
Pan-Cancer Analysis of Immune Complement Signature C3/C5/C3AR1/C5AR1 in Association with Tumor Immune Evasion and Therapy Resistance

 ,
,  , ,
, ,  , ,
, ,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Pan-Cancer Analysis of Differential Gene Expression of C3, C5, C3AR1, and C5AR1 between Tumor and Normal Tissue, Tumor Stages, and Tumor Subtypes
2.2. Pan-Cancer Analysis of Single Nucleotide Variations of C3, C5, C3AR1, and C5AR1
2.3. Pan-Cancer Methylation Analysis of C3, C5, C3AR1, and C5AR1
2.4. Pan-Cancer Analysis of Copy Number Variation in C3, C5, C3AR1, and C5AR1
2.5. Pan-Cancer Analysis of the C3, C5, C3AR1, and C5AR1 Association with Tumor Immune and Immune-Suppressive Cell Infiltrations, Dysfunctional T-Cell Phenotype, and T-Cell Exclusion
2.6. Functional Enrichment and PPI Network Analysis
2.7. Analysis of Gene Expression Correlation with Drug Sensitivity and Immunotherapy Response
2.8. Gene Prioritization of C3, C5, C3AR1, and C5AR1 across Four Immunosuppressive Indices
2.9. Comparative Biomarker Evaluation between Standardized Biomarkers and the C3, C5, C3AR1 and C5AR1 Gene Set
2.10. Gene Pathway Activity and Interaction Network
2.11. Data Analysis and Visualization
3. Results
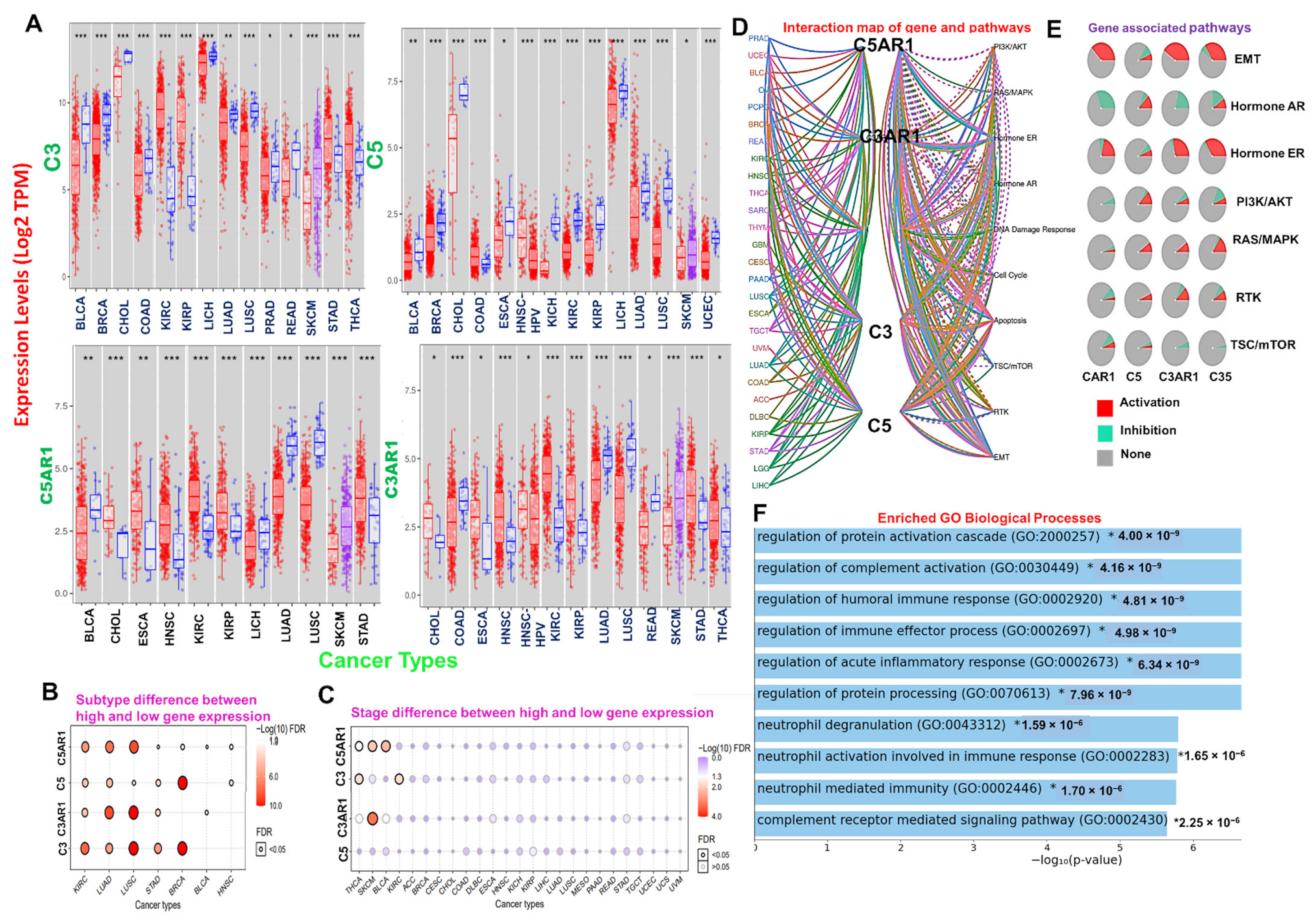
3.1. Complement Components C3, C5, C3AR1, and C5AR1 Demonstrated Context-Dependent Deregulatory Expression and Are Associated with Activation of Immune-Related Oncogenic Processes and Prognosis of the Cohort in Various Cancer Types
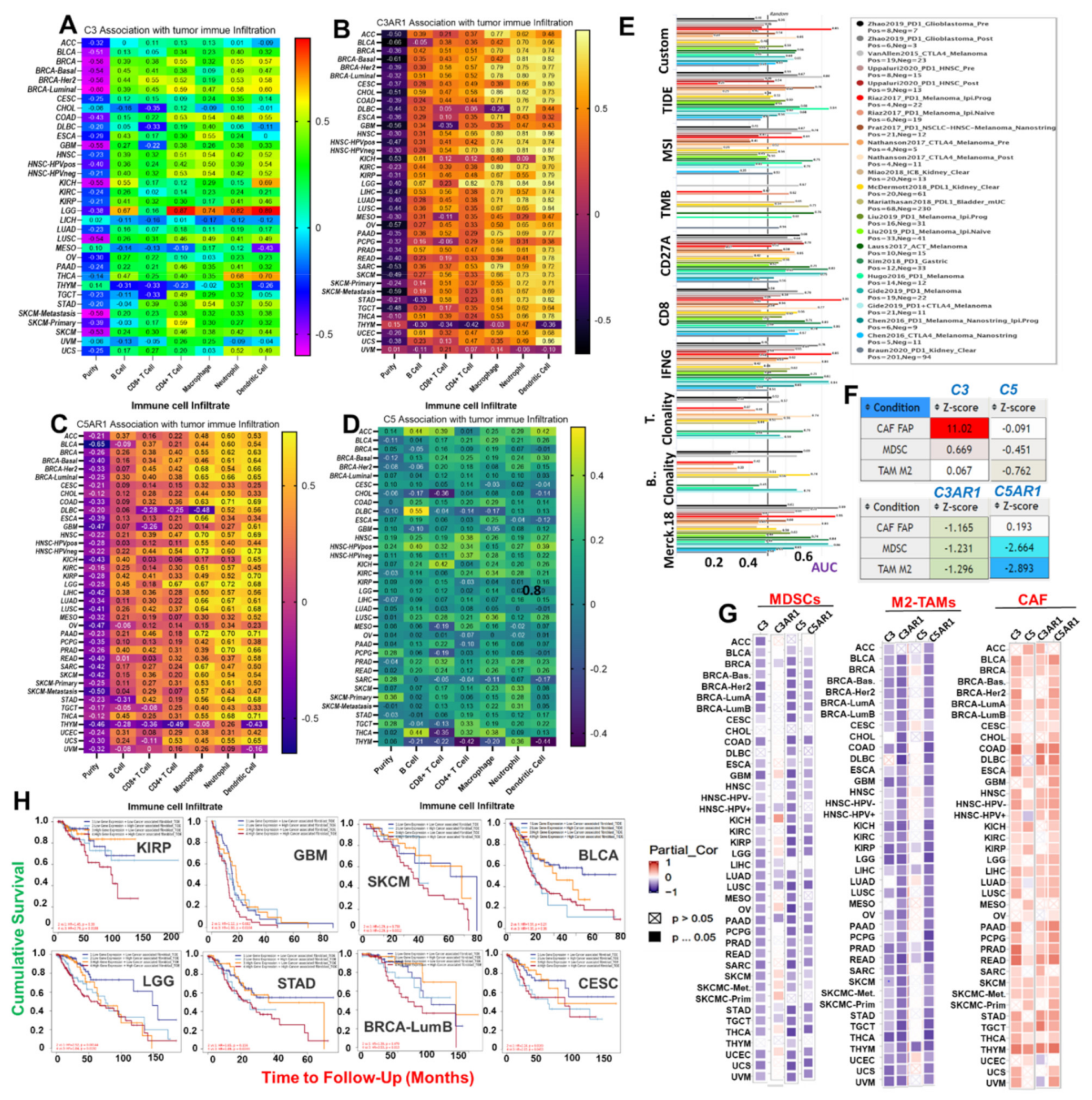
3.2. C3, C5, C3AR1, and C5AR1 Are Associated with Context-Dependent Tumor Immune Evasion via Dysfunctional T-Cell Phenotypes with a Lesser Contribution of T-Cell Exclusion
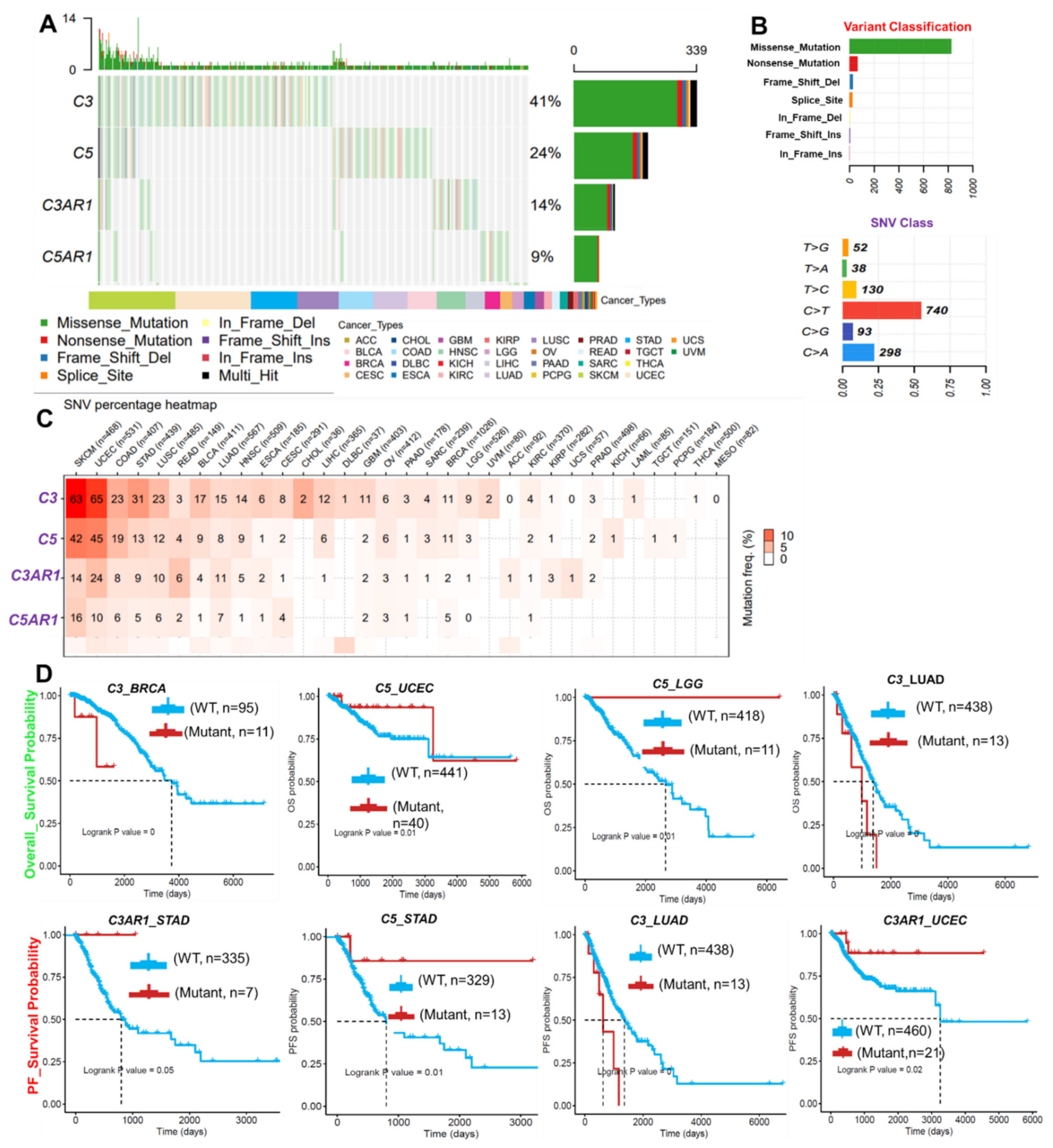
3.3. SNVs of C3, C5, C3AR1, and C5AR1 Are Associated with Prognosis and Co-Occurred with Other Oncogenic Mutations
3.4. C3, C5, C3AR1, and C5AR1 Expression Exhibited a Tumor-Context-Dependent Association with Copy Number Variation, Gene Methylation, and Dysfunctional T-Cell Phenotypes
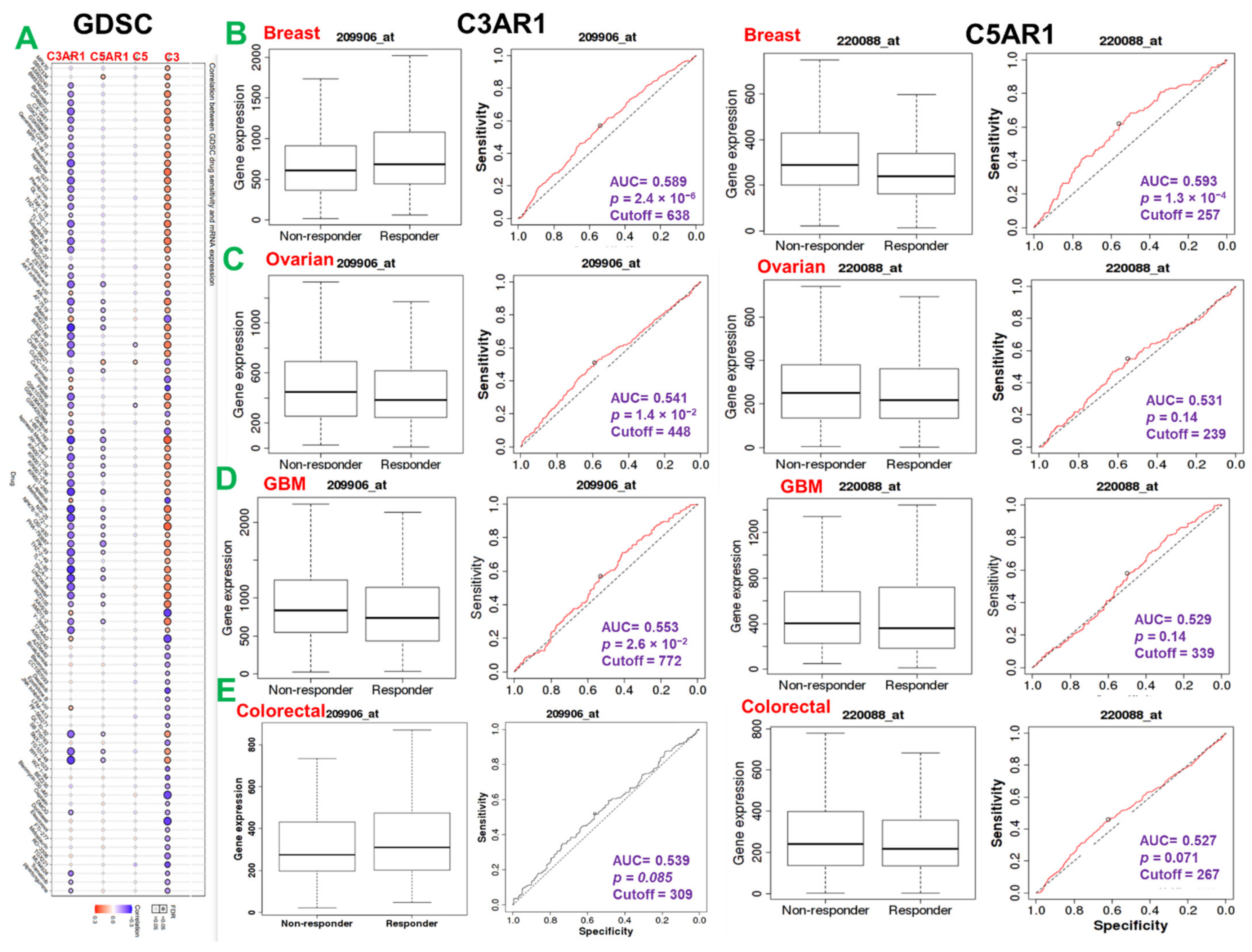
3.5. C3, C5, C3AR1, and C5AR1 Are Associated with Chemotherapy Outcome in Multiple Cancer Types
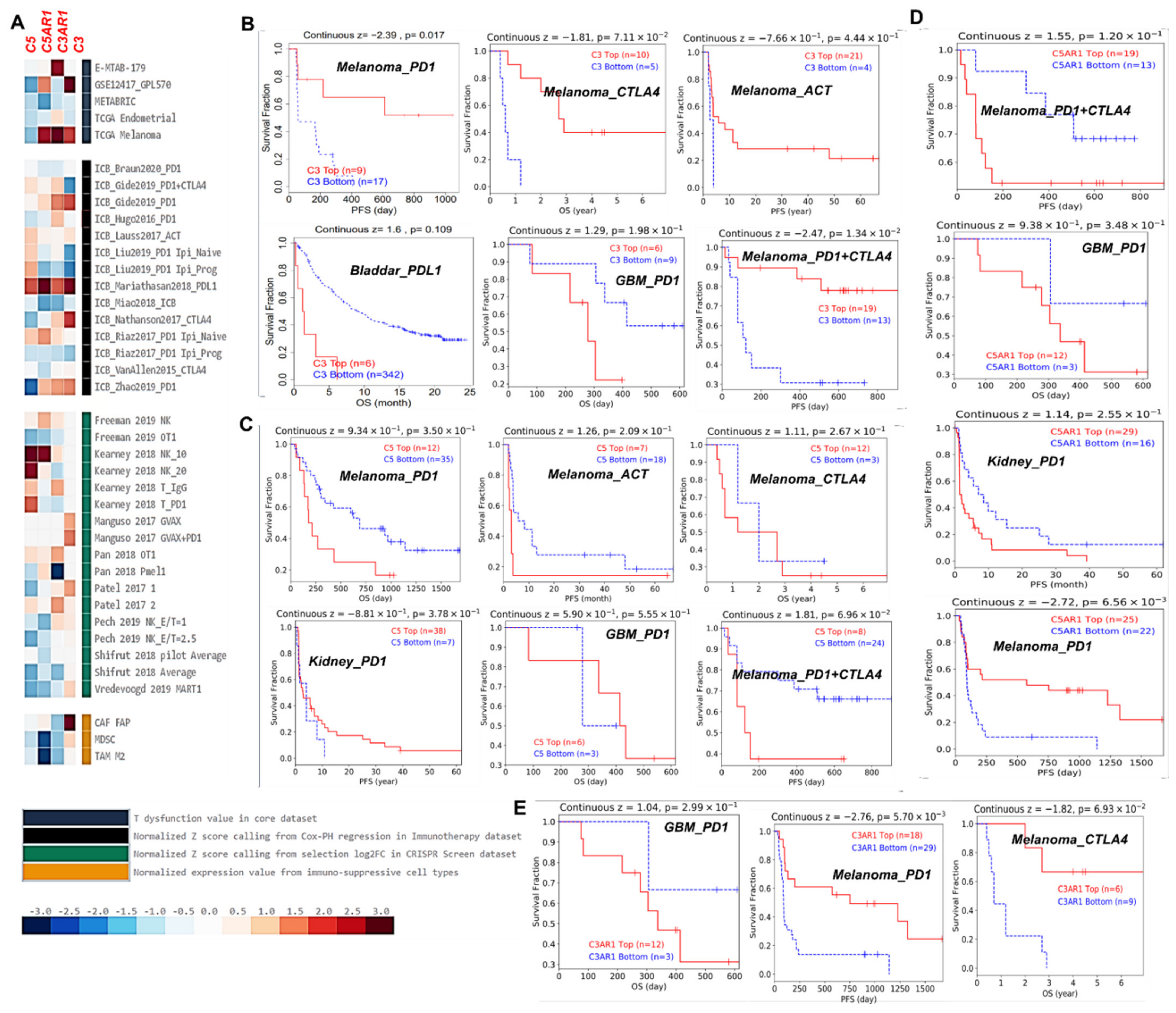
3.6. C3, C5, C3AR1, and C5AR1 Are Associated with Lymphocyte-Mediated Tumor Killing and Immunotherapy Outcome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Guo, S.; Deng, C.X. Effect of Stromal Cells in Tumor Microenvironment on Metastasis Initiation. Int. J. Biol. Sci. 2018, 14, 2083–2093. [Google Scholar] [CrossRef]
- Janeway, C.; Travers, P.; Walport, M.; Capra, J. Immunobiology: The Immune System in Health and Disease; Churchill Livingstone: New York, NY, USA, 1997. [Google Scholar]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Zwarthoff, S.A.; Berends, E.T.M.; Mol, S.; Ruyken, M.; Aerts, P.C.; Józsi, M.; de Haas, C.J.C.; Rooijakkers, S.H.M.; Gorham, R.D. Functional Characterization of Alternative and Classical Pathway C3/C5 Convertase Activity and Inhibition Using Purified Models. Front. Immunol. 2018, 9, 1691. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Lambris, J.D. Complement in immune and inflammatory disorders: Pathophysiological mechanisms. J. Immunol. 2013, 190, 3831–3838. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Mastellos, D.C.; Reis, E.S.; Lambris, J.D. The renaissance of complement therapeutics. Nat. Rev. Nephrol. 2018, 14, 26–47. [Google Scholar] [CrossRef]
- Morgan, B.P.; Harris, C.L. Complement, a target for therapy in inflammatory and degenerative diseases. Nat. Rev. Drug Discov. 2015, 14, 857–877. [Google Scholar] [CrossRef]
- Holers, V.M. Complement and its receptors: New insights into human disease. Annu. Rev. Immunol. 2014, 32, 433–459. [Google Scholar] [CrossRef]
- Horiuchi, T.; Tsukamoto, H. Complement-targeted therapy: Development of C5- and C5a-targeted inhibition. Inflamm. Regen. 2016, 36, 11. [Google Scholar] [CrossRef] [PubMed]
- Mastellos, D.C.; da Silva, B.G.P.; Fonseca, B.A.L.; Fonseca, N.P.; Auxiliadora-Martins, M.; Mastaglio, S.; Ruggeri, A.; Sironi, M.; Radermacher, P.; Chrysanthopoulou, A.; et al. Complement C3 vs C5 inhibition in severe COVID-19: Early clinical findings reveal differential biological efficacy. Clin. Immunol. 2020, 220, 108598. [Google Scholar] [CrossRef]
- Muenstermann, M.; Strobel, L.; Klos, A.; Wetsel, R.A.; Woodruff, T.M.; Köhl, J.; Johswich, K.O. Distinct roles of the anaphylatoxin receptors C3aR, C5aR1 and C5aR2 in experimental meningococcal infections. Virulence 2019, 10, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Strainic, M.G.; Shevach, E.M.; An, F.; Lin, F.; Medof, M.E. Absence of signaling into CD4⁺ cells via C3aR and C5aR enables autoinductive TGF-β1 signaling and induction of Foxp3⁺ regulatory T cells. Nat. Immunol. 2013, 14, 162–171. [Google Scholar] [CrossRef]
- Markiewski, M.M.; Lambris, J.D. Is complement good or bad for cancer patients? A new perspective on an old dilemma. Trends Immunol. 2009, 30, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Markiewski, M.M.; DeAngelis, R.A.; Benencia, F.; Ricklin-Lichtsteiner, S.K.; Koutoulaki, A.; Gerard, C.; Coukos, G.; Lambris, J.D. Modulation of the antitumor immune response by complement. Nat. Immunol. 2008, 9, 1225–1235. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liao, Q.; Zhao, Y. Chemotherapy and tumor microenvironment of pancreatic cancer. Cancer Cell Int. 2017, 17, 68. [Google Scholar] [CrossRef]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to Tumor Angiogenesis from Innate Immune Cells within the Tumor Microenvironment: Implications for Immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Johnson, T.S. Treg Destabilization and Reprogramming: Implications for Cancer Immunotherapy. Cancer Res. 2018, 78, 5191–5199. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, B.; Hu, Q.; Qin, Y.; Xu, W.; Liu, W.; Yu, X.; Xu, J. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics 2018, 8, 5072–5087. [Google Scholar] [CrossRef]
- Labidi-Galy, S.I.; Treilleux, I.; Goddard-Leon, S.; Combes, J.D.; Blay, J.Y.; Ray-Coquard, I.; Caux, C.; Bendriss-Vermare, N. Plasmacytoid dendritic cells infiltrating ovarian cancer are associated with poor prognosis. Oncoimmunology 2012, 1, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Lawal, B.; Lin, L.-C.; Lee, J.-C.; Chen, J.-H.; Bekaii-Saab, T.S.; Wu, A.T.H.; Ho, C.-L. Multi-Omics Data Analysis of Gene Expressions and Alterations, Cancer-Associated Fibroblast and Immune Infiltrations, Reveals the Onco-Immune Prognostic Relevance of STAT3/CDK2/4/6 in Human Malignancies. Cancers 2021, 13, 954. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liu, Q.; Li, T.; Liao, Q.; Zhao, Y. Role of the complement system in the tumor microenvironment. Cancer Cell Int. 2019, 19, 300. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Ajona, D.; Rafail, S.; Lasarte, J.J.; Riezu-Boj, J.I.; Lambris, J.D.; Rouzaut, A.; Pajares, M.J.; Montuenga, L.M.; Pio, R. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J. Immunol. 2012, 189, 4674–4683. [Google Scholar] [CrossRef]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Daugan, M.V.; Petitprez, F.; Sautès-Fridman, C.; Fridman, W.H. Context-dependent roles of complement in cancer. Nat. Rev. Cancer 2019, 19, 698–715. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef]
- Liu, C.-J.; Hu, F.-F.; Xia, M.-X.; Han, L.; Zhang, Q.; Guo, A.-Y. GSCALite: A web server for gene set cancer analysis. Bioinformatics 2018, 34, 3771–3772. [Google Scholar] [CrossRef] [PubMed]
- Mayakonda, A.; Koeffler, P. Maftools: Efficient analysis, visualization and summarization of MAF files from large-scale cohort based cancer studies. bioRxiv 2016. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Rivas, M.A.; Pirinen, M.; Conrad, D.F.; Lek, M.; Tsang, E.K.; Karczewski, K.J.; Maller, J.B.; Kukurba, K.R.; DeLuca, D.S.; Fromer, M.; et al. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science 2015, 348, 666. [Google Scholar] [CrossRef]
- Andersen, P.K.; Gill, R.D. Cox’s Regression Model for Counting Processes: A Large Sample Study. Ann. Stat. 1982, 10, 1100–1120. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Li, K.; Zhang, W.; Wan, C.; Zhang, J.; Jiang, P.; Liu, X.S. Large-scale public data reuse to model immunotherapy response and resistance. Genome Med. 2020, 12, 21. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef] [PubMed]
- Kaimal, V.; Bardes, E.E.; Tabar, S.C.; Jegga, A.G.; Aronow, B.J. ToppCluster: A multiple gene list feature analyzer for comparative enrichment clustering and network-based dissection of biological systems. Nucleic Acids Res. 2010, 38, W96–W102. [Google Scholar] [CrossRef]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef]
- Fekete, J.T.; Győrffy, B. ROCplot.org: Validating predictive biomarkers of chemotherapy/hormonal therapy/anti-HER2 therapy using transcriptomic data of 3104 breast cancer patients. Int. J. Cancer 2019, 145, 3140–3151. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Ng, P.K.S.; Werner, H.M.J.; Shahmoradgoli, M.; Zhang, F.; Ju, Z.; Liu, W.; Yang, J.-Y.; Yoshihara, K.; Li, J.; et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat. Commun. 2014, 5, 3887. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Zhang, J.; Zeng, D.; Sun, H.; Rong, X.; Shi, M.; Bin, J.; Liao, Y.; Liao, W. Immune cell infiltration as a biomarker for the diagnosis and prognosis of stage I–III colon cancer. Cancer Immunol. Immunother. 2019, 68, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Lawal, B.; Lee, C.-Y.; Mokgautsi, N.; Sumitra, M.R.; Khedkar, H.; Wu, A.T.H.; Huang, H.-S. mTOR/EGFR/iNOS/MAP2K1/FGFR/TGFB1 Are Druggable Candidates for N-(2,4-Difluorophenyl)-2′,4′-Difluoro-4-Hydroxybiphenyl-3-Carboxamide (NSC765598), with Consequent Anticancer Implications. Front. Oncol. 2021, 11, 656738. [Google Scholar] [CrossRef]
- Ajona, D.; Castaño, Z.; Garayoa, M.; Zudaire, E.; Pajares, M.J.; Martinez, A.; Cuttitta, F.; Montuenga, L.M.; Pio, R. Expression of complement factor H by lung cancer cells: Effects on the activation of the alternative pathway of complement. Cancer Res. 2004, 64, 6310–6318. [Google Scholar] [CrossRef]
- Blok, V.T.; Daha, M.R.; Tijsma, O.M.; Weissglas, M.G.; van den Broek, L.J.; Gorter, A. A possible role of CD46 for the protection in vivo of human renal tumor cells from complement-mediated damage. Lab. Investig. 2000, 80, 335–344. [Google Scholar] [CrossRef]
- Buettner, R.; Huang, M.; Gritsko, T.; Karras, J.; Enkemann, S.; Mesa, T.; Nam, S.; Yu, H.; Jove, R. Activated signal transducers and activators of transcription 3 signaling induces CD46 expression and protects human cancer cells from complement-dependent cytotoxicity. Mol. Cancer Res. MCR 2007, 5, 823–832. [Google Scholar] [CrossRef]
- Yang, J.; Lin, P.; Yang, M.; Liu, W.; Fu, X.; Liu, D.; Tao, L.; Huo, Y.; Zhang, J.; Hua, R. Integrated genomic and transcriptomic analysis reveals unique characteristics of hepatic metastases and pro-metastatic role of complement C1q in pancreatic ductal adenocarcinoma. Genome Biol. 2021, 22, 1–20. [Google Scholar] [CrossRef]
- Huang, H.; Tan, M.; Zheng, L.; Yan, G.; Li, K.; Lu, D.; Cui, X.; He, S.; Lei, D.; Zhu, B. Prognostic implications of the complement protein C1Q and its correlation with immune infiltrates in osteosarcoma. Onco Targets Ther. 2021, 14, 1737. [Google Scholar] [CrossRef]
- Rolfe, B.; Pio, R.; Woodruff, T.; Markiewski, M.; Manthey, H. Editorial: The Role of Complement in Tumors. Front. Immunol. 2020, 11, 139. [Google Scholar] [CrossRef]
- Wu, X.; Ragupathi, G.; Panageas, K.; Hong, F.; Livingston, P.O. Accelerated Tumor Growth Mediated by Sublytic Levels of Antibody-Induced Complement Activation Is Associated with Activation of the PI3K/AKT Survival Pathway. Clin. Cancer Res. 2013, 19, 4728–4739. [Google Scholar] [CrossRef]
- Niculescu, F.; Rus, H.; Van Biesen, T.; Shin, M.L. Activation of Ras and mitogen-activated protein kinase pathway by terminal complement complexes is G protein dependent. J. Immunol. 1997, 158, 4405–4412. [Google Scholar]
- Kraus, S.; Seger, R.; Fishelson, Z. Involvement of the ERK mitogen-activated protein kinase in cell resistance to complement-mediated lysis. Clin. Exp. Immunol. 2001, 123, 366–374. [Google Scholar] [CrossRef]
- Wan, J.; Zhou, X.; Cui, J.; Zou, Z.; Xu, Y.; You, D. Role of complement 3 in TNF-α-induced mesenchymal transition of renal tubular epithelial cells in vitro. Mol. Biotechnol. 2013, 54, 92–100. [Google Scholar] [CrossRef]
- Zhou, X.; Fukuda, N.; Matsuda, H.; Endo, M.; Wang, X.; Saito, K.; Ueno, T.; Matsumoto, T.; Matsumoto, K.; Soma, M. Complement 3 activates the renal renin-angiotensin system by induction of epithelial-to-mesenchymal transition of the nephrotubulus in mice. Am. J. Physiol. Renal Physiol. 2013, 305, F957–F967. [Google Scholar] [CrossRef]
- Chen, B.; Zhou, W.; Tang, C.; Wang, G.; Yuan, P.; Zhang, Y.; Bhushan, S.C.; Ma, J.; Leng, J. Down-Regulation of C3aR/C5aR Inhibits Cell Proliferation and EMT in Hepatocellular Carcinoma. Technol. Cancer Res. Treat. 2020, 19, 1533033820970668. [Google Scholar] [CrossRef]
- Toor, S.M.; Nair, V.S.; Decock, J.; Elkord, E. Immune checkpoints in the tumor microenvironment. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti–PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef]
- Grace, P.Y.; Chiang, D.; Song, S.J.; Hoyte, E.G.; Huang, J.; Vanishsarn, C.; Nadeau, K.C. Regulatory T cell dysfunction in subjects with common variable immunodeficiency complicated by autoimmune disease. Clin. Immunol. 2009, 131, 240–253. [Google Scholar]
- Spranger, S.; Gajewski, T.F. Tumor-intrinsic oncogene pathways mediating immune avoidance. Oncoimmunology 2016, 5, e1086862. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef]
- Kaderbhaï, C.; Tharin, Z.; Ghiringhelli, F. The Role of Molecular Profiling to Predict the Response to Immune Checkpoint Inhibitors in Lung Cancer. Cancers 2019, 11, 201. [Google Scholar] [CrossRef]
- Man, Y.G.; Stojadinovic, A.; Mason, J.; Avital, I.; Bilchik, A.; Bruecher, B.; Protic, M.; Nissan, A.; Izadjoo, M.; Zhang, X.; et al. Tumor-infiltrating immune cells promoting tumor invasion and metastasis: Existing theories. J. Cancer 2013, 4, 84–95. [Google Scholar] [CrossRef]
- Jackson, W.D.; Gulino, A.; Fossati-Jimack, L.; Seoane, R.C.; Tian, K.; Best, K.; Köhl, J.; Belmonte, B.; Strid, J.; Botto, M. C3 Drives Inflammatory Skin Carcinogenesis Independently of C5. J. Investig. Dermatol. 2021, 141, 404–414.e6. [Google Scholar] [CrossRef]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef]
- Nabizadeh, J.A.; Manthey, H.D.; Steyn, F.J.; Chen, W.; Widiapradja, A.; Md Akhir, F.N.; Boyle, G.M.; Taylor, S.M.; Woodruff, T.M.; Rolfe, B.E. The Complement C3a Receptor Contributes to Melanoma Tumorigenesis by Inhibiting Neutrophil and CD4+ T Cell Responses. J. Immunol. 2016, 196, 4783–4792. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- De Visser, K.E.; Korets, L.V.; Coussens, L.M. Early Neoplastic Progression Is Complement Independent. Neoplasia 2004, 6, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Eculizumab: A Review in Generalized Myasthenia Gravis. Drugs 2018, 78, 367–376. [Google Scholar] [CrossRef]
- Romi, F. Thymoma in myasthenia gravis: From diagnosis to treatment. Autoimmune Dis. 2011, 2011, 474512. [Google Scholar] [CrossRef]
- Vélez-Santamaría, V.; Nedkova, V.; Díez, L.; Homedes, C.; Alberti, M.A.; Casasnovas, C. Eculizumab as a promising treatment in thymoma-associated myasthenia gravis. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420932035. [Google Scholar] [CrossRef] [PubMed]
- Mastrandrea, L.D. An Overview of Organ-Specific Autoimmune Diseases Including Immunotherapy. Immunol. Investig. 2015, 44, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression. Vaccines 2016, 4, 36. [Google Scholar] [CrossRef]
- Allavena, P.; Mantovani, A. Immunology in the clinic review series; focus on cancer: Tumour-associated macrophages: Undisputed stars of the inflammatory tumour microenvironment. Clin. Exp. Immunol. 2012, 167, 195–205. [Google Scholar] [CrossRef]
- Galdiero, M.R.; Bonavita, E.; Barajon, I.; Garlanda, C.; Mantovani, A.; Jaillon, S. Tumor associated macrophages and neutrophils in cancer. Immunobiology 2013, 218, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A. The growing diversity and spectrum of action of myeloid-derived suppressor cells. Eur. J. Immunol. 2010, 40, 3317–3320. [Google Scholar] [CrossRef]
- Hsieh, C.-C.; Chou, H.-S.; Yang, H.-R.; Lin, F.; Bhatt, S.; Qin, J.; Wang, L.; Fung, J.J.; Qian, S.; Lu, L. The role of complement component 3 (C3) in differentiation of myeloid-derived suppressor cells. Blood 2013, 121, 1760–1768. [Google Scholar] [CrossRef] [PubMed]
- Zha, H.; Wang, X.; Zhu, Y.; Chen, D.; Han, X.; Yang, F.; Gao, J.; Hu, C.; Shu, C.; Feng, Y.; et al. Intracellular Activation of Complement C3 Leads to PD-L1 Antibody Treatment Resistance by Modulating Tumor-Associated Macrophages. Cancer Immunol. Res. 2019, 7, 193–207. [Google Scholar] [CrossRef]
- Piao, C.; Zhang, W.-M.; Li, T.-T.; Zhang, C.-C.; Qiu, S.; Liu, Y.; Liu, S.; Jin, M.; Jia, L.-X.; Song, W.-C.; et al. Complement 5a stimulates macrophage polarization and contributes to tumor metastases of colon cancer. Exp. Cell Res. 2018, 366, 127–138. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef]
- Surace, L.; Lysenko, V.; Fontana, A.O.; Cecconi, V.; Janssen, H.; Bicvic, A.; Okoniewski, M.; Pruschy, M.; Dummer, R.; Neefjes, J.; et al. Complement Is a Central Mediator of Radiotherapy-Induced Tumor-Specific Immunity and Clinical Response. Immunity 2015, 42, 767–777. [Google Scholar] [CrossRef]
- Davoli, T.; Xu, A.W.; Mengwasser, K.E.; Sack, L.M.; Yoon, J.C.; Park, P.J.; Elledge, S.J. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013, 155, 948–962. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355. [Google Scholar] [CrossRef]
- Duesberg, P.; Li, R. Multistep carcinogenesis: A chain reaction of aneuploidizations. Cell Cycle 2003, 2, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, R.; Schvartzman, J.M.; Socci, N.D.; Benezra, R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature 2010, 464, 436–440. [Google Scholar] [CrossRef]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Razin, A.; Cedar, H. DNA methylation and gene expression. Microbiol. Rev. 1991, 55, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Lim, P.S.; Li, J.; Holloway, A.F.; Rao, S. Epigenetic regulation of inducible gene expression in the immune system. Immunology 2013, 139, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Reddy, D.; Gupta, S. Global histone post-translational modifications and cancer: Biomarkers for diagnosis, prognosis and treatment? World J. Biol. Chem. 2015, 6, 333–345. [Google Scholar] [CrossRef]
- Mhaskar, D.; Goodman, M. On the molecular basis of transition mutations. Frequency of forming 2-aminopurine-cytosine base mispairs in the GXC—-AXT mutational pathway by T4 DNA polymerase in vitro. J. Biol. Chem. 1984, 259, 11713–11717. [Google Scholar] [CrossRef]
- Hahn, W.C.; Weinberg, R.A. Rules for making human tumor cells. N. Engl. J. Med. 2002, 347, 1593–1603. [Google Scholar] [CrossRef]
- Garnis, C.; Buys, T.P.H.; Lam, W.L. Genetic alteration and gene expression modulation during cancer progression. Mol. Cancer 2004, 3, 9. [Google Scholar] [CrossRef]
- Blakely, C.M.; Watkins, T.B.K.; Wu, W.; Gini, B.; Chabon, J.J.; McCoach, C.E.; McGranahan, N.; Wilson, G.A.; Birkbak, N.J.; Olivas, V.R.; et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat. Genet. 2017, 49, 1693–1704. [Google Scholar] [CrossRef]
- Hong, S.; Gao, F.; Fu, S.; Wang, Y.; Fang, W.; Huang, Y.; Zhang, L. Concomitant Genetic Alterations With Response to Treatment and Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Patients With EGFR-Mutant Advanced Non–Small Cell Lung Cancer. JAMA Oncol. 2018, 4, 739–742. [Google Scholar] [CrossRef]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Kearney, C.J.; Vervoort, S.J.; Hogg, S.J.; Ramsbottom, K.M.; Freeman, A.J.; Lalaoui, N.; Pijpers, L.; Michie, J.; Brown, K.K.; Knight, D.A.; et al. Tumor immune evasion arises through loss of TNF sensitivity. Sci. Immunol. 2018, 3, eaar3451. [Google Scholar] [CrossRef] [PubMed]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Sayah, S.; Ischenko, A.M.; Zhakhov, A.; Bonnard, A.S.; Fontaine, M. Expression of cytokines by human astrocytomas following stimulation by C3a and C5a anaphylatoxins: Specific increase in interleukin-6 mRNA expression. J. Neurochem. 1999, 72, 2426–2436. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xu, F.; Lu, T.; Duan, Z.; Zhang, Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat. Rev. 2012, 38, 904–910. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCGA Code | Cancer Type | Histology | Body Location |
|---|---|---|---|
| ACC | Adrenocortical carcinoma | Carcinoma | Endocrine |
| BLCA | Bladder urothelial carcinoma | Carcinoma | Genitourinary |
| BRCA | Breast invasive carcinoma | Carcinoma | Breast |
| CESC | Cervical squamous cell carcinoma and endocervical adenocarcinoma | Carcinoma | Gynecology |
| CHOL | Cholangiocarcinoma (bile duct) | Carcinoma | Digestive |
| COAD | Colon adenocarcinoma | Carcinoma | Digestive |
| DLBC | Lymphoid neoplasm diffuse large B-cell lymphoma | Lymphoma | Lymphoma |
| ESCA | Esophageal carcinoma | Carcinoma | Digestive |
| GBM | Glioblastoma multiforme | Sarcoma | Neurologic |
| HNSC | Head and neck squamous cell carcinoma | Carcinoma | Head and neck |
| KICH | Kidney chromophobe | Carcinoma | Genitourinary |
| KIRC | Kidney renal clear cell carcinoma | Carcinoma | Genitourinary |
| KIRP | Kidney renal papillary cell carcinoma | Carcinoma | Genitourinary |
| LAML | Acute myeloid leukemia | Leukemia | Hematologic |
| LGG | Brain lower grade glioma | Sarcoma | Neurologic |
| LIHC | Liver hepatocellular carcinoma | Carcinoma | Digestive |
| LUAD | Lung adenocarcinoma | Carcinoma | Respiratory |
| LUSC | Lung squamous cell carcinoma | Carcinoma | Respiratory |
| OV | Ovarian serous cystadenocarcinoma | Carcinoma | Gynecology |
| PAAD | Pancreatic adenocarcinoma | Carcinoma | Digestive |
| PCPG | Pheochromocytoma and paraganglioma (adrenal gland) | Endocrine | |
| PRAD | Prostate adenocarcinoma | Carcinoma | Genitourinary |
| READ | Rectum adenocarcinoma | Carcinoma | Digestive |
| SARC | Sarcoma | Sarcoma | Gynecology |
| SKCM | Skin cutaneous melanoma | Skin | |
| STAD | Stomach adenocarcinoma | Carcinoma | Digestive |
| TGCT | Testicular germ cell tumors | Carcinoma | Genitourinary |
| THCA | Thyroid carcinoma | Carcinoma | Endocrine |
| THYM | Thymoma | Lymphoma | Respiratory |
| UCEC | Uterine corpus endometrial carcinoma | Carcinoma | Gynecology |
| UCS | Uterine carcinosarcoma | Mixed type | Gynecology |
| UVM | Uveal melanoma | Carcinoma | Eye |
| H. | ID | Name | p-Value | FDR B&H | FDR B&Y | Bonferroni |
|---|---|---|---|---|---|---|
| GO:Molecular Function | GO:0004875 | complement receptor activity | 8.290 × 10−8 | 1.981 × 10−5 | 1.200 × 10−4 | 1.981 × 10−5 |
| GO:0004878 | complement component C5a receptor activity | 4.219 × 10−6 | 3.361 × 10−4 | 2.036 × 10−3 | 1.008 × 10−3 | |
| GO:0001856 | complement component C5a binding | 4.219 × 10−6 | 3.361 × 10−4 | 2.036 × 10−3 | 1.008 × 10−3 | |
| GO:0001848 | complement binding | 2.091 × 10−5 | 1.249 × 10−3 | 7.566 × 10−3 | 4.998 × 10−3 | |
| GO:0001847 | opsonin receptor activity | 4.194 × 10−5 | 2.005 × 10−3 | 1.214 × 10−2 | 1.002 × 10−2 | |
| ID | Name | p-Value | FDR B&H | FDR B&Y | Bonferroni | |
| GO: Biological Process | GO:0010575 | positive regulation of vascular endothelial growth factor production | 3.378 × 10−7 | 5.327 × 10−4 | 4.230 × 10−3 | 5.327 × 10−4 |
| GO:0002430 | complement receptor-mediated signaling pathway | 2.205 × 10−6 | 8.695 × 10−4 | 6.905 × 10−3 | 3.478 × 10−3 | |
| GO:0030449 | regulation of complement activation | 3.684 × 10−6 | 1.064 × 10−3 | 8.449 × 10−3 | 5.809 × 10−3 | |
| GO:0038178 | complement component C5a signaling pathway | 4.048 × 10−6 | 1.064 × 10−3 | 8.449 × 10−3 | 6.384 × 10−3 | |
| GO:0002688 | regulation of leukocyte chemotaxis | 4.911 × 10−6 | 1.106 × 10−3 | 8.786 × 10−3 | 7.745 × 10−3 | |
| GO:0002920 | regulation of humoral immune response | 8.615 × 10−6 | 1.601 × 10−3 | 1.271 × 10−2 | 1.359 × 10−2 | |
| GO:0010758 | regulation of macrophage chemotaxis | 3.723 × 10−5 | 5.338 × 10−3 | 4.239 × 10−2 | 5.872 × 10−2 | |
| GO:0090022 | regulation of neutrophil chemotaxis | 4.479 × 10−5 | 5.887 × 10−3 | 4.675 × 10−2 | 7.064 × 10−2 | |
| GO:0030593 | neutrophil chemotaxis | 8.376 × 10−5 | 7.812 × 10−3 | 6.204 × 10−2 | 1.321 × 10−1 | |
| GO:0002685 | regulation of leukocyte migration | 8.422 × 10−5 | 7.812 × 10−3 | 6.204 × 10−2 | 1.328 × 10−1 | |
| GO:0097529 | myeloid leukocyte migration | 1.101 × 10−4 | 8.171 × 10−3 | 6.488 × 10−2 | 1.736 × 10−1 | |
| GO:1905521 | regulation of macrophage migration | 1.117 × 10−4 | 8.171 × 10−3 | 6.488 × 10−2 | 1.762 × 10−1 | |
| GO:0002253 | activation of immune response | 1.162 × 10−4 | 8.171 × 10−3 | 6.488 × 10−2 | 1.832 × 10−1 | |
| ID | Name | p-Value | FDR B&H | FDR B&Y | Bonferroni | |
| Pathway | 1269250 | Regulation of Complement cascade | 3.005 × 10−9 | 1.337 × 10−6 | 8.929 × 10−6 | 1.337 × 10−6 |
| 1269241 | Complement cascade | 1.130 × 10−7 | 2.513 × 10−5 | 1.678 × 10−4 | 5.027 × 10−5 | |
| 172846 | Staphylococcus aureus infection | 6.804 × 10−6 | 1.009 × 10−3 | 6.739 × 10−3 | 3.028 × 10−3 | |
| M16894 | Complement and coagulation cascades | 1.570 × 10−5 | 1.747 × 10−3 | 1.166 × 10−2 | 6.986 × 10−3 | |
| 83073 | Complement and coagulation cascades | 2.687 × 10−5 | 2.391 × 10−3 | 1.597 × 10−2 | 1.196 × 10−2 | |
| 1269546 | Peptide ligand-binding receptors | 5.391 × 10−5 | 3.999 × 10−3 | 2.670 × 10−2 | 2.399 × 10−2 | |
| 1269203 | Innate Immune System | 6.409 × 10−5 | 4.075 × 10−3 | 2.720 × 10−2 | 2.852 × 10−2 | |
| 1269248 | Activation of C3 and C5 | 1.007 × 10−4 | 5.603 × 10−3 | 3.741 × 10−2 | 4.483 × 10−2 | |
| M22072 | Alternative Complement Pathway | 1.722 × 10−4 | 8.515 × 10−3 | 5.685 × 10−2 | 7.663 × 10−2 | |
| ID | Name | p-Value | FDR B&H | FDR B&Y | Bonferroni | |
| Diseases | C0025306 | Meningococcemia | 4.257 × 10−8 | 5.364 × 10−5 | 4.139 × 10−4 | 5.364 × 10−5 |
| C2931788 | Atypical Hemolytic Uremic Syndrome | 2.466 × 10−6 | 1.554 × 10−3 | 1.199 × 10−2 | 3.107 × 10−3 | |
| C0020951 | Immune Complex Diseases | 6.637 × 10−5 | 1.394 × 10−2 | 1.076 × 10−1 | 8.363 × 10−2 | |
| C0003907 | Arthus Reaction | 1.417 × 10−4 | 1.984 × 10−2 | 1.531 × 10−1 | 1.786 × 10−1 | |
| C2717961 | Thrombotic Microangiopathies | 1.729 × 10−4 | 2.001 × 10−2 | 1.544 × 10−1 | 2.178 × 10−1 | |
| C0740345 | Germ Cell Cancer | 3.289 × 10−4 | 2.072 × 10−2 | 1.599 × 10−1 | 4.144 × 10−1 | |
| C0027654 | Embryonal Neoplasm | 3.289 × 10−4 | 2.072 × 10−2 | 1.599 × 10−1 | 4.144 × 10−1 | |
| C0027658 | Neoplasms, Germ Cell, and Embryonal | 3.289 × 10−4 | 2.072 × 10−2 | 1.599 × 10−1 | 4.144 × 10−1 | |
| C0751364 | Cancer, Embryonal | 3.289 × 10−4 | 2.072 × 10−2 | 1.599 × 10−1 | 4.144 × 10−1 | |
| C0003257 | Antibody Deficiency Syndrome | 4.250 × 10−4 | 2.550 × 10−2 | 1.968 × 10−1 | 5.355 × 10−1 | |
| C0008149 | Chlamydia Infections | 5.355 × 10−4 | 2.550 × 10−2 | 1.968 × 10−1 | 6.747 × 10−1 | |
| C0021051 | Immunologic Deficiency Syndromes | 7.852 × 10−4 | 2.550 × 10−2 | 1.968 × 10−1 | 9.894 × 10−1 | |
| C1319860 | Sendai virus infection | 9.290 × 10−4 | 2.550 × 10−2 | 1.968 × 10−1 | 1.000 | |
| C0221238 | Mesangial proliferative glomerulonephritis | 1.252 × 10−3 | 2.550 × 10−2 | 1.968 × 10−1 | 1.000 | |
| C0272242 | Complement deficiency disease | 1.620 × 10−3 | 2.550 × 10−2 | 1.968 × 10−1 | 1.000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lawal, B.; Tseng, S.-H.; Olugbodi, J.O.; Iamsaard, S.; Ilesanmi, O.B.; Mahmoud, M.H.; Ahmed, S.H.; Batiha, G.E.-S.; Wu, A.T.H. Pan-Cancer Analysis of Immune Complement Signature C3/C5/C3AR1/C5AR1 in Association with Tumor Immune Evasion and Therapy Resistance. Cancers 2021, 13, 4124. https://doi.org/10.3390/cancers13164124
Lawal B, Tseng S-H, Olugbodi JO, Iamsaard S, Ilesanmi OB, Mahmoud MH, Ahmed SH, Batiha GE-S, Wu ATH. Pan-Cancer Analysis of Immune Complement Signature C3/C5/C3AR1/C5AR1 in Association with Tumor Immune Evasion and Therapy Resistance. Cancers. 2021; 13(16):4124. https://doi.org/10.3390/cancers13164124
Chicago/Turabian StyleLawal, Bashir, Sung-Hui Tseng, Janet Olayemi Olugbodi, Sitthichai Iamsaard, Omotayo B. Ilesanmi, Mohamed H. Mahmoud, Sahar H. Ahmed, Gaber El-Saber Batiha, and Alexander T. H. Wu. 2021. "Pan-Cancer Analysis of Immune Complement Signature C3/C5/C3AR1/C5AR1 in Association with Tumor Immune Evasion and Therapy Resistance" Cancers 13, no. 16: 4124. https://doi.org/10.3390/cancers13164124
APA StyleLawal, B., Tseng, S. -H., Olugbodi, J. O., Iamsaard, S., Ilesanmi, O. B., Mahmoud, M. H., Ahmed, S. H., Batiha, G. E. -S., & Wu, A. T. H. (2021). Pan-Cancer Analysis of Immune Complement Signature C3/C5/C3AR1/C5AR1 in Association with Tumor Immune Evasion and Therapy Resistance. Cancers, 13(16), 4124. https://doi.org/10.3390/cancers13164124