
APLN/APLNR Signaling Controls Key Pathological Parameters of Glioblastoma


Abstract
:Simple Summary
Abstract
1. Introduction
2. APLN and Its Receptor APLNR
3. APLN/APLNR in Vascular Development
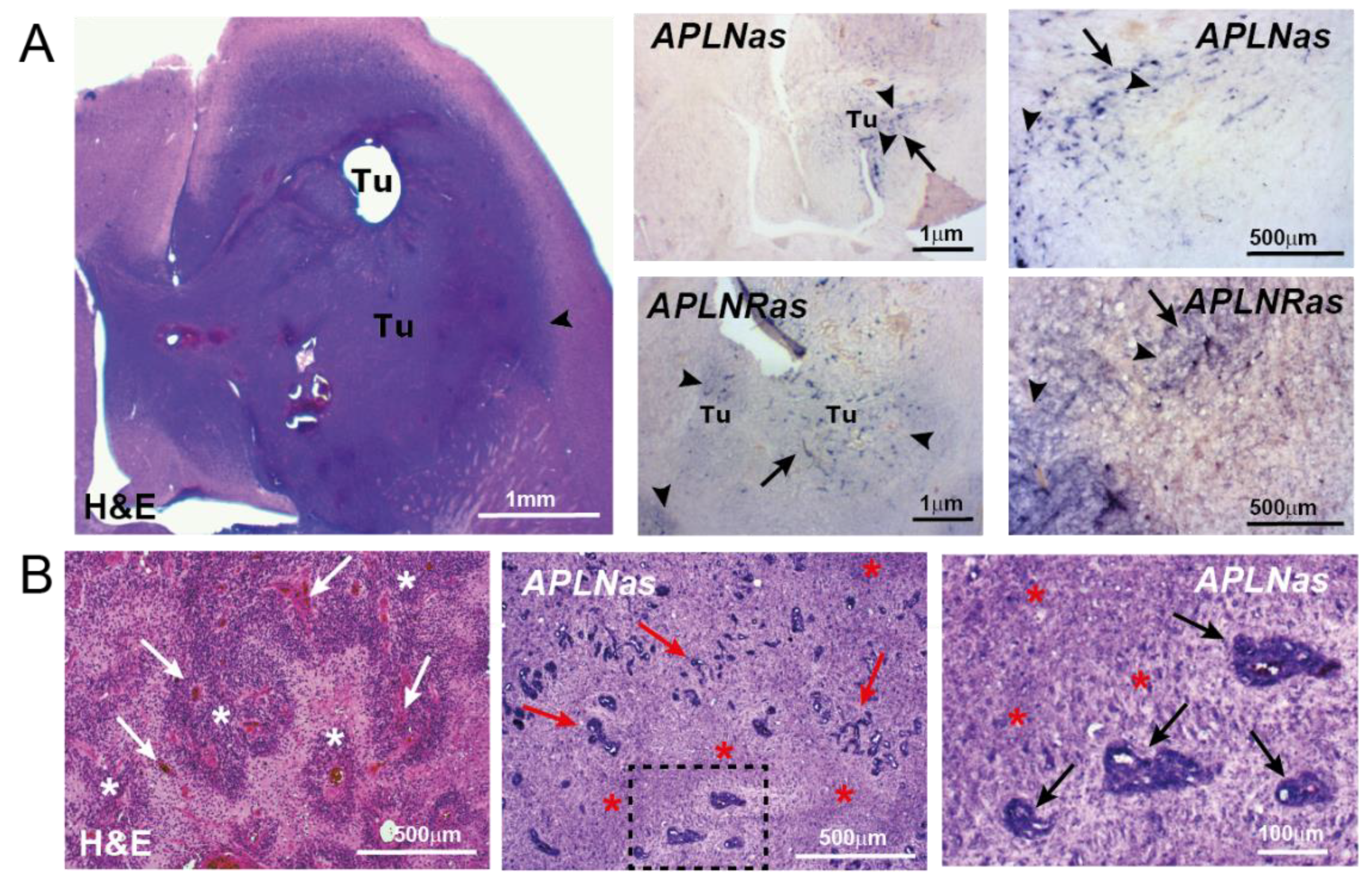
4. APLN in the Formation of the Glioblastoma Vasculature
5. The Role of APLN and APLNR in Glioblastoma Cell Invasion
6. Apelin-F13A Blocks Glioblastoma Invasion and Simultaneously Attenuates Tumor Angiogenesis
7. APLN and APLNR in Neurons and Astrocytes
8. APLN and APLNR in Pericytes
9. APLN/APLNR Signaling in Microglia, Macrophages, and T-Cells
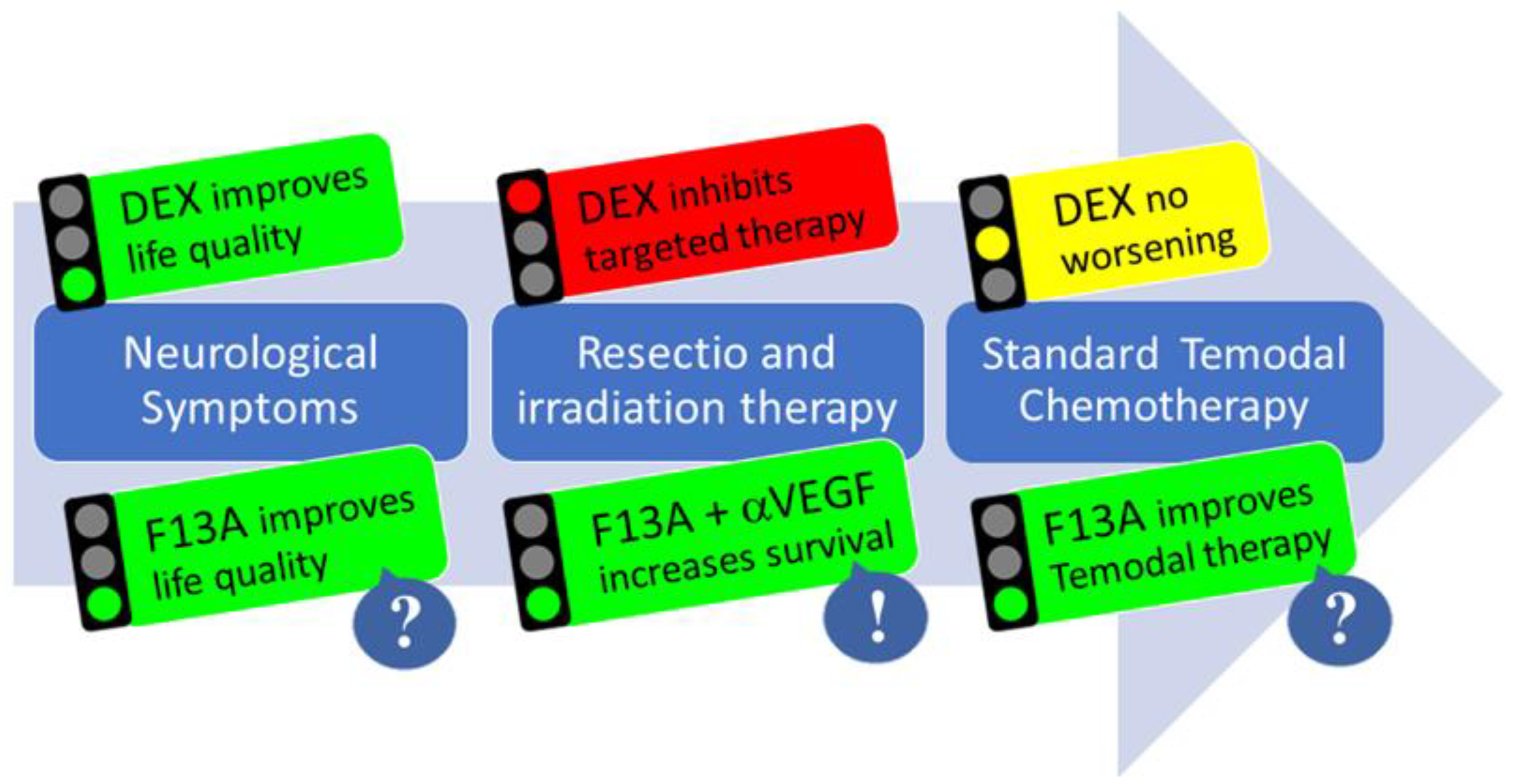
10. Perspectives for APLN-Mediated Multimodal Glioblastoma Therapy
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APJ | putative receptor protein related to the angiotensin II receptor-like 1 (Angtrl1) |
| APLN | Apelin |
| APLN-KD | APLN knockdown |
| APLN-KO | APLN knockout mouse |
| APLNR | Apelin receptor |
| BBB | blood-brain barrier |
| creER | cre recombinase estrogen receptor fusion gene |
| DEX | Dexamethasone |
| ECs | endothelial cells |
| ESCs | embryonic stem cells |
| GBM | glioblastoma |
| GPCR | G-protein-coupled receptor |
| HE | Hematoxylin/Eosin |
| HSC | hematopoietic stem cells |
| KD | knockdown |
| TAM | tumor associated cells |
| TME | tumor microenvironment |
| Msr | mesenchyme-associated serpentine receptor |
| NPCs | neural precursor cells |
| VEGFA | Vascular endothelial growth factor A |
| VEGFR | VEGF receptor |
| WT | wildtype mouse |
References
- Chen, J.; McKay, R.M.; Parada, L.F. Malignant glioma: Lessons from genomics, mouse models, and stem cells. Cell 2012, 149, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019, 16, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and niche concept. Cancers 2018, 11, 5. [Google Scholar] [CrossRef] [Green Version]
- Audia, A.; Conroy, S.; Glass, R.; Bhat, K.P.L. The impact of the tumor microenvironment on the properties of glioma stem-like cells. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, R.; Synowitz, M. CNS macrophages and peripheral myeloid cells in brain tumours. Acta Neuropathol. 2014, 128, 347–362. [Google Scholar] [CrossRef] [Green Version]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2015, 19, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Twyman-Saint, V.C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nat. Cell Biol. 2015, 520, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [Green Version]
- Chinot, O.L. Cilengitide in glioblastoma: When did it fail? The Lancet. Oncology 2014, 15, 1044–1045. [Google Scholar] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kälin, R.E.; Kretz, M.P.; Meyer, A.M.; Kispert, A.; Heppner, F.; Brändli, A.W. Paracrine and autocrine mechanisms of apelin signaling govern embryonic and tumor angiogenesis. Dev. Biol. 2007, 305, 599–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plate, K.H.; Breier, G.; Weich, H.A.; Risau, W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nat. Cell Biol. 1992, 359, 845–848. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, B.F.; Heiber, M.; Chan, A.; Heng, H.H.; Tsui, L.C.; Kennedy, J.L.; Shi, X.; Petronis, A.; George, S.R.; Nguyen, T. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene 1993, 136, 355–360. [Google Scholar] [CrossRef]
- Kälin, S.; Kälin, R.E. Apelin and Cancer. In Adipocitokines, Energy Balance and Cancer; Reizes, O., Berger, N.A., Eds.; Springer International Publishing Switzerland: Cham, Switzerland, 2017; Volume 12, pp. 137–166. [Google Scholar]
- Devic, E.; Paquereau, L.; Vernier, P.; Knibiehler, B.; Audigier, Y. Expression of a new G protein-coupled receptor X-msr is associated with an endothelial lineage in Xenopus laevis. Mech. Dev. 1996, 59, 129–140. [Google Scholar] [CrossRef]
- Cleaver, O.; Tonissen, K.F.; Saha, M.S.; Krieg, P.A. Neovascularization of the Xenopus embryo. Dev. Dyn. 1997, 210, 66–77. [Google Scholar] [CrossRef]
- Kälin, R.E.; Bänziger-Tobler, N.E.; Detmar, M.; Brändli, A.W. An in vivo chemical library screen in Xenopus tadpoles reveals novel pathways involved in angiogenesis and lymphangiogenesis. Blood 2009, 114, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Devic, E.; Rizzoti, K.; Bodin, S.; Knibiehler, B.; Audigier, Y. Amino acid sequence and embryonic expression of msr/apj, the mouse homolog of Xenopus X-msr and human APJ. Mech. Dev. 1999, 84, 199–203. [Google Scholar] [CrossRef]
- De Mota, N.; Lenkei, Z.; Llorens-Cortès, C. Cloning, pharmacological characterization and brain distribution of the rat apelin receptor. Neuroendocrinology 2000, 72, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, M.; Kawamata, Y.; Fukusumi, S.; Fujii, R.; Habata, Y.; Hinuma, S.; Kitada, C.; Honda, S.; Kurokawa, T.; Onda, H.; et al. Molecular and functional characteristics of APJ. Tissue distribution of mRNA and interaction with the endogenous ligand apelin. J. Biol. Chem. 2000, 275, 21061–21067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Carroll, A.-M.; Selby, T.L.; Palkovits, M.; Lolait, S.J. Distribution of mRNA encoding B78/apj, the rat homologue of the human APJ receptor, and its endogenous ligand apelin in brain and peripheral tissues. Biochim. Et Biophys. Acta (BBA) Gene Struct. Expr. 2000, 1492, 72–80. [Google Scholar] [CrossRef]
- Tatemoto, K.; Hosoya, M.; Habata, Y.; Fujii, R.; Kakegawa, T.; Zou, M.-X.; Kawamata, Y.; Fukusumi, S.; Hinuma, S.; Kitada, C.; et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem. Biophys. Res. Commun. 1998, 251, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Reaux, A.; De Mota, N.; Skultetyova, I.; Lenkei, Z.; El Messari, S.; Gallatz, K.; Corvol, P.; Palkovits, M.; Llorens-Cortès, C. Physiological role of a novel neuropeptide, apelin, and its receptor in the rat brain. J. Neurochem. 2001, 77, 1085–1096. [Google Scholar] [CrossRef]
- Saint-Geniez, M.; Masri, B.; Malecaze, F.; Knibiehler, B.; Audigier, Y. Expression of the murine msr/apj receptor and its ligand apelin is upregulated during formation of the retinal vessels. Mech. Dev. 2002, 110, 183–186. [Google Scholar] [CrossRef]
- Kasai, A.; Shintani, N.; Oda, M.; Kakuda, M.; Hashimoto, H.; Matsuda, T.; Hinuma, S.; Baba, A. Apelin is a novel angiogenic factor in retinal endothelial cells. Biochem. Biophys. Res. Commun. 2004, 325, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.M.; D’Agostino, S.L.; Miller, M.K.; Heimark, R.L.; Krieg, P.A. Apelin, the ligand for the endothelial G-protein-coupled receptor, APJ, is a potent angiogenic factor required for normal vascular development of the frog embryo. Dev. Biol. 2006, 296, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Kasai, A.; Ishimaru, Y.; Kinjo, T.; Satooka, T.; Matsumoto, N.; Yoshioka, Y.; Yamamuro, A.; Gomi, F.; Shintani, N.; Baba, A.; et al. Apelin is a crucial factor for hypoxia-induced retinal angiogenesis. Arter. Thromb. Vasc. Biol. 2010, 30, 2182–2187. [Google Scholar] [CrossRef]
- Kidoya, H.; Ueno, M.; Yamada, Y.; Mochizuki, N.; Nakata, M.; Yano, T.; Fujii, R.; Takakura, N. Spatial and temporal role of the apelin/APJ system in the caliber size regulation of blood vessels during angiogenesis. EMBO J. 2008, 27, 522–534. [Google Scholar] [CrossRef] [Green Version]
- Kidoya, H.; Naito, H.; Takakura, N.; Bee, T.; Swiers, G.; Muroi, S.; Pozner, A.; Nottingham, W.; Santos, A.C.; Li, P.-S.; et al. Apelin induces enlarged and nonleaky blood vessels for functional recovery from ischemia. Blood 2010, 115, 3166–3174. [Google Scholar] [CrossRef] [Green Version]
- Del Toro, R.; Prahst, C.; Mathivet, T.; Siegfried, G.; Kaminker, J.S.; Larrivee, B.; Breant, C.; Duarte, A.; Takakura, N.; Fukamizu, A.; et al. Identification and functional analysis of endothelial tip cell–enriched genes. Blood 2010, 116, 4025–4033. [Google Scholar] [CrossRef] [Green Version]
- Strasser, G.A.; Kaminker, J.S.; Tessier-Lavigne, M. Microarray analysis of retinal endothelial tip cells identifies CXCR4 as a mediator of tip cell morphology and branching. Blood 2010, 115, 5102–5110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helker, C.S.; Eberlein, J.; Wilhelm, K.; Sugino, T.; Malchow, J.; Schuermann, A.; Baumeister, S.; Kwon, H.-B.; Maischein, H.-M.; Potente, M.; et al. Apelin signaling drives vascular endothelial cells toward a pro-angiogenic state. eLife 2020, 9. [Google Scholar] [CrossRef]
- Liu, Q.; Hu, T.; He, L.; Huang, X.; Tian, X.; Zhang, H.; He, L.; Pu, W.; Zhang, L.; Sun, H.; et al. Genetic targeting of sprouting angiogenesis using Apln-CreER. Nat. Commun. 2015, 6, 6020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Seaman, S.; Stevens, J.; Yang, M.Y.; Logsdon, D.; Graff-Cherry, C.; St Croix, B. Genes that distinguish physiological and pathological angiogenesis. Cancer Cell 2007, 11, 539–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masiero, M.; Simões, F.; Han, H.D.; Snell, C.; Peterkin, T.; Bridges, E.; Mangala, L.S.; Wu, S.Y.; Pradeep, S.; Li, D.; et al. A core human primary tumor angiogenesis signature identifies the endothelial orphan receptor ELTD1 as a key regulator of angiogenesis. Cancer Cell 2013, 24, 229–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakariassen, P.O.; Prestegarden, L.; Wang, J.; Skaftnesmo, K.O.; Mahesparan, R.; Molthoff, C.; Sminia, P.; Sundlisaeter, E.; Misra, A.; Tysnes, B.B.; et al. Angiogenesis-independent tumor growth mediated by stem-like cancer cells. Proc. Nat. Acad. Sci. USA 2006, 103, 16466–16471. [Google Scholar] [CrossRef] [Green Version]
- Talasila, K.M.; Soentgerath, A.; Euskirchen, P.; Rosland, G.V.; Wang, J.; Huszthy, P.C.; Prestegarden, L.; Skaftnesmo, K.O.; Sakariassen, P.O.; Eskilsson, E.; et al. EGFR wild-type amplification and activation promote invasion and development of glioblastoma independent of angiogenesis. Acta Neuropathol. 2013, 125, 683–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrella, G.; Hou, M.; Li, M.; Stoecklein, V.M.; Zdouc, N.; Volmar, M.N.M.; Miletic, H.; Reinhard, S.; Herold-Mende, C.C.; Kleber, S.; et al. Targeting APLN/APLNR Improves antiangiogenic efficiency and blunts proinvasive side effects of VEGFA/VEGFR2 blockade in glioblastoma. Cancer Res. 2019, 79, 2298–2313. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Tian, X.; He, L.; Li, Y.; Pu, W.; Liu, Q.; Tang, J.; Wu, J.; Cheng, X.; Liu, Y.; et al. Apj+ vessels drive tumor growth and represent a tractable therapeutic target. Cell Rep. 2018, 25, 1241–1254.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, A.; Kälin, S.; Monk, R.; Radke, J.; Heppner, F.L.; Kälin, R.E. Apelin controls angiogenesis-dependent glioblastoma growth. Int. J. Mol. Sci. 2020, 21, 4179. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Zhang, L.; Imai, Y.; Arab, S.; Chen, M.; Maekawa, Y.; Leschnik, M.; Leibbrandt, A.; Markovic, M.; Schwaighofer, J.; et al. Impaired heart contractility in Apelin gene-deficient mice associated with aging and pressure overload. Circ. Res. 2007, 101, e32–e42. [Google Scholar] [CrossRef] [Green Version]
- Uribesalgo, I.; Hoffmann, D.; Zhang, Y.; Kavirayani, A.; Lazovic, J.; Berta, J.; Novatchkova, M.; Pai, T.P.; Wimmer, R.A.; Laszlo, V.; et al. Apelin inhibition prevents resistance and metastasis associated with anti-angiogenic therapy. EMBO Mol. Med. 2019, 11, e9266. [Google Scholar] [CrossRef] [PubMed]
- Sorli, S.C.; Le Gonidec, S.; Knibiehler, B.; Audigier, Y. Apelin is a potent activator of tumour neoangiogenesis. Oncogene 2007, 26, 7692–7699. [Google Scholar] [CrossRef] [Green Version]
- Harford-Wright, E.; Andre-Gregoire, G.; Jacobs, K.A.; Treps, L.; Le Gonidec, S.; Leclair, H.M.; Gonzalez-Diest, S.; Roux, Q.; Guillonneau, F.; Loussouarn, D.; et al. Pharmacological targeting of apelin impairs glioblastoma growth. Brain 2017, 140, 2939–2954. [Google Scholar] [CrossRef] [Green Version]
- Chng, S.C.; Ho, L.; Tian, J.; Reversade, B. ELABELA: A hormone essential for heart development signals via the apelin receptor. Dev. Cell 2013, 27, 672–680. [Google Scholar] [CrossRef] [Green Version]
- Pauli, A.; Norris, M.L.; Valen, E.; Chew, G.L.; Gagnon, J.A.; Zimmerman, S.; Mitchell, A.; Ma, J.; Dubrulle, J.; Reyon, D.; et al. Toddler: An embryonic signal that promotes cell movement via Apelin receptors. Science 2014, 343, 1248636. [Google Scholar] [CrossRef]
- Ganguly, D.; Cai, C.; Sims, M.M.; Yang, C.H.; Thomas, M.; Cheng, J.; Saad, A.G.; Pfeffer, L.M. APELA expression in glioma, and its association with patient survival and tumor grade. Pharmaceuticals 2019, 12, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodyanik, M.A.; Yu, J.; Zhang, X.; Tian, S.; Stewart, R.; Thomson, J.A.; Slukvin, I.I. A mesoderm-derived precursor for mesenchymal stem and endothelial cells. Cell Stem Cell 2010, 7, 718–729. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.C.; Hirst, C.E.; Costa, M.; Ng, E.S.; Schiesser, J.V.; Gertow, K.; Stanley, E.G.; Elefanty, A.G. APELIN promotes hematopoiesis from human embryonic stem cells. Blood 2012, 119, 6243–6254. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nat. Cell Biol. 2010, 468, 829–833. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef]
- Li, M.; Gou, H.; Tripathi, B.K.; Huang, J.; Jiang, S.; Dubois, W.; Waybright, T.; Lei, M.; Shi, J.; Zhou, M.; et al. AN apela rna-containing negative feedback loop regulates p53-mediated apoptosis in embryonic stem cells. Cell Stem Cell 2015, 16, 669–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masri, B.; Lahlou, H.; Mazarguil, H.; Knibiehler, B.; Audigier, Y. Apelin (65–77) activates extracellular signal-regulated kinases via a PTX-sensitive g protein. Biochem. Biophys. Res. Commun. 2002, 290, 539–545. [Google Scholar] [CrossRef]
- Habata, Y.; Fujii, R.; Hosoya, M.; Fukusumi, S.; Kawamata, Y.; Hinuma, S.; Kitada, C.; Nishizawa, N.; Murosaki, S.; Kurokawa, T.; et al. Apelin, the natural ligand of the orphan receptor APJ, is abundantly secreted in the colostrum. Biochim. Et Biophys. Acta 1999, 1452, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Masri, B.; Morin, N.; Pedebernade, L.; Knibiehler, B.; Audigier, Y. The apelin receptor is coupled to Gi1 or Gi2 protein and is differentially desensitized by apelin fragments. J. Biol. Chem. 2006, 281, 18317–18326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choe, W.; Albright, A.; Sulcove, J.; Jaffer, S.; Hesselgesser, J.; Lavi, E.; Crino, P.; Kolson, D.L. Functional expression of the seven-transmembrane HIV-1 co-receptor APJ in neural cells. J. Neurovirol. 2000, 6 (Suppl. 1), S61–S69. [Google Scholar] [PubMed]
- Lo, H.-W. Targeting Ras-RAF-ERK and its interactive pathways as a novel therapy for malignant gliomas. Curr. Cancer Drug Targets 2010, 10, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Masri, B.; Morin, N.; Cornu, M.; Knibiehler, B.; Audigier, Y. Apelin (65–77) activates p70 S6 kinase and is mitogenic for umbilical endothelial cells. FASEB J. 2004, 18, 1909–1911. [Google Scholar] [CrossRef]
- Tatemoto, K.; Takayama, K.; Zou, M.-X.; Kumaki, I.; Zhang, W.; Kumano, K.; Fujimiya, M. The novel peptide apelin lowers blood pressure via a nitric oxide-dependent mechanism. Regul. Pept. 2001, 99, 87–92. [Google Scholar] [CrossRef]
- Ho, L.; Tan, S.Y.; Wee, S.; Wu, Y.; Tan, S.J.; Ramakrishna, N.B.; Chng, S.C.; Nama, S.; Szczerbinska, I.; Chan, Y.S.; et al. ELABELA Is an endogenous growth factor that sustains hESC self-renewal via the PI3K/AKT pathway. Cell Stem Cell 2015, 17, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amoozgar, Z.; Jain, R.K.; Duda, D.G. Role of apelin in glioblastoma vascularization and invasion after anti-vegf therapy: What is the impact on the immune system? Cancer Res. 2019, 79, 2104–2106. [Google Scholar] [CrossRef]
- Lu-Emerson, C.; Duda, D.G.; Emblem, K.E.; Taylor, J.W.; Gerstner, E.R.; Loeffler, J.S.; Batchelor, T.T.; Jain, R.K. Lessons from anti–vascular endothelial growth factor and anti–vascular endothelial growth factor receptor trials in patients with glioblastoma. J. Clin. Oncol. 2015, 33, 1197–1213. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.K.; Saldivia, V.R.; Nguyen, T.; Cheng, R.; George, S.R.; O’Dowd, B.F. Modification of the terminal residue of Apelin-13 antagonizes its hypotensive action. Endocrinology 2005, 146, 231–236. [Google Scholar] [CrossRef]
- Macaluso, N.J.M.; Pitkin, S.L.; Maguire, J.J.; Davenport, A.P.; Glen, R.C. Discovery of a competitive Apelin receptor (APJ) antagonist. ChemMedChem 2011, 6, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.J.; Yu, S.P.; Zhang, L.; Wei, L. Neuroprotective effect of the endogenous neural peptide apelin in cultured mouse cortical neurons. Exp. Cell Res. 2010, 316, 1773–1783. [Google Scholar] [CrossRef] [Green Version]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhauser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Sakimoto, S.; Kidoya, H.; Naito, H.; Kamei, M.; Sakaguchi, H.; Goda, N.; Fukamizu, A.; Nishida, K.; Takakura, N. A role for endothelial cells in promoting the maturation of astrocytes through the apelin/APJ system in mice. Development 2012, 139, 1327–1335. [Google Scholar] [CrossRef] [Green Version]
- Silver, J.; Miller, J.H. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 2004, 5, 146–156. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armulik, A.; Genove, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Develop. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef]
- Hamilton-Whitaker, N.B.; Attwell, D.; Hall, C.N. Pericyte-mediated regulation of capillary diameter: A component of neurovascular coupling in health and disease. Front. Neuroenergetics 2010, 2. [Google Scholar] [CrossRef] [Green Version]
- Peppiatt, C.M.; Howarth, C.; Mobbs, P.; Attwell, D. Bidirectional control of CNS capillary diameter by pericytes. Nat. Cell Biol. 2006, 443, 700–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.K.; Cheng, R.; Nguyen, T.; Fan, T.; Kariyawasam, A.P.; Liu, Y.; Osmond, D.H.; George, S.R.; O’Dowd, B.F. Characterization of Apelin, the ligand for the APJ receptor. J. Neurochem. 2001, 74, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Sonmez, A.; Celebi, G.; Erdem, G.; Tapan, S.; Genc, H.; Tasci, I.; Ercin, C.N.; Dogru, T.; Kilic, S.; Uckaya, G.; et al. Plasma apelin and ADMA Levels in patients with essential hypertension. Clin. Experim. Hypertens. 2010, 32, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Maguire, J.J.; Kleinz, M.J.; Pitkin, S.L.; Davenport, A.P. [Pyr1]apelin-13 identified as the predominant apelin isoform in the human heart: Vasoactive mechanisms and inotropic action in disease. Hypertension 2009, 54, 598–604. [Google Scholar] [CrossRef] [Green Version]
- Ishida, J.; Hashimoto, T.; Hashimoto, Y.; Nishiwaki, S.; Iguchi, T.; Harada, S.; Sugaya, T.; Matsuzaki, H.; Yamamoto, R.; Shiota, N.; et al. Regulatory roles for APJ, a seven-transmembrane receptor related to angiotensin-type 1 receptor in blood pressure in vivo. J. Biol. Chem. 2004, 279, 26274–26279. [Google Scholar] [CrossRef] [Green Version]
- Kagiyama, S.; Fukuhara, M.; Matsumura, K.; Lin, Y.; Fujii, K.; Iida, M. Central and peripheral cardiovascular actions of apelin in conscious rats. Regul. Pept. 2005, 125, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Kasai, A.; Ishimaru, Y.; Higashino, K.; Kobayashi, K.; Yamamuro, A.; Yoshioka, Y.; Maeda, S. Inhibition of apelin expression switches endothelial cells from proliferative to mature state in pathological retinal angiogenesis. Angiogenesis 2013, 16, 723–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, D.; Trenkwalder, T.; Lee, S.; Chillo, O.; Deindl, E.; Kupatt, C.; Hinkel, R. Early vessel destabilization mediated by angiopoietin-2 and subsequent vessel maturation via angiopoietin-1 induce functional neovasculature after ischemia. PLoS ONE 2013, 8, e61831. [Google Scholar] [CrossRef] [Green Version]
- Kidoya, H.; Kunii, N.; Naito, H.; Muramatsu, F.; Okamoto, Y.; Nakayama, T.; Takakura, N. The apelin/APJ system induces maturation of the tumor vasculature and improves the efficiency of immune therapy. Oncogene 2011, 31, 3254–3264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinz, M.J.; Davenport, A.P. Immunocytochemical localization of the endogenous vasoactive peptide apelin to human vascular and endocardial endothelial cells. Regul. Pept. 2004, 118, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Tao, Y.; Feng, J.; Jiang, Y.R. Apelin protects primary rat retinal pericytes from chemical hypoxia-induced apoptosis. J. Ophthalmol. 2015, 2015, 1–14. [Google Scholar] [CrossRef]
- Olson, J.K.; Miller, S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef] [Green Version]
- Glass, R.; Synowitz, M.; Kronenberg, G.; Walzlein, J.-H.; Markovic, D.S.; Wang, L.-P.; Gast, D.; Kiwit, J.; Kempermann, G.; Kettenmann, H. Glioblastoma-induced attraction of endogenous neural precursor cells is associated with improved survival. J. Neurosci. 2005, 25, 2637–2646. [Google Scholar] [CrossRef] [Green Version]
- Markovic, D.S.; Glass, R.; Synowitz, M.; Van Rooijen, N.; Kettenmann, H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J. Neuropathol. Exp. Neurol. 2005, 64, 754–762. [Google Scholar] [CrossRef] [Green Version]
- Watters, J.J.; Schartner, J.M.; Badie, B. Microglia function in brain tumors. J. Neurosci. Res. 2005, 81, 447–455. [Google Scholar] [CrossRef]
- Markovic, D.S.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehmann, S.; Kälin, R.; van Rooijen, N.; et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535. [Google Scholar] [CrossRef] [Green Version]
- Fantin, A.; Vieira, J.M.; Gestri, G.; Denti, L.; Schwarz, Q.; Prykhozhij, S.; Peri, F.; Wilson, S.W.; Ruhrberg, C. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 2010, 116, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Coffelt, S.; Hughes, R.; Lewis, C.E. Tumor-associated macrophages: Effectors of angiogenesis and tumor progression. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2009, 1796, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandenburg, S.; Muller, A.; Turkowski, K.; Radev, Y.T.; Rot, S.; Schmidt, C.; Bungert, A.D.; Acker, G.; Schorr, A.; Hippe, A.; et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 2016, 131, 365–378. [Google Scholar] [CrossRef]
- Kälin, R.E.; Cai, L.; Li, Y.; Zhao, D.; Zhang, H.; Cheng, J.; Zhang, W.; Wu, Y.; Eisenhut, K.; Janssen, P.; et al. TAMEP are brain tumor parenchymal cells controlling neoplastic angiogenesis and progression. Cell Syst. 2021, 12, 248–262.e7. [Google Scholar] [CrossRef] [PubMed]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When immune cells turn bad—Tumor-associated microglia/macrophages in glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [Green Version]
- Kerber, M.; Reiss, Y.; Wickersheim, A.; Jugold, M.; Kiessling, F.; Heil, M.; Tchaikovski, V.; Waltenberger, J.; Shibuya, M.; Plate, K.H.; et al. Flt-1 signaling in macrophages promotes glioma growth in vivo. Cancer Res. 2008, 68, 7342–7351. [Google Scholar] [CrossRef] [Green Version]
- Antunes, A.R.P.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H.; et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef]
- Zhao, D.; Zhang, H.; Uyar, R.; Hossain, J.A.; Miletic, H.; Tonn, J.-C.; Glass, R.; Kälin, R.E. Comparing tumor cell invasion and myeloid cell composition in compatible primary and relapsing glioblastoma. Cancers 2021, 13, 3636. [Google Scholar] [CrossRef]
- Eskilsson, E.; Rosland, G.V.; Talasila, K.M.; Knappskog, S.; Keunen, O.; Sottoriva, A.; Foerster, S.; Solecki, G.; Taxt, T.; Jirik, R.; et al. EGFRvIII mutations can emerge as late and heterogenous events in glioblastoma development and promote angiogenesis through Src activation. Neuro-Oncol. 2016, 18, 1644–1655. [Google Scholar] [CrossRef] [Green Version]
- Sandmann, T.; Bourgon, R.; Garcia, J.; Li, C.; Cloughesy, T.F.; Chinot, O.L.; Wick, W.; Nishikawa, R.; Mason, W.P.; Henriksson, R.; et al. Patients with proneural glioblastoma may derive overall survival benefit from the addition of bevacizumab to first-line radiotherapy and temozolomide: Retrospective analysis of the AVAglio trial. J. Clin. Oncol. 2015, 33, 2735–2744. [Google Scholar] [CrossRef]
- Leeper, N.J.; Tedesco, M.M.; Kojima, Y.; Schultz, G.M.; Kundu, R.K.; Ashley, E.A.; Tsao, P.S.; Dalman, R.L.; Quertermous, T. Apelin prevents aortic aneurysm formation by inhibiting macrophage inflammation. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1329–H1335. [Google Scholar] [CrossRef] [Green Version]
- Sawane, M.; Kidoya, H.; Muramatsu, F.; Takakura, N.; Kajiya, K. Apelin attenuates UVB-induced edema and inflammation by promoting vessel function. Am. J. Pathol. 2011, 179, 2691–2697. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Ishida, J.; Yamamoto, R.; Fujiwara, K.; Asada, S.; Kasuya, Y.; Mochizuki, N.; Fukamizu, A. G protein-coupled APJ receptor signaling induces focal adhesion formation and cell motility. Int. J. Mol. Med. 2005, 16, 787–792. [Google Scholar] [CrossRef]
- Choe, H.; Farzan, M.; Konkel, M.; Martin, K.; Sun, Y.; Marcon, L.; Cayabyab, M.; Berman, M.; Dorf, M.E.; Gerard, N.; et al. The orphan seven-transmembrane receptor apj supports the entry of primary T-cell-line-tropic and dualtropic human immunodeficiency virus type 1. J. Virol. 1998, 72, 6113–6118. [Google Scholar] [CrossRef] [Green Version]
- Edinger, A.L.; Hoffman, T.L.; Sharron, M.; Lee, B.; Yi, Y.; Choe, W.; Kolson, D.L.; Mitrovic, B.; Zhou, Y.; Faulds, D.; et al. An orphan seven-transmembrane domain receptor expressed widely in the brain functions as a coreceptor for human immunodeficiency virus type 1 and simian immunodeficiency virus. J. Virol. 1998, 72, 7934–7940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of essential genes for cancer immunotherapy. Nat. Cell Biol. 2017, 548, 537–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnell, O.; Tonn, J.C. Treatment of edema formation in oncology. In Brain Edema: From Molecular Mechanisms to Clinical Practice; Badaut, J., Plesnila, N., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 477–495. [Google Scholar]
- Galicich, J.H.; French, L.A.; Melby, J.C. Use of dexamethasone in treatment of cerebral edema associated with brain tumors. Lancet 1961, 81, 46–53. [Google Scholar]
- Vecht, C.J.; Hovestadt, A.; Verbiest, H.B.; van Vliet, J.J.; van Putten, W.L. Dose-effect relationship of dexamethasone on Karnofsky performance in metastatic brain tumors: A randomized study of doses of 4, 8, and 16 mg per day. Neurology 1994, 44, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Cenciarini, M.; Valentino, M.; Belia, S.; Sforna, L.; Rosa, P.; Ronchetti, S.; D’Adamo, M.C.; Pessia, M. Dexamethasone in glioblastoma multiforme therapy: Mechanisms and controversies. Front. Mol. Neurosci. 2019, 12, 65. [Google Scholar] [CrossRef]
- Hui, C.Y.; Rudra, S.; Ma, S.; Campian, J.L.; Huang, J. Impact of overall corticosteroid exposure during chemoradiotherapy on lymphopenia and survival of glioblastoma patients. J. Neuro-Oncol. 2019, 143, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Schmidt, C.; Roth, W.; Dichgans, J. Chemotherapy of human malignant glioma: Prevention of efficacy by dexamethasone? Neurology 1997, 48, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Pitter, K.L.; Tamagno, I.; Alikhanyan, K.; Hosni-Ahmed, A.; Pattwell, S.S.; Donnola, S.; Dai, C.; Ozawa, T.; Chang, M.; Chan, T.A.; et al. Corticosteroids compromise survival in glioblastoma. Brain 2016, 139, 1458–1471. [Google Scholar] [CrossRef] [Green Version]
- Obradovic, M.M.S.; Hamelin, B.; Manevski, N.; Couto, J.P.; Sethi, A.; Coissieux, M.-M.; Muenst, S.; Okamoto, R.; Kohler, H.; Schmidt, A.; et al. Glucocorticoids promote breast cancer metastasis. Nat. Cell Biol. 2019, 567, 540–544. [Google Scholar] [CrossRef]
- Chitadze, G.; Flüh, C.; Quabius, E.S.; Freitag-Wolf, S.; Peters, C.; Lettau, M.; Bhat, J.; Wesch, D.; Oberg, H.-H.; Luecke, S.; et al. In-depth immunophenotyping of patients with glioblastoma multiforme: Impact of steroid treatment. OncoImmunology 2017, 6, e1358839. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.T.; Lok, E.; Gautam, S.; Swanson, K.D. Dexamethasone exerts profound immunologic interference on treatment efficacy for recurrent glioblastoma. Br. J. Cancer 2015, 113, 1642. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nat. Cell Biol. 2018, 565, 234–239. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Trans. Med. 2017, 9, 399. [Google Scholar] [CrossRef] [Green Version]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; Lopez-Janeiro, A.; Porciuncula, A.; Idoate-Gastearena, M.; Inogés, S.; De Andrea, C.; Lopez-Diaz De Cerio, A.L.-D.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2020, 18, 170–186. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.V.; Bergers, G. Mechanisms of evasive resistance to anti-VEGF therapy in glioblastoma. CNS Oncol. 2013, 2, 49–65. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Annotation Cluster | Representative Annotation Term | Enrichment Score |
|---|---|---|
| 1 | immune (acute inflammatory) response (lymphocyte-mediated immunity) | 5.31 |
| 2 | keratinization/epidermal cell differentiation | 2.94 |
| 3 | Natural killer cell-mediated cytotoxicity (autophagy, Toll-like receptor/JakSTAT/interferon signaling) | 2.63 |
| 4 | lysosome/lytic vacuole | 2.23 |
| 5 | ATP binding | 2.19 |
| 6 | Helicase and RNase D C-terminal, HRDC | 2.19 |
| 7 | G1/S transition of mitotic cell cycle/interphase | 2.07 |
| 8 | BRCT | 2.06 |
| 9 | nuclear division/cell division/M phase of mitotic cell cycle | 1.93 |
| 10 | SH3 | 1.91 |
| 11 | ATP-dependent helicase activity | 1.88 |
| 12 | MHC class II protein complex/antigen processing and presentation | 1.78 |
| 13 | integral to plasma membrane | 1.73 |
| 14 | T cell selection and differentiation/leukocyte activation | 1.70 |
| 15 | endocytosis/membrane invagination | 1.67 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kälin, R.E.; Glass, R. APLN/APLNR Signaling Controls Key Pathological Parameters of Glioblastoma. Cancers 2021, 13, 3899. https://doi.org/10.3390/cancers13153899
Kälin RE, Glass R. APLN/APLNR Signaling Controls Key Pathological Parameters of Glioblastoma. Cancers. 2021; 13(15):3899. https://doi.org/10.3390/cancers13153899
Chicago/Turabian StyleKälin, Roland E., and Rainer Glass. 2021. "APLN/APLNR Signaling Controls Key Pathological Parameters of Glioblastoma" Cancers 13, no. 15: 3899. https://doi.org/10.3390/cancers13153899
APA StyleKälin, R. E., & Glass, R. (2021). APLN/APLNR Signaling Controls Key Pathological Parameters of Glioblastoma. Cancers, 13(15), 3899. https://doi.org/10.3390/cancers13153899