs, M.; Sundar, R.; Wilson, R.; Spicer, J.; Jones, R.; Brada, M.; Talbot, D.; Steele, N.; Garces, A.I.; et al. Vistusertib (dual m-TORC1/2 inhibitor) in combination with paclitaxel in patients with high-grade serous ovarian and squamous non-small-cell lung cancer. Ann. Oncol. 2018, 29, 1918–1925. [Google Scholar] [CrossRef]
- Kudoh, A.; Oishi, T.; Itamochi, H.; Sato, S.; Naniwa, J.; Sato, S.; Shimada, M.; Kigawa, J.; Harada, T. Dual Inhibition of Phosphatidylinositol 3′-Kinase and Mammalian Target of Rapamycin Using NVP-BEZ235 as a Novel Therapeutic Approach for Mucinous Adenocarcinoma of the Ovary. Int. J. Gynecol. Cancer 2014, 24, 444–453. [Google Scholar] [CrossRef]
- Moore, K.N.; Bauer, T.M.; Falchook, G.S.; Chowdhury, S.; Patel, C.; Neuwirth, R.; Enke, A.; Zohren, F.; Patel, M.R. Phase I study of the investigational oral mTORC1/2 inhibitor sapanisertib (TAK-228): Tolerability and food effects of a milled formulation in patients with advanced solid tumours. ESMO Open 2018, 3, e000291. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Cell Type | Main Genomic Aberrations |
|---|---|---|
| HeLa | Endometrial carcinoma | |
| JHOS2 | High grade ovarian serous adenocarcinoma. | TP53 mut; BRCA1 mut [28,29] |
| Kuramochi | High grade ovarian serous adenocarcinoma | TP53 mut; BRCA2 mut; MYC amp; KRAS amp |
| COV362 | High grade ovarian serous adenocarcinoma | TP53 mut; BRCA1 mut; MYC amp; RB1 del |
| Ovsaho | High grade ovarian serous adenocarcinoma | TP53 mut; RB1 del; BRCA1 del [28,29] |
| OVCAR3 | High grade ovarian serous adenocarcinoma | TP53 mut [30] |
| OVCAR3cis | High grade ovarian serous adenocarcinoma, resistant to cisplatin | TP53 mut [30,31] |
| A2780 | Ovarian endometrioid adenocarcinoma. | PIK3CA mut; PTEN mut; BRAF mut; ARD1A mut [28,29] |
| A2780cis | Ovarian endometrioid adenocarcinoma, resistant to cisplatin | |
| PDC #1 | Patient-derived cell culture (PDCs) from high grade serous OC tumor | TP53 mut p.R175H; CCNE1 amp; PAX8 positive |
| PDC #2 | Patient-derived cell culture (PDCs) from high grade serous OC tumor | TP53 mut p. R283P; CCNE1 amp; PAX8 positive |
| PDC #3 | Patient-derived cell culture (PDCs) from high grade serous OC tumor | TP53 fs, MYC amp; KRAS amp; PAX8 positive |
| PDC #4 | Patient-derived cell culture (PDCs) from low grade serous OC tumor | TP53 WT; CDKN2A homozygous loss; PAX8 positive |
| PDC #5 | Patient-derived cell culture (PDCs) from low grade serous OC tumor | TP53 WT; CDKN2A homozygous loss; PAX8 positive |
| Drug Name | Mechanism/Targets | Biochemical Class | Clinical Trials (OC) |
|---|---|---|---|
| Daporinad [36] | Targets nicotinamide phosphoribosyltransferase (NAMPT), an intermediate in the biosynthesis of nicotinamide adenine dinucleotide (NAD) | Metabolic modifier | Melanoma (Phase II), B-cell Chronic Lymphocytic Leukemia (Phase I/II), T-cell Lymphoma (Phase II) |
| Pevonedistat [36] | Inhibitor of Nedd8 activating enzyme (NAE) | Metabolic modifier | Myelodysplastic Syndrome (Phase I), Acute Myeloid Leukemia (Phase I/2), Advanced/Solid Tumors (Phase I), Multiple Myeloma (Phase I), Melanoma (Phase I) |
| AVN944 [37] | Inhibitor of inosine monosphosphate dehydrogenase (IMPDH), an enzyme involved in the de novo synthesis of GTP | Metabolic modifier | Hematological Malignancies (Phase I) |
| Erastin [38] | Activator of ferroptosis by acting as a voltage-dependent anion channel (VDAC) inhibitor; depletes cellular cysteine and glutathione, inducing excessive lipid peroxidation and cell death | Metabolic modifier | - |
| Methotrexate [39] | Inhibits dihydrofolate reductase enzyme, resulting in inhibition of purine nucleotide and thymidylate synthesis | Metabolic modifier | Ovarian cancer (Phase II), Acute Lymphoblastic Leukemia (Phase IV), Rheumatoid arthritis (Phase IV); Psoriasis (Phase IV), Breast Cancer (Phase II) |
| Atorvastatin [40] | Statin, inhibits hepatic hydroxymethyl-glutaryl coenzyme A (HMG-CoA) reductase involved in cholesterol synthesis | Metabolic modifier | Cardiovascular Disease (Phase IV), Cholesterol LDL (Phase IV), Type 2 Diabetes Mellitus (Phase IV), Alzheimer’s Disease (Phase III) |
| Triapine [41] | Inhibitor of ribonucleotide reductase (RNR); DNA synthesis inhibitor | Metabolic modifier | Ovarian Epithelial Cancer (Phase II), Leukemia/ Myelodysplastic Syndromes (Phase I/II) Lung Cancer (Phase II), Prostate Cancer (Phase II), Adenocarcinoma (Phase II) |
| TH588 [42] | Inhibitor of mut-T homolog-1 (MTH1, also known as NUDT1) that eliminates oxidized dNTP pools to prevent incorporation of damaged bases during DNA replication; impairs mitotic progression and mitotic DNA synthesis | Metabolic modifier | - |
| AZD3965 [43] | Monocarboxylate transporter 1 (MCT1) inhibitor; impairs lactate efflux leading to accumulation of glycolytic intermediates | Metabolic modifier | - |
| Disulfiram (+CuCl2) [44] | Alcohol dehydrogenase inhibitor chelating Cu+ selectively accumulated in cancer cells; generates reactive oxygen species (ROS) and inhibits proteasome activity | Metabolic modifier | Alcohol Dependence (Phase IV), Opioid Dependence (Phase II), Melanomas (Phase II), Glioblastoma (Phase II/III), HIV Infections (Phase I/II) |
| Pemetrexed [45] | Inhibitor of thymidylate synthase (TS) enzyme involved in DNA synthesis | Metabolic modifier | Ovarian cancer (Phase II), Lung cancer (Phase I), Breast cancer (Phase I), Prostate cancer (Phase II). |
| Ridaforolimus [46] | mTORC1 inhibitor, binds peptidyl-prolyl cis-trans isomerase FKBP12 | Rapalog | Ovarian Cancer (Phase I), Metastatic sarcomas (Phase III), Breast cancer (Phase II), Prostate cancer (Phase II), Lymphoma/Myeloma (Phase II), Lung Cancer (Phase II) |
| Temsirolimus [46] | mTORC1 inhibitor, binds peptidyl-prolyl cis-trans isomerase FKBP12 | Rapalog | Ovarian cancer (Phase II), Pancreatic Cancer (Phase II), Acute Myeloid Leukemia (Phase II), Glioblastoma (Phase II), Sarcomas (Phase II), Renal Cancers (Phase I), Breast cancer (Phase I/II) |
| Everolimus [46] | mTORC1 inhibitor, binds peptidyl-prolyl cis-trans isomerase FKBP12 | Rapalog | Ovarian Cancer (Phase II), Breast cancer (Phase IV), Allograft rejection (Phase IV), |
| Sirolimus [47] | mTORC1 inhibitor, binds peptidyl-prolyl cis-trans isomerase FKBP12 | Rapalog | Ovarian cancer (Phase II), Allograft rejection (Phase IV), Leukemia (Phase III), Hepatocellular Carcinoma (Phase III), Breast Cancer (Phase II) |
| AZD8055 [48] | mTOR inhibitor | Kinase inhibitor | Advanced Solid Malignancies (Phase I) |
| Vistusertib [48] | mTOR inhibitor, ATP-competitive | Kinase inhibitor | Breast Cancer (Phase I), Prostate Cancer (Phase I), Lung Cancer (Phase I) |
| Dactolisib [49] | mTOR/(PI3K) inhibitor | Kinase inhibitor | Breast Cancer (Phase I), Prostate Cancer (Phase I) |
| Sapanisertib [50] | mTOR inhibitor | Kinase inhibitor | Breast Cancer (Phase I), Prostate Cancer (Phase I), Lung cancer (Phase I) |
| CC-115 [51] | mTOR/DNA-dependent protein kinase (DNA-PK) inhibitor | Kinase inhibitor | - |
| CC-223 [52] | mTOR inhibitor | Kinase inhibitor | Hepatocellular Carcinoma (Phase I/II), B-Cell Lymphoma (Phase I/II), Glioblastoma (Phase I/II), Lung Cancer (Phase I/II) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacome Sanz, D.; Raivola, J.; Karvonen, H.; Arjama, M.; Barker, H.; Murumägi, A.; Ungureanu, D. Evaluating Targeted Therapies in Ovarian Cancer Metabolism: Novel Role for PCSK9 and Second Generation mTOR Inhibitors. Cancers 2021, 13, 3727. https://doi.org/10.3390/cancers13153727
Jacome Sanz D, Raivola J, Karvonen H, Arjama M, Barker H, Murumägi A, Ungureanu D. Evaluating Targeted Therapies in Ovarian Cancer Metabolism: Novel Role for PCSK9 and Second Generation mTOR Inhibitors. Cancers. 2021; 13(15):3727. https://doi.org/10.3390/cancers13153727
Chicago/Turabian StyleJacome Sanz, Dafne, Juuli Raivola, Hanna Karvonen, Mariliina Arjama, Harlan Barker, Astrid Murumägi, and Daniela Ungureanu. 2021. "Evaluating Targeted Therapies in Ovarian Cancer Metabolism: Novel Role for PCSK9 and Second Generation mTOR Inhibitors" Cancers 13, no. 15: 3727. https://doi.org/10.3390/cancers13153727
APA StyleJacome Sanz, D., Raivola, J., Karvonen, H., Arjama, M., Barker, H., Murumägi, A., & Ungureanu, D. (2021). Evaluating Targeted Therapies in Ovarian Cancer Metabolism: Novel Role for PCSK9 and Second Generation mTOR Inhibitors. Cancers, 13(15), 3727. https://doi.org/10.3390/cancers13153727