
Drug Repurposing to Identify a Synergistic High-Order Drug Combination to Treat Sunitinib-Resistant Renal Cell Carcinoma

 ,
,  ,
,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Drugs
2.3. Heterotypic 3D Co-Cultures
2.4. Cell Viability Assay
2.5. RNA Sequencing
2.6. Metacore Analysis
2.7. Analysis of the Lipid Classes
2.8. Flow Cytometry Analysis
2.9. Organoid Generation
3. Results
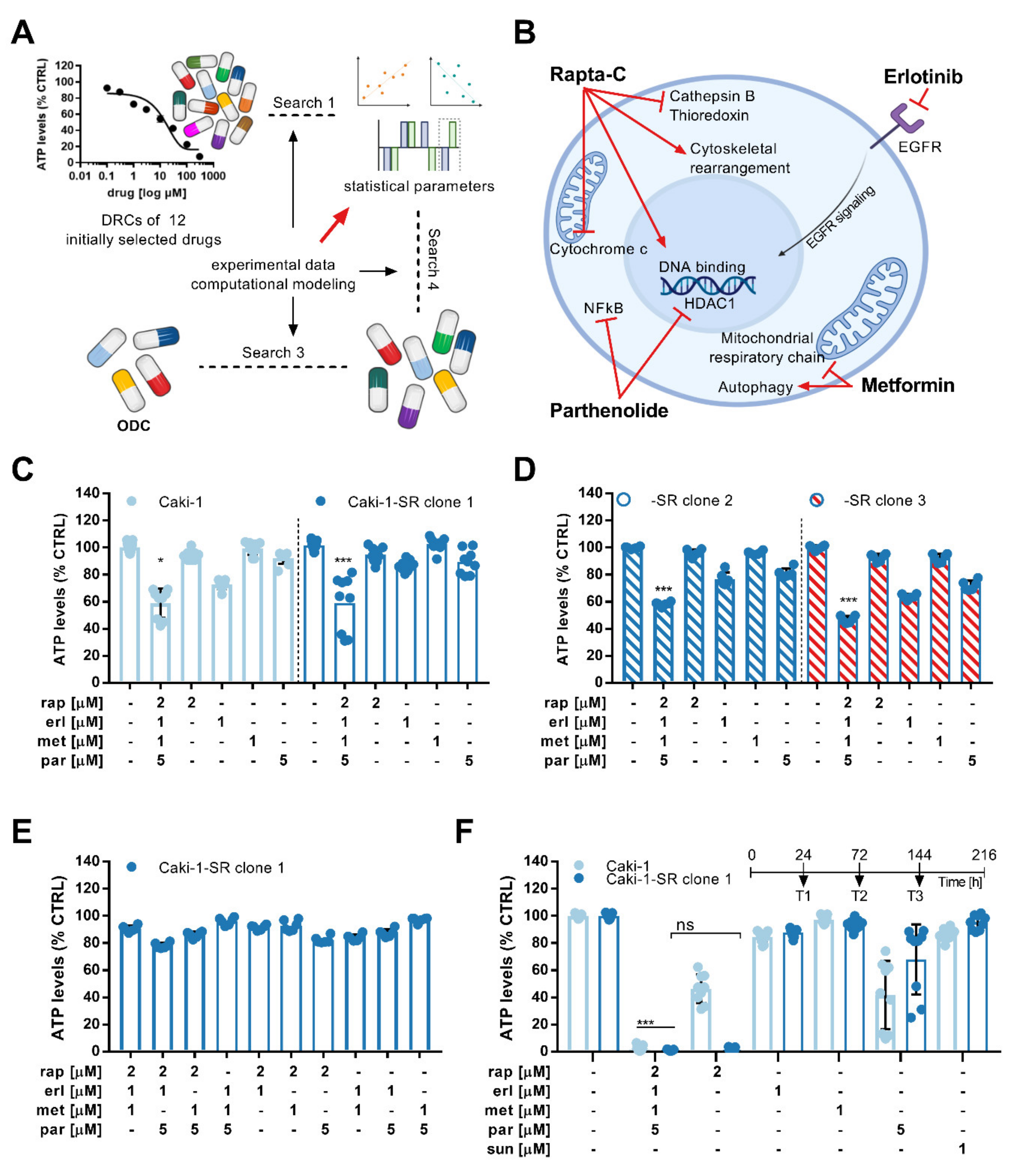
3.1. Phenotypic Synergy Screen Identifies Drug Hits for Combinatory Treatment in ccRCC Cell Lines
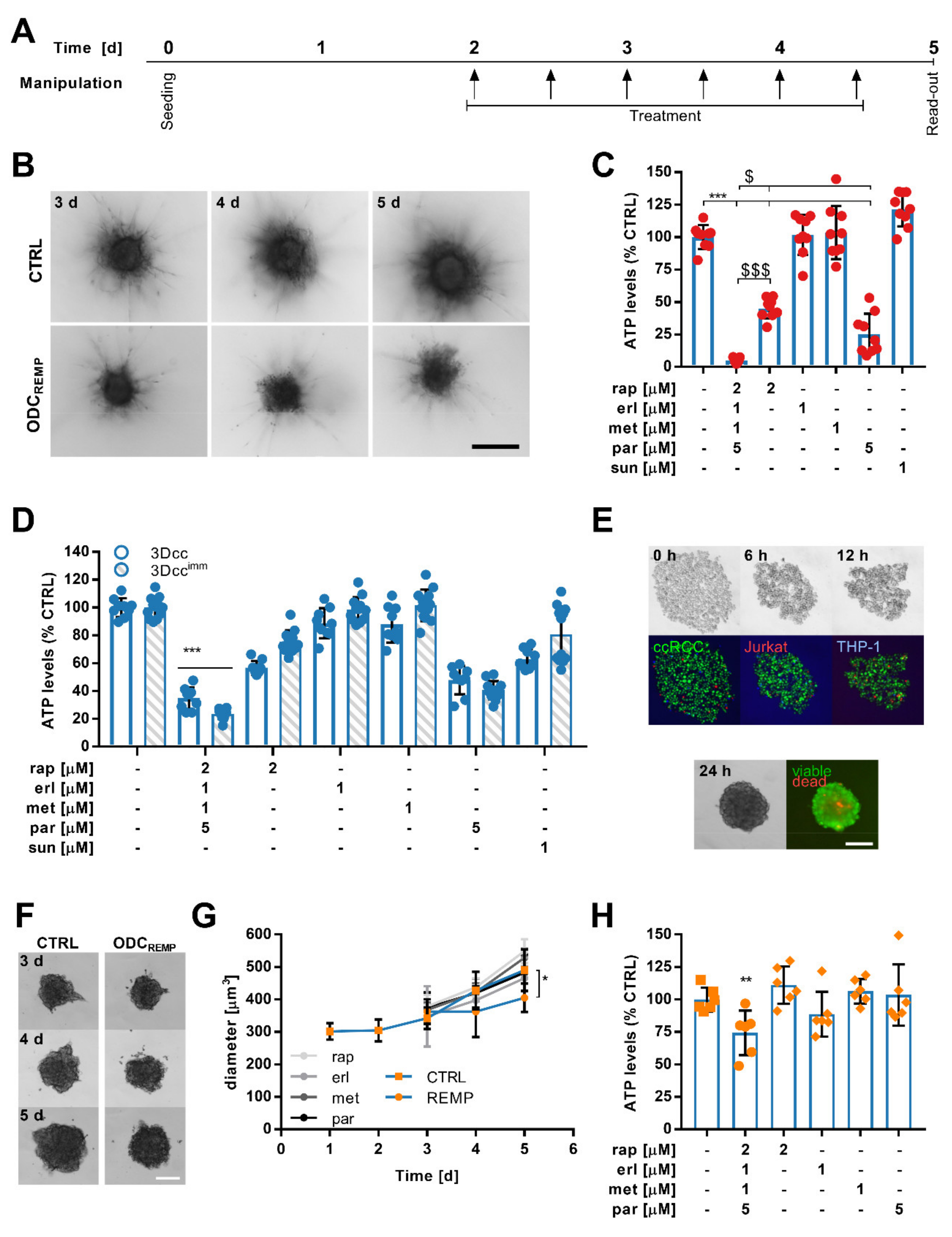
3.2. Activity Validation of the ODC
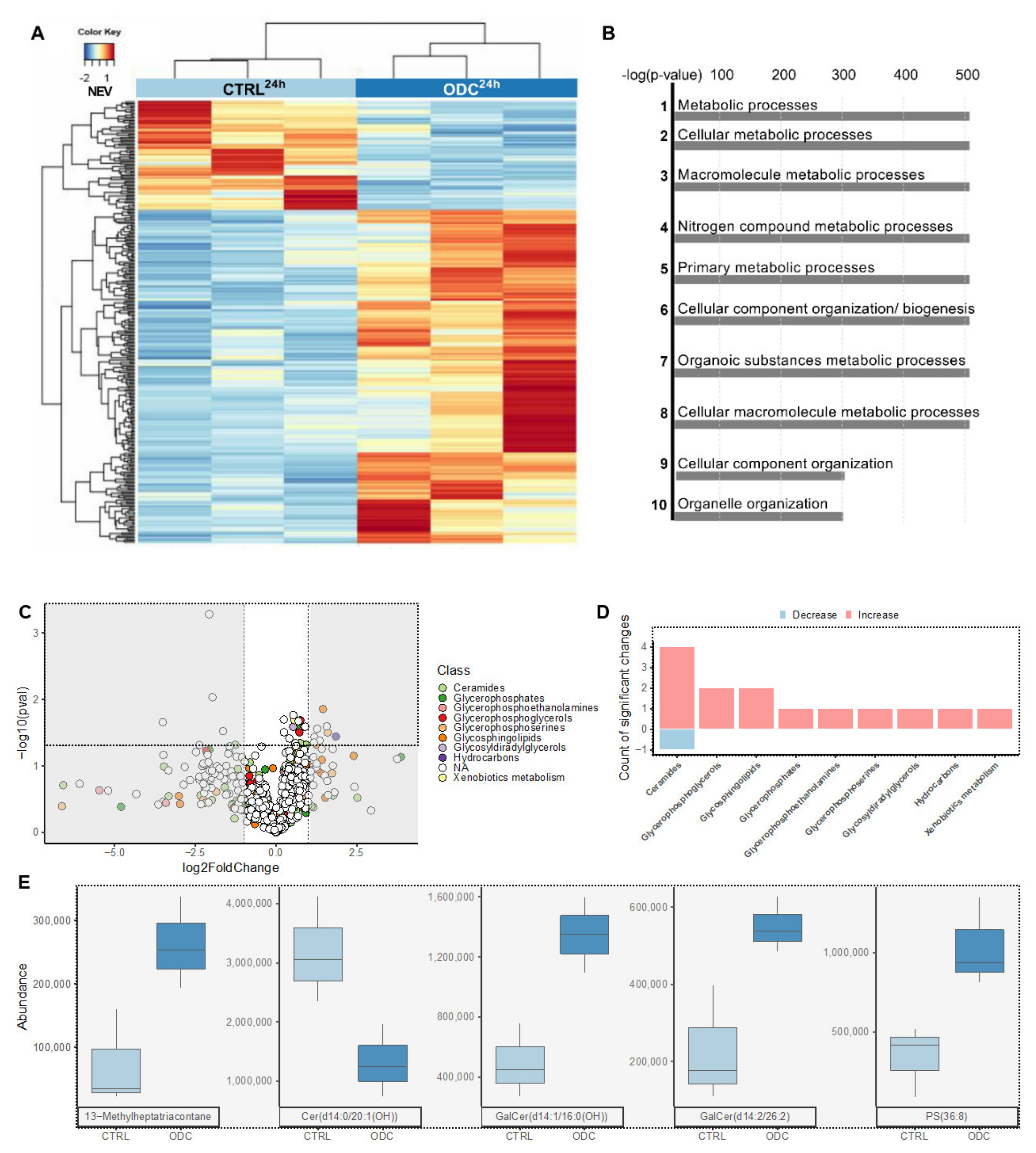
3.3. RNA Sequencing Reveals the Upregulation of Genes Related to Apoptosis, Cell Adherence and Metabolism upon ODCREMP Treatment
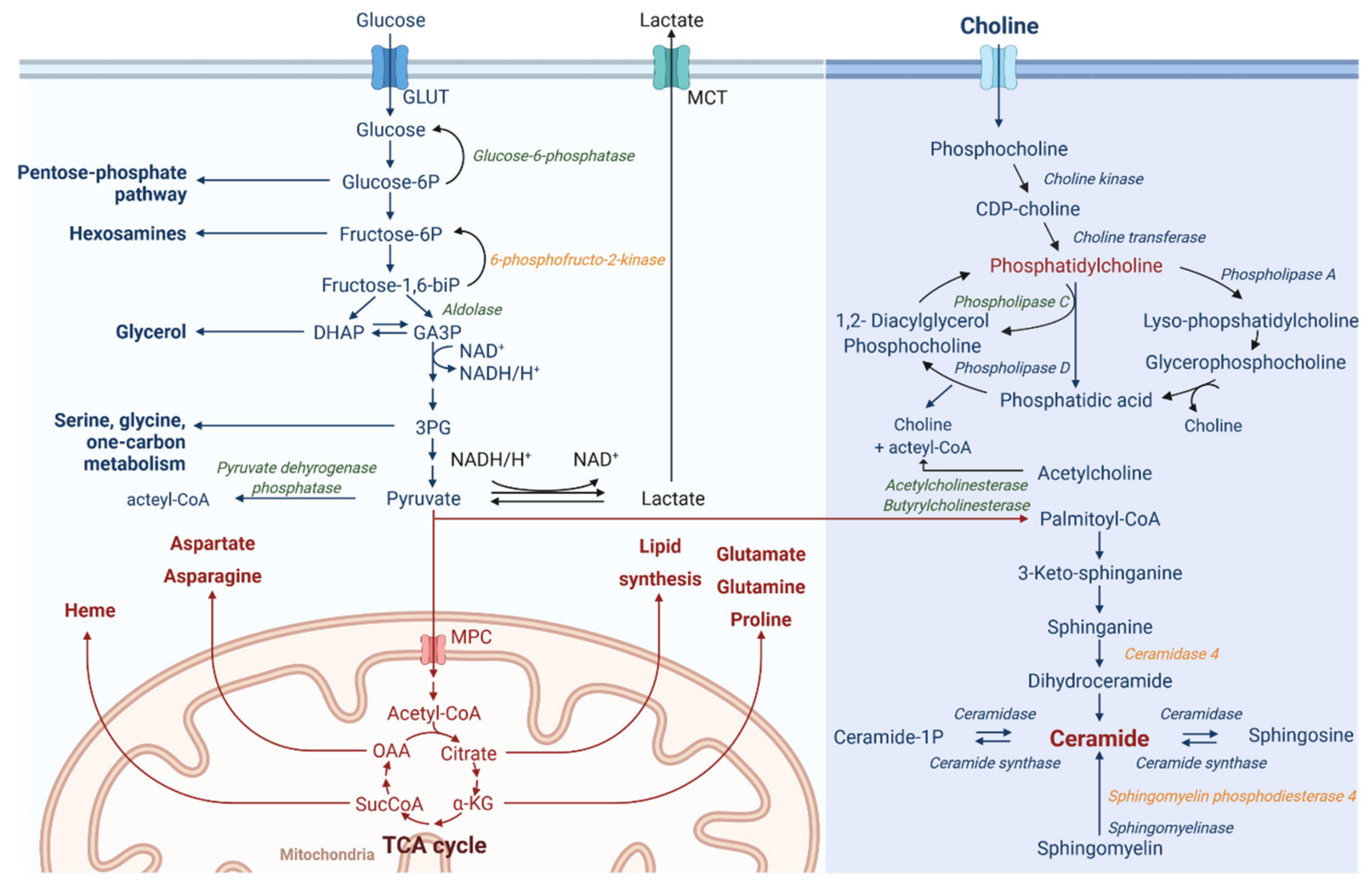
3.4. ODCREMP Treatment Has an Impact on the Metabolism of Ceramide and Glycerophospholipids in ccRCC Cells
3.5. ODCREMP Activity in Complex Heterotypic 3D Co-Cultures
3.6. Flow Cytometry Measurements Reveal Selective ODCREMP Targeting in Cancer Cells
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takebe, T.; Imai, R.; Ono, S. The Current Status of Drug Discovery and Development as Originated in United States Academia: The Influence of Industrial and Academic Collaboration on Drug Discovery and Development. Clin. Transl. Sci. 2018, 11, 597–606. [Google Scholar] [CrossRef]
- Talevi, A.; Bellera, C.L. Challenges and opportunities with drug repurposing: Finding strategies to find alternative uses of therapeutics. Expert Opin. Drug Discov. 2020, 15, 397–401. [Google Scholar] [CrossRef] [Green Version]
- Cha, Y.; Erez, T.; Reynolds, I.J.; Kumar, D.; Ross, J.; Koytiger, G.; Kusko, R.; Zeskind, B.; Risso, S.; Kagan, E.; et al. Drug repurposing from the perspective of pharmaceutical companies. Br. J. Pharm. 2018, 175, 168–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langedijk, J.; Mantel-Teeuwisse, A.K.; Slijkerman, D.S.; Schutjens, M.H. Drug repositioning and repurposing: Terminology and definitions in literature. Drug Discov. Today 2015, 20, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Armando, R.G.; Mengual Gómez, D.L.; Gomez, D.E. New drugs are not enough-drug repositioning in oncology: An update. Int. J. Oncol. 2020, 56, 651–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak-Sliwinska, P.; Scapozza, L.; Ruiz, I.; Altaba, A. Drug repurposing in oncology: Compounds, pathways, phenotypes and computational approaches for colorectal cancer. Biochim. Biophys. Acta. Rev. Cancer 2019, 1871, 434–454. [Google Scholar] [CrossRef] [PubMed]
- Sleire, L.; Forde, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.O. Drug repurposing in cancer. Pharm. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef]
- Tannir, N.M.; Schwab, G.; Grünwald, V. Cabozantinib: An Active Novel Multikinase Inhibitor in Renal Cell Carcinoma. Curr. Oncol. Rep. 2017, 19, 14. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.X.; Maher, V.E.; Zhang, L.; Tang, S.; Sridhara, R.; Ibrahim, A.; Kim, G.; Pazdur, R. FDA Approval Summary: Nivolumab in Advanced Renal Cell Carcinoma After Anti-Angiogenic Therapy and Exploratory Predictive Biomarker Analysis. Oncologist 2017, 22, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; McDermott, D.F. Ipilimumab in combination with nivolumab for the treatment of renal cell carcinoma. Expert Opin. Biol. 2018, 18, 947–957. [Google Scholar] [CrossRef]
- Caruso, C. Checkpoint Inhibitor-TKI Combos Effective in RCC. Cancer Discov. 2019, 9, 460. [Google Scholar] [CrossRef] [Green Version]
- Heckman-Stoddard, B.M.; DeCensi, A.; Sahasrabuddhe, V.V.; Ford, L.G. Repurposing metformin for the prevention of cancer and cancer recurrence. Diabetologia 2017, 60, 1639–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, J.J.; Pryszlak, M.; Smith, L.; Yanchus, C.; Kurji, N.; Shahani, V.M.; Molinski, S.V. Giving Drugs a Second Chance: Overcoming Regulatory and Financial Hurdles in Repurposing Approved Drugs As Cancer Therapeutics. Front. Oncol. 2017, 7, 273. [Google Scholar] [CrossRef] [Green Version]
- Pantziarka, P.; Bouche, G.; Meheus, L.; Sukhatme, V.; Sukhatme, V.P.; Vikas, P. The Repurposing Drugs in Oncology (ReDO) Project. Ecancermedicalscience 2014, 8, 442. [Google Scholar] [CrossRef]
- Verbaanderd, C.; Meheus, L.; Huys, I.; Pantziarka, P. Repurposing Drugs in Oncology: Next Steps. Trends. Cancer 2017, 3, 543–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerbini, L.F.; Bhasin, M.K.; de Vasconcellos, J.F.; Paccez, J.D.; Gu, X.; Kung, A.L.; Libermann, T.A. Computational repositioning and preclinical validation of pentamidine for renal cell cancer. Mol. Cancer 2014, 13, 1929–1941. [Google Scholar] [CrossRef] [Green Version]
- Jeong, D.E.; Song, H.J.J.; Lim, S.; Lee, S.J.J.; Lim, J.E.; Nam, D.-H.; Joo, K.M.; Jeong, B.C.; Jeon, S.S.; Choi, H.Y.; et al. Repurposing the anti-malarial drug artesunate as a novel therapeutic agent for metastatic renal cell carcinoma due to its attenuation of tumor growth, metastasis, and angiogenesis. Oncotarget 2015, 6, 33046–33064. [Google Scholar] [CrossRef] [PubMed]
- Koudijs, K.K.M.; Terwisscha van Scheltinga, A.G.T.; Böhringer, S.; Schimmel, K.J.M.; Guchelaar, H.-J. The impact of estimated tumour purity on gene expression-based drug repositioning of Clear Cell Renal Cell Carcinoma samples. Sci. Rep. 2019, 9, 2495. [Google Scholar] [CrossRef]
- Koudijs, K.K.M.; Terwisscha van Scheltinga, A.G.T.; Böhringer, S.; Schimmel, K.J.M.; Guchelaar, H.-J. Personalised drug repositioning for Clear Cell Renal Cell Carcinoma using gene expression. Sci. Rep. 2018, 8, 5250. [Google Scholar] [CrossRef] [Green Version]
- Czarnecka, A.M.; Niedzwiedzka, M.; Porta, C.; Szczylik, C. Hormone signaling pathways as treatment targets in renal cell cancer (Review). Int. J. Oncol. 2016, 48, 2221–2235. [Google Scholar] [CrossRef] [Green Version]
- Song, A.; Zhang, C.; Meng, X. Mechanism and application of metformin in kidney diseases: An update. Biomed. Pharmacother. 2021, 138, 111454. [Google Scholar] [CrossRef] [PubMed]
- Pasha, M.; Sivaraman, S.K.; Frantz, R.; Agouni, A.; Munusamy, S. Metformin Induces Different Responses in Clear Cell Renal Cell Carcinoma Caki Cell Lines. Biomolecules 2019, 9, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Zhong, X.; Gao, P.; Shi, J.; Wu, Z.; Guo, Z.; Wang, Z.; Song, Y. The Potential Effect of Metformin on Cancer: An Umbrella Review. Front. Endocrinol. 2019, 10, 617. [Google Scholar] [CrossRef]
- Nowak-Sliwinska, P.; Weiss, A.; Ding, X.; Dyson, P.J.; van den Bergh, H.; Griffioen, A.W.; Ho, C.M. Optimization of drug combinations using Feedback System Control. Nat. Protoc. 2016, 11, 302–315. [Google Scholar] [CrossRef]
- Weiss, A.; Berndsen, R.H.; Ding, X.; Ho, C.M.; Dyson, P.J.; van den Bergh, H.; Griffioen, A.W.; Nowak-Sliwinska, P. A streamlined search technology for identification of synergistic drug combinations. Sci. Rep. 2015, 5, 14508. [Google Scholar] [CrossRef] [PubMed]
- Lehar, J.; Krueger, A.S.; Zimmermann, G.R.; Borisy, A.A. Therapeutic selectivity and the multi-node drug target. Discov. Med. 2009, 8, 185–190. [Google Scholar]
- Kummar, S.; Chen, H.X.; Wright, J.; Holbeck, S.; Millin, M.D.; Tomaszewski, J.; Zweibel, J.; Collins, J.; Doroshow, J.H. Utilizing targeted cancer therapeutic agents in combination: Novel approaches and urgent requirements. Nat. Rev. Drug Discov. 2010, 9, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Fernandes Neto, J.M.; Nadal, E.; Ooft, S.N.; Bosdriesz, E.; Farre, L.; McLean, C.; Klarenbeek, S.; Jurgens, A.; Hagen, H.; Wang, L.; et al. Multiple Low Dose (MLD) therapy: An effective strategy to treat EGFR inhibitor-resistant NSCLC tumours. bioRxiv 2019, 821975. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Aren Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthelemy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of Drug Repositioning Approaches and Resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef] [Green Version]
- Setoain, J.; Franch, M.; Martinez, M.; Tabas-Madrid, D.; Sorzano, C.O.; Bakker, A.; Gonzalez-Couto, E.; Elvira, J.; Pascual-Montano, A. NFFinder: An online bioinformatics tool for searching similar transcriptomics experiments in the context of drug repositioning. Nucleic Acids Res. 2015, 43, W193–W199. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Kundu, S.; Bhattacharyya, A.; Hartinger, C.G.; Dyson, P.J. The ruthenium(II)-arene compound RAPTA-C induces apoptosis in EAC cells through mitochondrial and p53-JNK pathways. J. Biol. Inorg. Chem. 2008, 13, 1149–1155. [Google Scholar] [CrossRef]
- Scolaro, C.; Hartinger, C.G.; Allardyce, C.S.; Keppler, B.K.; Dyson, P.J. Hydrolysis study of the bifunctional antitumour compound RAPTA-C, [Ru(eta6-p-cymene)Cl2(pta)]. J. Inorg. Biochem. 2008, 102, 1743–1748. [Google Scholar] [CrossRef]
- Berndsen, R.H.; Weiss, A.; Abdul, U.K.; Wong, T.J.; Meraldi, P.; Griffioen, A.W.; Dyson, P.J.; Nowak-Sliwinska, P. Combination of ruthenium(II)-arene complex [Ru(eta(6)-p-cymene)Cl2(pta)] (RAPTA-C) and the epidermal growth factor receptor inhibitor erlotinib results in efficient angiostatic and antitumor activity. Sci. Rep. 2017, 7, 43005. [Google Scholar] [CrossRef]
- Kilpin, K.J.; Cammack, S.M.; Clavel, C.M.; Dyson, P.J. Ruthenium(II) arene PTA (RAPTA) complexes: Impact of enantiomerically pure chiral ligands. Dalton. Trans. 2013, 42, 2008–2014. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Sliwinska, P.; van Beijnum, J.R.; Casini, A.; Nazarov, A.A.; Wagnieres, G.; van den Bergh, H.; Dyson, P.J.; Griffioen, A.W. Organometallic ruthenium(II) arene compounds with antiangiogenic activity. J. Med. Chem. 2011, 54, 3895–3902. [Google Scholar] [CrossRef]
- Rausch, M.; Dyson, P.J.; Nowak-Sliwinska, P. Recent Considerations in the Application of RAPTA-C for Cancer Treatment and Perspectives for Its Combination with Immunotherapies. Adv. Ther. 2019, 2, 1900042. [Google Scholar] [CrossRef]
- Riedel, T.; Demaria, O.; Zava, O.; Joncic, A.; Gilliet, M.; Dyson, P.J. Drug Repurposing Approach Identifies a Synergistic Drug Combination of an Antifungal Agent and an Experimental Organometallic Drug for Melanoma Treatment. Mol. Pharm. 2018, 15, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Rausch, M.; Weiss, A.; Achkhanian, J.; Rotari, A.; Nowak-Sliwinska, P. Identification of low-dose multidrug combinations for sunitinib-naive and pre-treated renal cell carcinoma. Br. J. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Rausch, M.; Weiss, A.; Zoetemelk, M.; Piersma, S.R.; Jimenez, C.R.; van Beijnum, J.R.; Nowak-Sliwinska, P. Optimized Combination of HDACI and TKI Efficiently Inhibits Metabolic Activity in Renal Cell Carcinoma and Overcomes Sunitinib Resistance. Cancers 2020, 12, 3172. [Google Scholar] [CrossRef]
- Zoetemelk, M.; Ramzy, G.M.; Rausch, M.; Koessler, T.; van Beijnum, J.R.; Weiss, A.; Mieville, V.; Piersma, S.R.; de Haas, R.R.; Delucinge-Vivier, C.; et al. Optimized low-dose combinatorial drug treatment boosts selectivity and efficacy of colorectal carcinoma treatment. Mol. Oncol. 2020. [Google Scholar] [CrossRef]
- Allardyce, C.S.; Dyson, P.J.; Ellis, D.J.; Heath, S.L. [Ru(η--cymene)Cl(pta)] (pta = 1,3,5-triaza-7-phosphatricyclo- [3.3.1.1]decane): A water soluble compound that exhibits pH dependent DNA binding providing selectivity for diseased cells. Chem. Commun. 2001, 15, 1396–1397. [Google Scholar] [CrossRef]
- Rausch, M.; Blanc, L.; Silva, O.D.S.; Dormond, O.; Griffioen, A.W.; Nowak-Sliwinska, P. Characterization of Renal Cell Carcinoma Heterotypic 3D Co-Cultures with Immune Cell Subsets. Cancers 2021, 13, 2551. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enrichr. Available online: http://amp.pharm.mssm.edu/Enrichr (accessed on 12 January 2020).
- Gene Expression Omnibus. Available online: www.ncbi.nlm.nih.gov/geo (accessed on 12 January 2020).
- Clarivate. Available online: https://clarivate.com/ (accessed on 4 August 2021).
- Rausch, M.; Rutz, A.; Allard, P.M.; Delucinge-Vivier, C.; Docquier, M.; Dormond, O.; Wolfender, J.L.; Nowak-Sliwinska, P. Molecular and functional analysis of sunitinib resistance induction in human renal cell carcinoma cells. Int. J. Mol. Sci. 2021, 22, 6467. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [Green Version]
- Kondo, J.; Ekawa, T.; Endo, H.; Yamazaki, K.; Tanaka, N.; Kukita, Y.; Okuyama, H.; Okami, J.; Imamura, F.; Ohue, M.; et al. High-throughput screening in colorectal cancer tissue-originated spheroids. Cancer Sci. 2019, 110, 345–355. [Google Scholar] [CrossRef]
- Zeng, L.; Gupta, P.; Chen, Y.; Wang, E.; Ji, L.; Chao, H.; Chen, Z.-S. The development of anticancer ruthenium(ii) complexes: From single molecule compounds to nanomaterials. Chem. Soc. Rev. 2017, 46, 5771–5804. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, C.Y.; Nam, T.-G. Ruthenium Complexes as Anticancer Agents: A Brief History and Perspectives. Drug Des. Dev. Ther. 2020, 14, 5375–5392. [Google Scholar] [CrossRef]
- Babak, M.V.; Meier, S.M.; Huber, K.V.M.; Reynisson, J.; Legin, A.A.; Jakupec, M.A.; Roller, A.; Stukalov, A.; Gridling, M.; Bennett, K.L.; et al. Target profiling of an antimetastatic RAPTA agent by chemical proteomics: Relevance to the mode of action. Chem. Sci. 2015, 6, 2449–2456. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.L.; Wang, X.; Xing, J.S.; Yu, Z.L.; Lou, J.C.; Ma, X.C.; Zhang, B. Anti-cancer effects of dopamine in human glioma: Involvement of mitochondrial apoptotic and anti-inflammatory pathways. Oncotarget 2017, 8, 88488–88500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Liu, Q.; Liao, Q.; Zhao, Y. Potential Roles of Peripheral Dopamine in Tumor Immunity. J. Cancer 2017, 8, 2966–2973. [Google Scholar] [CrossRef] [Green Version]
- Pantziarka, P.; Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)-diclofenac as an anti-cancer agent. Ecancermedicalscience 2016, 10, 610. [Google Scholar] [CrossRef] [PubMed]
- Breuer, S.; Maimon, O.; Appelbaum, L.; Peretz, T.; Hubert, A. TL-118-anti-angiogenic treatment in pancreatic cancer: A case report. Med. Oncol. 2013, 30, 585. [Google Scholar] [CrossRef]
- Ding, N.; Zhu, Q. Disulfiram combats cancer via crippling valosin-containing protein/p97 segregase adaptor NPL4. Transl. Cancer Res. 2018, 7, S495–S499. [Google Scholar] [CrossRef]
- Parshad, R.; Kapoor, S.; Gupta, S.D.; Kumar, A.; Chattopadhyaya, T.K. Does famotidine enhance tumor infiltrating lymphocytes in breast cancer? Results of a randomized prospective pilot study. Acta. Oncol. 2002, 41, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Nambara, S.; Masuda, T.; Nishio, M.; Kuramitsu, S.; Tobo, T.; Ogawa, Y.; Hu, Q.; Iguchi, T.; Kuroda, Y.; Ito, S.; et al. Antitumor effects of the antiparasitic agent ivermectin via inhibition of Yes-associated protein 1 expression in gastric cancer. Oncotarget 2017, 8, 107666–107677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zi, F.; Zi, H.; Li, Y.; He, J.; Shi, Q.; Cai, Z. Metformin and cancer: An existing drug for cancer prevention and therapy. Oncol. Lett. 2018, 15, 683–690. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.C.; Kim, S.L.; Park, Y.R.; Lee, S.-T.; Kim, S.W. Parthenolide promotes apoptotic cell death and inhibits the migration and invasion of SW620 cells. Intest. Res. 2017, 15, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Ghantous, A.; Sinjab, A.; Herceg, Z.; Darwiche, N. Parthenolide: From plant shoots to cancer roots. Drug Discov. Today 2013, 18, 894–905. [Google Scholar] [CrossRef]
- Zhao, L.; Zhao, Y.; Schwarz, B.; Mysliwietz, J.; Hartig, R.; Camaj, P.; Bao, Q.; Jauch, K.W.; Guba, M.; Ellwart, J.W.; et al. Verapamil inhibits tumor progression of chemotherapy-resistant pancreatic cancer side population cells. Int. J. Oncol. 2016, 49, 99–110. [Google Scholar] [CrossRef] [Green Version]
- Elwood, P.C.; Morgan, G.; Pickering, J.E.; Galante, J.; Weightman, A.L.; Morris, D.; Kelson, M.; Dolwani, S. Aspirin in the Treatment of Cancer: Reductions in Metastatic Spread and in Mortality: A Systematic Review and Meta-Analyses of Published Studies. PLoS ONE 2016, 11, e0152402. [Google Scholar] [CrossRef] [Green Version]
- Cavalla, D. Using human experience to identify drug repurposing opportunities: Theory and practice. Br. J. Clin. Pharmacol. 2019, 85, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug. Discov. 2018. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Le Roux-Bourdieu, M.; Zoetemelk, M.; Ramzy, G.M.; Rausch, M.; Harry, D.; Miljkovic-Licina, M.; Falamaki, K.; Wehrle-Haller, B.; Meraldi, P.; et al. Identification of a Synergistic Multi-Drug Combination Active in Cancer Cells via the Prevention of Spindle Pole Clustering. Cancers 2019, 11, 1612. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic. Acids. Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [Green Version]
- Artner, C.; Holtkamp, H.U.; Hartinger, C.G.; Meier-Menches, S.M. Characterizing activation mechanisms and binding preferences of ruthenium metallo-prodrugs by a competitive binding assay. J. Inorg. Biochem. 2017, 177, 322–327. [Google Scholar] [CrossRef]
- Holtkamp, H.U.; Movassaghi, S.; Morrow, S.J.; Kubanik, M.; Hartinger, C.G. Metallomic study on the metabolism of RAPTA-C and cisplatin in cell culture medium and its impact on cell accumulation. Metallomics 2018, 10, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Fahy, E.; Subramaniam, S. RefMet: A reference nomenclature for metabolomics. Nat. Methods 2020, 17, 1173–1174. [Google Scholar] [CrossRef]
- Pradas, I.; Rovira-Llopis, S.; Naudí, A.; Bañuls, C.; Rocha, M.; Hernandez-Mijares, A.; Pamplona, R.; Victor, V.M.; Jové, M. Metformin induces lipid changes on sphingolipid species and oxidized lipids in polycystic ovary syndrome women. Sci. Rep. 2019, 9, 16033. [Google Scholar] [CrossRef]
- Van Stee, M.F.; de Graaf, A.A.; Groen, A.K. Actions of metformin and statins on lipid and glucose metabolism and possible benefit of combination therapy. Cardiovasc. Diabetol. 2018, 17, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.Y.; Kang, B.; Hong, J.; Choi, H.-S. Parthenolide inhibits lipid accumulation via activation of Nrf2/Keap1 signaling during adipocyte differentiation. Food Sci. Biotechnol. 2019, 29, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Posocco, B.; Buzzo, M.; Giodini, L.; Crotti, S.; D’Aronco, S.; Traldi, P.; Agostini, M.; Marangon, E.; Toffoli, G. Analytical aspects of sunitinib and its geometric isomerism towards therapeutic drug monitoring in clinical routine. J. Pharm. Biomed. Anal. 2018, 160, 360–367. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends. Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Huinen, Z.R.; Huijbers, E.J.M.; van Beijnum, J.R.; Nowak-Sliwinska, P.; Griffioen, A.W. Anti-angiogenic agents—overcoming tumour endothelial cell anergy and improving immunotherapy outcomes. Nat. Rev. Clin. Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Suissa, S.; Azoulay, L. Metformin and Cancer: Mounting Evidence Against an Association. Diabetes Care 2014, 37, 1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malek, M.; Aghili, R.; Emami, Z.; Khamseh, M.E. Risk of Cancer in Diabetes: The Effect of Metformin. ISRN Endocrinol. 2013, 2013, 636927. [Google Scholar] [CrossRef]
- Ugwueze, C.V.; Ogamba, O.J.; Young, E.E.; Onyenekwe, B.M.; Ezeokpo, B.C. Metformin: A Possible Option in Cancer Chemotherapy. Anal. Cell. Pathol. 2020, 2020, 7180923. [Google Scholar] [CrossRef]
- Richard, S.M.; Martinez Marignac, V.L. Sensitization to oxaliplatin in HCT116 and HT29 cell lines by metformin and ribavirin and differences in response to mitochondrial glutaminase inhibition. J. Cancer Res. 2015, 11, 336–340. [Google Scholar] [CrossRef]
- Sun, L.; Yuan, W.; Wen, G.; Yu, B.; Xu, F.; Gan, X.; Tang, J.; Zeng, Q.; Zhu, L.; Chen, C.; et al. Parthenolide inhibits human lung cancer cell growth by modulating the IGF-1R/PI3K/Akt signaling pathway. Oncol. Rep. 2020, 44, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Ding, X.; van Beijnum, J.R.; Wong, I.; Wong, T.J.; Berndsen, R.H.; Dormond, O.; Dallinga, M.; Shen, L.; Schlingemann, R.O.; et al. Rapid optimization of drug combinations for the optimal angiostatic treatment of cancer. Angiogenesis 2015, 18, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Ratanaphan, A.; Temboot, P.; Dyson, P.J. In vitro ruthenation of human breast cancer suppressor gene 1 (BRCA1) by the antimetastasis compound RAPTA-C and its analogue CarboRAPTA-C. Chem. Biodivers. 2010, 7, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Jafari, N.; Nazeri, S.; Enferadi, S.T. Parthenolide reduces metastasis by inhibition of vimentin expression and induces apoptosis by suppression elongation factor α − 1 expression. Phytomedicine 2018, 41, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Makhov, P.; Naito, S.; Haifler, M.; Kutikov, A.; Boumber, Y.; Uzzo, R.G.; Kolenko, V.M. The convergent roles of NF-κB and ER stress in sunitinib-mediated expression of pro-tumorigenic cytokines and refractory phenotype in renal cell carcinoma. Cell Death Dis. 2018, 9, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Wang, Z.; Zhang, D.; Wang, J. Metabonomic study of the intervention effects of Parthenolide on anti-thyroid cancer activity. J. Chromatogr. B 2020, 1150, 122179. [Google Scholar] [CrossRef] [PubMed]
- Meacham, W.D.; Antoon, J.W.; Burow, M.E.; Struckhoff, A.P.; Beckman, B.S. Sphingolipids as Determinants of Apoptosis and Chemoresistance in the MCF-7 Cell Model System. Exp. Biol. Med. 2009, 234, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.C.; Wallington-Beddoe, C.T.; Powell, J.A.; Pitson, S.M. Targeting sphingolipid metabolism as an approach for combination therapies in haematological malignancies. Cell Death Discov. 2018, 4, 72. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Pharmaceutical Class | Indication | Targets | Clinical Stage | Potential Use in Cancer |
|---|---|---|---|---|---|
| Rapta-C | Chemotherapy; Anti-cancer [56]; Anti-angiogenic | - | p53-JNK pathway [33], protein; histones in DNA | preclinical | - |
| 3-Hydroxy-tyraminium chloride | Anti-hypertension | Hemodynamic imbalance | System-wideagonist of dopamine type 1–5 receptors | approved | Glioma [57], colorectal cancer, tumor immunity ** [58] |
| Diclofenac sodium | Analgesics | Pain | Inhibition of prostaglandin synthesis by inhibition of the transiently expressed prostaglandin-endo-peroxide synthase-2 (PGES-2), also known as Cycloxygenase-2 (COX-2) | approved | Colorectal cancer, neuroblastoma, post-operative [59],pancreatic cancer ** [60] |
| Disulfiram | Acetaldehyde dehydrogenase inhibitor; Proteasome inhibitor | Alcoholism | Inhibits enzyme acetaldehyde dehydrogenase | approved | Any cancer type [61] |
| Erlotinib·HCl | Anti-cancer agent | Non-small cell lung cancer | Inhibition of the epidermal growth factor receptor | approved | |
| Famotidine | Anti-ulcer agent | Gastro-esophageal reflux disease | H2 Blocker and Histamine Receptor Antagonist | approved | Pre-operative, tumor immunity ** [62] |
| Haloprogin | Anti-infective agent; Anti-fungal agent | Skin infections (athlete’s foot, jock itch, ringworm, and tinea versicolor (a fungus) | GPCRs,dopamine and adrenergic receptors | discont.(side effects and available new drugs) | Melanoma [39],lung and pancreatic |
| Ivermectin | Anti-infective agent | Parasite infections | Glutamate-gated chloride channels central nervous system depression, and consequent ataxia, potentiation of inhibitory GABA-ergic synapses | approved | Cancer stem-like cells (breast), gastric cancer [63] |
| Metformin·HCl | Hypoglycemic agent | Diabetes type II | Decrease hepatic glucose production, mostly through a mild and transient inhibition of the mitochondrial respiratory chain complex I, the resulting decrease in hepatic energy status activates AMPK (AMP-activated protein kinase), a cellular metabolic sensor, providing a generally accepted mechanism for the action of metformin on hepatic gluconeogenesis | approved | Breast cancer stem cells [64], pancreatic cancer **, head and neck squamous cancer ** |
| Parthenolide | Analgesics; Anti-inflammatory agent | HDAC1 inhibition; modulation of NF-kB-mediated inflammation | - | Under clinical investigation | (Breast) cancer stem(-like) cells, colorectal cancer [65,66] |
| Verapamil·HCl | Anti-hypertension | high blood pressure, chest pain from not enough blood flow to the heart muscle | Efflux pump inhibitor; voltage-dependent calcium channels | approved | Pancreatic tumor side population (stem-like) cells [67] |
| Acetylsalicylic acid | Analgesics; | Pain, fever | COX1 and COX2 | approved | Gastrointestinal tract and lungs **, colorectal cancer ** [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rausch, M.; Rutz, A.; Allard, P.-M.; Delucinge-Vivier, C.; Docquier, M.; Dormond, O.; Dyson, P.J.; Wolfender, J.-L.; Nowak-Sliwinska, P. Drug Repurposing to Identify a Synergistic High-Order Drug Combination to Treat Sunitinib-Resistant Renal Cell Carcinoma. Cancers 2021, 13, 3978. https://doi.org/10.3390/cancers13163978
Rausch M, Rutz A, Allard P-M, Delucinge-Vivier C, Docquier M, Dormond O, Dyson PJ, Wolfender J-L, Nowak-Sliwinska P. Drug Repurposing to Identify a Synergistic High-Order Drug Combination to Treat Sunitinib-Resistant Renal Cell Carcinoma. Cancers. 2021; 13(16):3978. https://doi.org/10.3390/cancers13163978
Chicago/Turabian StyleRausch, Magdalena, Adriano Rutz, Pierre-Marie Allard, Céline Delucinge-Vivier, Mylène Docquier, Olivier Dormond, Paul J. Dyson, Jean-Luc Wolfender, and Patrycja Nowak-Sliwinska. 2021. "Drug Repurposing to Identify a Synergistic High-Order Drug Combination to Treat Sunitinib-Resistant Renal Cell Carcinoma" Cancers 13, no. 16: 3978. https://doi.org/10.3390/cancers13163978
APA StyleRausch, M., Rutz, A., Allard, P. -M., Delucinge-Vivier, C., Docquier, M., Dormond, O., Dyson, P. J., Wolfender, J. -L., & Nowak-Sliwinska, P. (2021). Drug Repurposing to Identify a Synergistic High-Order Drug Combination to Treat Sunitinib-Resistant Renal Cell Carcinoma. Cancers, 13(16), 3978. https://doi.org/10.3390/cancers13163978