
Challenges and Future Perspectives of Immunotherapy in Pancreatic Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
2. Mechanisms of Immunosuppression and Immune Evasion in PDAC
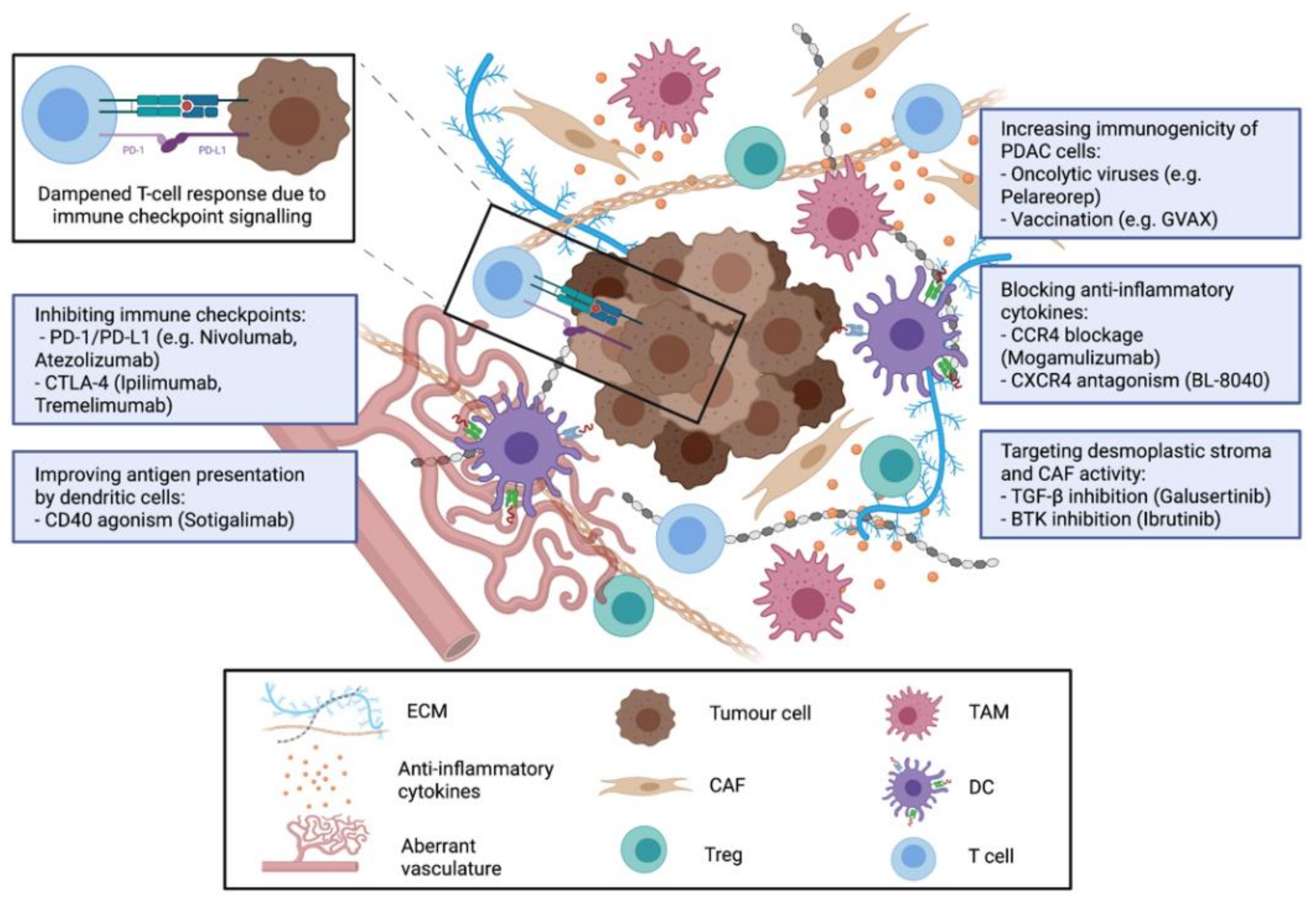
3. Strategies for Immunotherapy in PDAC
3.1. Cancer Vaccines for Treatment of PDAC
3.2. Immune Checkpoint Inhibitors for PDAC Treatment
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APC | Antigen presenting cell |
| BTK | Bruton Tyrosine Kinase |
| CAF | Carcinoma associated fibroblasts |
| CD40 | Cluster of Differentiation 40 |
| CTL | Cytotoxic T lymphocytes |
| CTLA-4 | Cytotoxic T-Lymphocyte-Associated Protein 4 |
| DCR | Disease control rate |
| ECM | Extracellular matrix |
| FOLFIRINOX | 5-FU, Irinotecan, Leucovorin, Oxaliplatin |
| 5-FU | 5-Fluorouracil |
| GM-CSF | Granulocyte-Macrophage Colony Stimulating Factor |
| ICOS | Inducible T-cell costimulator kinase inhibition |
| IHC | immunohistochemistry |
| INF-γ | Interferon-gamma |
| KIF20A | Kinesine-like Protein KIF 20A |
| MDSC | Myeloid derived suppressor cells |
| mOS | Mean overall survival |
| ORR | Overall response rate |
| PD-1 | Programmed cell death protein 1 |
| PDAC | Pancreatic ductal adenocarcinoma |
| PD-L1 | Programmed cell death 1 ligand 1 |
| STING | Stimulator of interferon genes |
| TAM | Tumour-associated macrophages |
| TCR | T-cell receptor |
| TGF-β | Transforming Growth Factor-beta |
| TIL | Tumour infiltrating lymphocytes |
| TME | Tumour microenvironment |
| Treg | regulatory T-cells |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Henley, S.J.; Ward, E.M.; Scott, S.; Ma, J.; Anderson, R.N.; Firth, A.U.; Thomas, C.C.; Islami, F.; Weir, H.K.; Lewis, D.R.; et al. Annual Report to the Nation on the Status of Cancer, Part I: National Cancer Statistics. Cancer 2020, 126, 2225–2249. [Google Scholar] [CrossRef]
- Conroy, T.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de La Fouchardière, C.; Bennouna, J.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Gillen, S.; Schuster, T.; zum Büschenfelde, C.M.; Friess, H.; Kleeff, J. Preoperative/Neoadjuvant Therapy in Pancreatic Cancer: A Systematic Review and Meta-Analysis of Response and Resection Percentages. PLoS Med. 2010, 7, e1000267. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Hammel, P.; Hebbar, M.; ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, E.S.; Hue, J.J.; Bajor, D.L.; Ocuin, L.M.; Ammori, J.B.; Hardacre, J.M.; Winter, J.M. A Comprehensive Analysis of Clinical Trials in Pancreatic Cancer: What Is Coming down the Pike? Oncotarget 2020, 11, 3489–3501. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The Tumour Microenvironment in Pancreatic Cancer—Clinical Challenges and Opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef]
- Orhan, A.; Vogelsang, R.P.; Andersen, M.B.; Madsen, M.T.; Hölmich, E.R.; Raskov, H.; Gögenur, I. The Prognostic Value of Tumour-Infiltrating Lymphocytes in Pancreatic Cancer: A Systematic Review and Meta-Analysis. Eur. J. Cancer 2020, 132, 71–84. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of Unique Neoantigen Qualities in Long-Term Survivors of Pancreatic Cancer. Nature 2017, 551, S12–S16. [Google Scholar] [CrossRef]
- Zhang, J.; Wolfgang, C.L.; Zheng, L. Precision Immuno-Oncology: Prospects of Individualized Immunotherapy for Pancreatic Cancer. Cancers 2018, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Lawlor, R.T.; Mattiolo, P.; Mafficini, A.; Hong, S.-M.; Piredda, M.L.; Taormina, S. v.; Malleo, G.; Marchegiani, G.; Pea, A.; Salvia, R.; et al. Tumor Mutational Burden as a Potential Biomarker for Immunotherapy in Pancreatic Cancer: Systematic Review and Still-Open Questions. Cancers 2021, 13, 3119. [Google Scholar] [CrossRef]
- Pandha, H.; Rigg, A.; John, J.; Lemoine, N. Loss of Expression of Antigen-Presenting Molecules in Human Pancreatic Cancer and Pancreatic Cancer Cell Lines. Clin. Exp. Immunol. 2007, 148, 127–135. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; Lobello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; von Hoff, D.D.; Han, H. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, K.; Ino, Y.; Yamazaki-Itoh, R.; Naito, C.; Shimasaki, M.; Takahashi, M.; Esaki, M.; Nara, S.; Kishi, Y.; Shimada, K.; et al. IAP Inhibitor, Embelin Increases VCAM-1 Levels on the Endothelium, Producing Lymphocytic Infiltration and Antitumor Immunity. OncoImmunology 2020, 9, 1838812. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the Immune Reaction to Pancreatic Cancer from Inception to Invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saka, D.; Gökalp, M.; Piyade, B.; Cevik, N.C.; Sever, E.A.; Unutmaz, D.; Ceyhan, G.O.; Demir, I.E.; Asimgil, H. Mechanisms of T-Cell Exhaustion in Pancreatic Cancer. Cancers 2020, 12, 2274. [Google Scholar] [CrossRef] [PubMed]
- Rahn, S.; Krüger, S.; Röcken, C.; Helm, O.; Sebens, S. Response to: “Patterns of PD-L1 Expression and CD8 T Cell Infiltration in Gastric Adenocarcinomas and Associated Immune Stroma”. Gut 2019, 68, 179–180. [Google Scholar] [CrossRef] [PubMed]
- Rahn, S.; Krüger, S.; Mennrich, R.; Goebel, L.; Wesch, D.; Oberg, H.-H.; Vogel, I.; Ebsen, M.; Röcken, C.; Helm, O.; et al. POLE Score: A Comprehensive Profiling of Programmed Death 1 Ligand 1 Expression in Pancreatic Ductal Adenocarcinoma. Oncotarget 2019, 10, 1572–1588. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Eresen, A.; Shangguan, J.; Ma, Q.; Zhang, Z.; Yaghmai, V. Effect of Route of Administration on the Efficacy of Dendritic Cell Vaccine in PDAC Mice. Am. J. Cancer Res. 2020, 10, 3911–3919. [Google Scholar]
- Miyazawa, M.; Katsuda, M.; Maguchi, H.; Katanuma, A.; Ishii, H.; Ozaka, M.; Yamao, K.; Imaoka, H.; Kawai, M.; Hirono, S.; et al. Phase II Clinical Trial Using Novel Peptide Cocktail Vaccine as a Postoperative Adjuvant Treatment for Surgically Resected Pancreatic Cancer Patients. Int. J. Cancer 2017, 140, 973–982. [Google Scholar] [CrossRef]
- Palmer, D.H.; Valle, J.W.; Ting Ma, Y.; Faluyi, O.; Neoptolemos, J.P.; Jensen Gjertsen, T.; Iversen, B.; Amund Eriksen, J.; Møller, A.S.; Aksnes, A.K.; et al. TG01/GM-CSF and Adjuvant Gemcitabine in Patients with Resected RAS-Mutant Adenocarcinoma of the Pancreas (CT TG01-01): A Single-Arm, Phase 1/2 Trial. Br. J. Cancer 2020, 122, 971–977. [Google Scholar] [CrossRef] [Green Version]
- Nishida, S.; Ishikawa, T.; Egawa, S.; Koido, S.; Yanagimoto, H.; Ishii, J.; Kanno, Y.; Kokura, S.; Yasuda, H.; Oba, M.S.; et al. Combination Gemcitabine and WT1 Peptide Vaccination Improves Progression-Free Survival in Advanced Pancreatic Ductal Adenocarcinoma: A Phase II Randomized Study. Cancer Immunol. Res. 2018, 6, 320–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieweg, J.; Rosenthal, F.M.; Bannerji, R.; Heston, W.D.W.; Fair, W.R.; Gansbacher, B.; Gilboa2, E. Immunotherapy of Prostate Cancer in the Dunning Rat Model: Use of Cytokine Gene Modified Tumor Vaccines. Cancer Res. 1994, 54, 1760–1765. [Google Scholar] [PubMed]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Jackson, V.; Hamada, H.; Pardoll, D.; Mulligan, R.C. Vaccination with Irradiated Tumor Cells Engineered to Secrete Murine Granulocyte-Macrophage Colony-Stimulating Factor Stimulates Potent, Specific, and Long-Lasting Anti-Tumor Immunity. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar] [CrossRef] [Green Version]
- Lutz, E.R.; Wu, A.A.; Bigelow, E.; Sharma, R.; Mo, G.; Soares, K.; Solt, S.; Dorman, A.; Wamwea, A.; Yager, A.; et al. Immunotherapy Converts Nonimmunogenic Pancreatic Tumors into Immunogenic Foci of Immune Regulation. Cancer Immunol. Res. 2014, 2, 616–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Ding, D.; Edil, B.H.; Judkins, C.; Durham, J.N.; Thomas, D.L.; Bever, K.M.; Mo, G.; Solt, S.E.; Hoare, J.A.; et al. Vaccine-Induced Intratumoral Lymphoid Aggregates Correlate with Survival Following Treatment with a Neoadjuvant and Adjuvant Vaccine in Patients with Resectable Pancreatic Adenocarcinoma. Clin. Cancer Res. 2021, 27, 1278–1286. [Google Scholar] [CrossRef]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and Survival with GVAX Pancreas Prime and Listeria Monocytogenes-Expressing Mesothelin (CRS-207) Boost Vaccines for Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Picozzi, V.J.; Ko, A.H.; Wainberg, Z.A.; Kindler, H.; Wang-Gillam, A.; Oberstein, P.; Morse, M.A.; Zeh, H.J.; Weekes, C.; et al. Results from a Phase IIb, Randomized, Multicenter Study of GVAX Pancreas and CRS-207 Compared with Chemotherapy in Adults with Previously Treated Metastatic Pancreatic Adenocarcinoma (ECLIPSE Study). Clin. Cancer Res. 2019, 25, 5493–5502. [Google Scholar] [CrossRef] [PubMed]
- Nair, N.; Chen, S.Y.; Lemmens, E.; Chang, S.; Le, D.T.; Jaffee, E.M.; Murphy, A.; Whiting, C.; Müller, T.; Brockstedt, D.G. Single-Cell Immune Competency Signatures Associate with Survival in Phase II GVAX and CRS-207 Randomized Studies in Patients with Metastatic Pancreatic Cancer. Cancer Immunol. Res. 2020, 8, 609–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brock Hewitt, D.; Nissen, N.; Hatoum, H.; Musher, B.; Seng, J.; Coveler, A.L.; Al-Rajabi, R.; Yeo, C.J.; Leiby, B.; Banks, J.; et al. A Phase 3 Randomized Clinical Trial of Chemotherapy with or without Algenpantucel-L (HyperAcute®-Pancreas) Immunotherapy in Subjects with Borderline Resectable or Locally Advanced Unresectable Pancreatic Cancer. Ann. Surg. 2020. Online ahead of print. [Google Scholar] [CrossRef]
- Hardacre, J.M.; Mulcahy, M.; Small, W.; Talamonti, M.; Obel, J.; Krishnamurthi, S.; Rocha-Lima, C.S.; Safran, H.; Lenz, H.J.; Chiorean, E.G. Addition of Algenpantucel-L Immunotherapy to Standard Adjuvant Therapy for Pancreatic Cancer: A Phase 2 Study. J. Gastrointest. Surg. 2013, 17, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.K.; Lee, K.M.; McKolanis, J.; Hitbold, E.; Schraut, W.; Moser, A.J.; Warnick, E.; Whiteside, T.; Osborne, J.; Kim, H.; et al. Phase I Study of a MUC1 Vaccine Composed of Different Doses of MUC1 Peptide with SB-AS2 Adjuvant in Resected and Locally Advanced Pancreatic Cancer. Cancer Immunol. Immunother. 2005, 54, 254–264. [Google Scholar] [CrossRef]
- Middleton, G.; Silcocks, P.; Cox, T.; Valle, J.; Wadsley, J.; Propper, D.; Coxon, F.; Ross, P.; Madhusudan, S.; Roques, T.; et al. Gemcitabine and Capecitabine with or without Telomerase Peptide Vaccine GV1001 in Patients with Locally Advanced or Metastatic Pancreatic Cancer (TeloVac): An Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2014, 15, 829–840. [Google Scholar] [CrossRef]
- Schmitz-Winnenthal, F.H.; Hohmann, N.; Niethammer, A.G.; Friedrich, T.; Lubenau, H.; Springer, M.; Breiner, K.M.; Mikus, G.; Weitz, J.; Ulrich, A.; et al. Anti-Angiogenic Activity of VXM01, an Oral T-Cell Vaccine against VEGF Receptor 2, in Patients with Advanced Pancreatic Cancer: A Randomized, Placebo-Controlled, Phase 1 Trial. OncoImmunology 2015, 4, e1001217. [Google Scholar] [CrossRef] [Green Version]
- Plate, J.M.D.; Plate, A.E.; Shott, S.; Bograd, S.; Harris, J.E. Effect of Gemcitabine on Immune Cells in Subjects with Adenocarcinoma of the Pancreas. Cancer Immunol. Immunother. 2005, 54, 915–925. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive Correlates of Response to the Anti-PD-L1 Antibody MPDL3280A in Cancer Patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, A.; Kang, S.P.; Rasco, D.; Papadopoulos, K.P.; Elassaiss-Schaap, J.; Beeram, M.; Drengler, R.; Chen, C.; Smith, L.; Espino, G.; et al. Phase I Study of Pembrolizumab (MK-3475; Anti-PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 4286–4293. [Google Scholar] [CrossRef] [Green Version]
- Homma, Y.; Taniguchi, K.; Nakazawa, M.; Matsuyama, R.; Mori, R.; Takeda, K.; Ichikawa, Y.; Tanaka, K.; Endo, I. Changes in the Immune Cell Population and Cell Proliferation in Peripheral Blood after Gemcitabine-Based Chemotherapy for Pancreatic Cancer. Clin. Transl. Oncol. 2014, 16, 330–335. [Google Scholar] [CrossRef]
- Eriksson, E.; Wenthe, J.; Irenaeus, S.; Loskog, A.; Ullenhag, G. Gemcitabine Reduces MDSCs, Tregs and TGFβ-1 While Restoring the Teff/Treg Ratio in Patients with Pancreatic Cancer. J. Transl. Med. 2016, 14, 282. [Google Scholar] [CrossRef]
- Shibuya, K.C.; Goel, V.K.; Xiong, W.; Sham, J.G.; Pollack, S.M.; Leahy, A.M.; Whiting, S.H.; Yeh, M.M.; Yee, C.; Riddell, S.R.; et al. Pancreatic Ductal Adenocarcinoma Contains an Effector and Regulatory Immune Cell Infiltrate That Is Altered by Multimodal Neoadjuvant Treatment. PLoS ONE 2014, 9, e1333210. [Google Scholar] [CrossRef] [PubMed]
- Aglietta, M.; Barone, C.; Sawyer, M.B.; Moore, M.J.; Miller, W.H.; Bagalà, C.; Colombi, F.; Cagnazzo, C.; Gioeni, L.; Wang, E.; et al. A Phase I Dose Escalation Trial of Tremelimumab (CP-675,206) in Combination with Gemcitabine in Chemotherapy-Naive Patients with Metastatic Pancreatic Cancer. Ann. Oncol. 2014, 25, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.A.; Hochster, H.S.; Kim, E.J.; George, B.; Kaylan, A.; Chiorean, E.G.; Waterhouse, D.M.; Guiterrez, M.; Parikh, A.; Jain, R.; et al. Open-Label, Phase I Study of Nivolumab Combined with Nab-Paclitaxel plus Gemcitabine in Advanced Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4814–4822. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schütz, E.; Khemka, V. Phase Ib/II Study of Gemcitabine, Nab-Paclitaxel, and Pembrolizumab in Metastatic Pancreatic Adenocarcinoma. Investig. New Drugs 2018, 36, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Duffy, A.G.; Brar, G.; Fioravanti, S.; Mabry-Hrones, D.; Walker, M.; Bonilla, C.M.; Wood, B.J.; Citrin, D.E.; Gil Ramirez, E.M.; et al. Immune Checkpoint Blockade in Combination with Stereotactic Body Radiotherapy in Patients with Metastatic Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2020, 26, 2318–2326. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab with or without Tremelimumab for Patients with Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Principe, D.R.; DeCant, B.; Mascariñas, E.; Wayne, E.A.; Diaz, A.M.; Akagi, N.; Hwang, R.; Pasche, B.; Dawson, D.W.; Fang, D.; et al. TGFβ Signaling in the Pancreatic Tumor Microenvironment Promotes Fibrosis and Immune Evasion to Facilitate Tumorigenesis. Cancer Res. 2016, 76, 2525–2539. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Oh, D.Y.; Hollebecque, A.; Calvo, E.; Varghese, A.; Borazanci, E.; Macarulla, T.; Merz, V.; Zecchetto, C.; Zhao, Y.; et al. Safety and Activity of the TGFβ Receptor I Kinase Inhibitor Galunisertib plus the Anti-PD-L1 Antibody Durvalumab in Metastatic Pancreatic Cancer. J. Immunother. Cancer 2021, 9, e002068. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Q.; Liao, Q. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Origin, Polarization, Function, and Reprogramming. Front. Cell Dev. Biol. 2021, 8, 607209. [Google Scholar] [CrossRef] [PubMed]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truit, M.; Olson, P.; et al. Bruton Tyrosine Kinase–Dependent Immune Cell Cross-Talk Drives Pancreas Cancer. Cancer Discov. 2016, 6, 270–285. [Google Scholar] [CrossRef] [Green Version]
- Masso-Valles, D.; Jauset, T.; Serrano, E.; Sodir, N.M.; Pedersen, K.; Affara, N.I.; Whitfield, J.R.; Beaulieu, M.E.; Evan, G.I.; Elias, L.; et al. Ibrutinib Exerts Potent Antifibrotic and Antitumor Activities in Mouse Models of Pancreatic Adenocarcinoma. Cancer Res. 2015, 75, 1675–1681. [Google Scholar] [CrossRef] [Green Version]
- Tempero, M.; Oh, D.Y.; Tabernero, J.; Reni, M.; van Cutsem, E.; Hendifar, A.; Waldschmidt, D.T.; Starling, N.; Bachet, J.B.; Chang, H.M.; et al. Ibrutinib in Combination with Nab-Paclitaxel and Gemcitabine for First-Line Treatment of Patients with Metastatic Pancreatic Adenocarcinoma: Phase III RESOLVE Study. Ann. Oncol. 2021, 32, 600–608. [Google Scholar] [CrossRef]
- Overman, M.; Javle, M.; Davis, R.E.; Vats, P.; Kumar-Sinha, C.; Xiao, L.; Mettu, N.B.; Parra, E.R.; Benson, A.B.; Lopez, C.D.; et al. Randomized Phase II Study of the Bruton Tyrosine Kinase Inhibitor Acalabrutinib, Alone or with Pembrolizumab in Patients with Advanced Pancreatic Cancer. J. Immunother. Cancer 2020, 8, e000587. [Google Scholar] [CrossRef] [Green Version]
- Gujar, S.; Pol, J.G.; Kim, Y.; Lee, P.W.; Kroemer, G. Antitumor Benefits of Antiviral Immunity: An Underappreciated Aspect of Oncolytic Virotherapies. Trends Immunol. 2018, 39, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Corns, R.; Mathew, R.K.; Fuller, M.J.; et al. Intravenous Delivery of Oncolytic Reovirus to Brain Tumor Patients Immunologically Primes for Subsequent Checkpoint Blockade. Sci. Transl. Med. 2018, 10, eaam7577. [Google Scholar] [CrossRef]
- Gujar, S.A.; Marcato, P.; Pan, D.; Lee, P.W.K. Reovirus Virotherapy Overrides Tumor Antigen Presentation Evasion and Promotes Protective Antitumor Immunity. Mol. Cancer Ther. 2010, 9, 2924–2933. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, D.; Goel, S.; Aparo, S.; Arora, S.P.; Noronha, N.; Tran, H.; Chakrabarty, R.; Selvaggi, G.; Gutierrez, A.; Coffey, M.; et al. A Phase II Study of Pelareorep (REOLYSIN®) in Combination with Gemcitabine for Patients with Advanced Pancreatic Adenocarcinoma. Cancers 2018, 10, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahalingam, D.; Wilkinson, G.A.; Eng, K.H.; Fields, P.; Raber, P.; Moseley, J.L.; Cheetham, K.; Coffey, M.; Nuovo, G.; Kalinski, P.; et al. Pembrolizumab in Combination with the Oncolytic Virus Pelareorep and Chemotherapy in Patients with Advanced Pancreatic Adenocarcinoma: A Phase Ib Study. Clin. Cancer Res. 2020, 26, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, D. Results Pembrolizumab and Pelareorep in Treating Patients with Advanced Pancreatic Cancer. Available online: https://clinicaltrials.gov/ct2/show/results/NCT03723915 (accessed on 11 June 2021).
- Hiraoka, N.; Onozato, K.; Kosuge, T.; Hirohashi, S. Prevalence of FOXP3+ Regulatory T Cells Increases during the Progression of Pancreatic Ductal Adenocarcinoma and Its Premalignant Lesions. Clin. Cancer Res. 2006, 12, 5423–5434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.; Brehm, C.U.; Gress, T.M.; Buchholz, M.; Alhamwe, B.A.; von Strandmann, E.P.; Slater, E.P.; Bartsch, J.W.; Bauer, C.; Lauth, M. The Immune Microenvironment in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 7307. [Google Scholar] [CrossRef] [PubMed]
- Marshall, L.A.; Marubayashi, S.; Jorapur, A.; Jacobson, S.; Zibinsky, M.; Robles, O.; Hu, D.X.; Jackson, J.J.; Pookot, D.; Sanchez, J.; et al. Tumors Establish Resistance to Immunotherapy by Regulating T Reg Recruitment via CCR4. J. ImmunoTherapy Cancer 2020, 8, e000764. [Google Scholar] [CrossRef] [PubMed]
- Kurose, K.; Ohue, Y.; Wada, H.; Iida, S.; Ishida, T.; Kojima, T.; Doi, T.; Suzuki, S.; Isobe, M.; Funakoshi, T.; et al. Phase Ia Study of FoxP3+ CD4 Treg Depletion by Infusion of a Humanized Anti-CCR4 Antibody, KW-0761, in Cancer Patients. Clin. Cancer Res. 2015, 21, 4327–4336. [Google Scholar] [CrossRef] [Green Version]
- Doi, T.; Muro, K.; Ishii, H.; Kato, T.; Tsushima, T.; Takenoyama, M.; Oizumi, S.; Gemmoto, K.; Suna, H.; Enokitani, K.; et al. A Phase I Study of the Anti-CC Chemokine Receptor 4 Antibody, Mogamulizumab, in Combination with Nivolumab in Patients with Advanced or Metastatic Solid Tumors. Clin. Cancer Res. 2019, 25, 6614–6622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamarin, D.; Hamid, O.; Nayak-Kapoor, A.; Sahebjam, S.; Sznol, M.; Collaku, A.; Fox, F.E.; Marshall, M.A.; Hong, D.S. Mogamulizumab in Combination with Durvalumab or Tremelimumab in Patients with Advanced Solid Tumors: A Phase I Study. Clin. Cancer Res. 2020, 26, 4531–4541. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes with Anti-PD-L1 Immunotherapy in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.D.; Jiang, X.; Sullivan, K.M.; Jalikis, F.G.; Smythe, K.S.; Abbasi, A.; Vignali, M.; Park, J.O.; Daniel, S.K.; Pollack, S.M.; et al. Mobilization of CD8+ T Cells via CXCR4 Blockade Facilitates PD-1 Checkpoint Therapy in Human Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 3934–3945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 Antagonist, in Combination with Pembrolizumab and Chemotherapy for Pancreatic Cancer: The COMBAT Trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.A.; Bever, K.M.; Ho, W.J.; Fertig, E.J.; Niu, N.; Zheng, L.; Parkinson, R.M.; Durham, J.N.; Onners, B.; Ferguson, A.K.; et al. A Phase II Study of Allogeneic GM-CSF–Transfected Pancreatic Tumor Vaccine (GVAX) with Ipilimumab as Maintenance Treatment for Metastatic Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 5129–5139. [Google Scholar] [CrossRef]
- Tsujikawa, T.; Crocenzi, T.; Durham, J.N.; Sugar, E.A.; Wu, A.A.; Onners, B.; Nauroth, J.M.; Anders, R.A.; Fertig, E.J.; Laheru, D.A.; et al. Evaluation of Cyclophosphamide/GVAX Pancreas Followed by Listeria-Mesothelin (CRS-207) with or without Nivolumab in Patients with Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 3578–3588. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, A.C.; Yarchoan, M.; Durham, J.N.; Yusko, E.C.; Rytlewski, J.A.; Robins, H.S.; Laheru, D.A.; Le, D.T.; Lutz, E.R.; Jaffee, E.M. T Cell Receptor Repertoire Features Associated with Survival in Immunotherapy-Treated Pancreatic Ductal Adenocarcinoma. JCI Insight 2018, 3, e122092. [Google Scholar] [CrossRef] [Green Version]
- Vonderheide, R.H. The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell 2018, 33, 563–569. [Google Scholar] [CrossRef] [Green Version]
- Vonderheide, R.H. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020, 71, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy against Pancreatic Carcinoma in Mice and Humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, A.H.; Diamond, M.S.; Hay, C.A.; Byrne, K.T.; Vonderheide, R.H. Sufficiency of CD40 Activation and Immune Checkpoint Blockade for T Cell Priming and Tumor Immunity. Proc. Natl. Acad. Sci. USA 2020, 117, 8022–8031. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, M.H.; O’Reilly, E.M.; Varadhachary, G.; Wolff, R.A.; Wainberg, Z.A.; Ko, A.H.; Fisher, G.; Rahma, O.; Lyman, J.P.; Cabanski, C.R.; et al. CD40 Agonistic Monoclonal Antibody APX005M (Sotigalimab) and Chemotherapy, with or without Nivolumab, for the Treatment of Metastatic Pancreatic Adenocarcinoma: An Open-Label, Multicentre, Phase 1b Study. Lancet Oncol. 2021, 22, 118–131. [Google Scholar] [CrossRef]
- O’Hara, M.H.; O’Reilly, E.M.; Wolff, R.A.; Wainberg, Z.A.; Ko, A.H.; Rahma, O.E.; Fisher, G.A.; Lyman, J.P.; Cabanski, C.R.; Karakunnel, J.J.; et al. Gemcitabine (Gem) and Nab-Paclitaxel (NP) ± Nivolumab (Nivo) ± CD40 Agonistic Monoclonal Antibody APX005M (Sotigalimab), in Patients (Pts) with Untreated Metastatic Pancreatic Adenocarcinoma (MPDAC): Phase (Ph) 2 Final Results. J. Clin. Oncol. 2021, 39, 4019. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid Models of Human and Mouse Ductal Pancreatic Cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holokai, L.; Chakrabarti, J.; Lundy, J.; Croagh, D.; Adhikary, P.; Richards, S.S.; Woodson, C.; Steele, N.; Kuester, R.; Scott, A.; et al. Murine-and Human-Derived Autologous Organoid/Immune Cell Co-Cultures as Pre-Clinical Models of Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 3816. [Google Scholar] [CrossRef]
- Misra, S.; Moro, C.F.; del Chiaro, M.; Pouso, S.; Sebestyén, A.; Löhr, M.; Björnstedt, M.; Verbeke, C.S. Ex Vivo Organotypic Culture System of Precision-Cut Slices of Human Pancreatic Ductal Adenocarcinoma. Sci. Rep. 2019, 9, 2133. [Google Scholar] [CrossRef]
- Sethi, V.; Kurtom, S.; Tarique, M.; Lavania, S.; Malchiodi, Z.; Hellmund, L.; Zhang, L.; Sharma, U.; Giri, B.; Garg, B.; et al. Gut Microbiota Promotes Tumor Growth in Mice by Modulating Immune Response. Gastroenterology 2018, 155, 33–37.e6. [Google Scholar] [CrossRef]
- Pushalkar, S.; Hundeyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Elements of Cancer Immunity and the Cancer-Immune Set Point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.H.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-Exome Sequencing of Pancreatic Cancer Defines Genetic Diversity and Therapeutic Targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual Microdissection Identifies Distinct Tumor- and Stroma-Specific Subtypes of Pancreatic Ductal Adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of Pancreatic Ductal Adenocarcinoma and Their Differing Responses to Therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole Genomes Redefine the Mutational Landscape of Pancreatic Cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamitopoulou, E. Tumour Microenvironment of Pancreatic Cancer: Immune Landscape Is Dictated by Molecular and Histopathological Features. Br. J. Cancer 2019, 121, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Balli, D.; Rech, A.J.; Stanger, B.Z.; Vonderheide, R.H. Immune Cytolytic Activity Stratifies Molecular Subsets of Human Pancreatic Cancer. Clin. Cancer Res. 2017, 23, 3129–3138. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Mechanism | Target | Compound | Combination | Phase | Identifier |
|---|---|---|---|---|---|
| Chemokine antagonism | IL-2 | XB2001 | Irinotecan, 5-FU, Leucovorin | I/II | NCT04825288 |
| IL-6 | Siltuximab | Spartalizumab | I/II | NCT04191421 | |
| CSF1 | ARRY-382 | Pembrolizumab | I/II | NCT02880371 | |
| IL-1β | Canakinumab | Nab-Pacli, Gem, Spartalizumab | I | NCT04581343 | |
| CD73 | CPI-006 | Ciforadenant or Nivolumab | I | NCT03454451 | |
| Immunostimulatory agonism | CD40 | CDX-1140 | CDX-301 | II | NCT04536077 |
| CD40 | ABBV-927 | Budigalimab, mFOLFORINOX | II | NCT04807972 | |
| IL-12 | M9241 | M7824 or M7824, RT | I/II | NCT03849469 | |
| STING | MK-1454 | Pembrolizumab | I | NCT03010176 | |
| Vaccination | Individual TA | RO7198457 | Atezolizumab, mFOLFIRINOX | I | NCT04161755 |
| Multiple Tas | GVAX | CY, Pembrolizumab, IMC-CS4 | Early I | NCT03153410 | |
| Multiple TAs | GVAX | CY or CY, Nivolumab or CY, Nivolumab, Urelumab | I/II | NCT02451982 | |
| Multiple TAs | GVAX | CY, Nivolumab, RT | II | NCT03161379 | |
| Multiple TAs | GVAX | CY, Pembrolizumab, RT | II | NCT02648282 | |
| KRAS | KRAS peptide vaccine | Nivolumab, Ipilimumab | I | NCT04117087 | |
| Individual TA | Personalized neoantigen DNA vaccine | I | NCT03122106 | ||
| Multiple TAs | PANC 10.05 pcDNA-1/GM-Neo and PANC 6.03 pcDNA-1 neo vaccine | Alone or CY i.v. or CY orally | II | NCT01088789 | |
| Ten TAs | OSE2101 | Alone or Nivolumab | II | NCT03806309 | |
| Oncolytic virus | Cancer Cell | OH2 Herpes simplex virus | I/II | NCT04637698 | |
| Cancer Cell | Talimogene laherparepvec | I | NCT03086642 | ||
| Cancer Cell | TBI-1401(HF10) | Nab-Pacli, Gem, S-1 | I | NCT03252808 | |
| Immune checkpoint inhibition | PD-1 | Durvalumab | Plerixafor | II | NCT04177810 |
| PD-1 | Dostarlimab | Niraparib, RT | II | NCT04409002 | |
| PD-1 | Camrelizumab | Gem, MIS-MWA | II | NCT04156087 | |
| PD-1 | Cemiplimab | Nab-Pacli, Gem | II | NCT04498689 | |
| PD-1 | Pembrolizumab | RT | I/II | NCT02305186 | |
| PD-1 | Pembrolizumab | Lenvatinib | II | NCT04887805 | |
| PD-1 | Pembrolizumab | Epacadostat, CRS-207 or Epacadostat, CRS-207, GVAX, CY | II | NCT03006302 | |
| PD-1 | Pembrolizumab | Azacitidine | II | NCT03264404 | |
| PD-1 | Nivolumab | Losartan, FOLFIRINOX, RT | II | NCT03563248 | |
| PD-1 | Nivolumab | IRE | II | NCT04212026 | |
| PD-1 | Nivolumab | None or IRE or IRE, TLR-9 | I | NCT04612530 | |
| PD-1 | Nivolumab | Nab-Pacli, Gem, Paricalcitol | I | NCT03519308 | |
| PD-1 | Nivolumab | Gem, S1 | II | NCT04377048 | |
| PD-1 | Nivolumab | RT or RT, Ipilimumab | II | NCT02866383 | |
| PD-1+CTLA-4 | Nivolumab, Ipilimumab | RT | II | NCT04361162 | |
| PD-1+CTLA-4 | Nivolumab, Ipilimumab | RT, Tocilizumab | II | NCT04258150 | |
| PD-1+CTLA-4 | Nivolumab, Ipilimumab | RT, Nab-Pacli, Gem | I/II | NCT04247165 | |
| PD-1+CTLA-4 | Nivolumab, Ipilimumab | RT | II | NCT03104439 | |
| PD-1+CTLA-4 | Nivolumab, Ipilimumab | CRS-207 or CRS-207, GVAX, CY | II | NCT03190265 | |
| PD-1+CTLA-4 | Nivolumab, Ipilimumab | Maraviroc | I | NCT04721301 | |
| PD-1+CTLA-4 | Durvalumab, Tremelimumab | RT, Gem | II | NCT03572400 | |
| PD-L1 | Atezolizumab | Nab-Pacli, Gem, Selicrelumab or Nab-Pacli, Gem, Bevacizumab or Nab-Pacli, Gem, AB928 or Nab-Pacli, Gem, Tiragolumab or Cobimetinib or PEGPH20 or BL-8040 or RO6874281 or Nab-Pacli, Gem, Tocilizumab | I/II | NCT03193190 | |
| PD-L1 | Atezolizumab | Cabozantinib | II | NCT04820179 | |
| PD-L1+TGF-β | SHR-1701 | Nab-Pacli, Gem | Ib/II | NCT04624217 | |
| ICOS | KY1044 | Alone or Atezolizumab | I/II | NCT03829501 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wandmacher, A.M.; Letsch, A.; Sebens, S. Challenges and Future Perspectives of Immunotherapy in Pancreatic Cancer. Cancers 2021, 13, 4235. https://doi.org/10.3390/cancers13164235
Wandmacher AM, Letsch A, Sebens S. Challenges and Future Perspectives of Immunotherapy in Pancreatic Cancer. Cancers. 2021; 13(16):4235. https://doi.org/10.3390/cancers13164235
Chicago/Turabian StyleWandmacher, Anna Maxi, Anne Letsch, and Susanne Sebens. 2021. "Challenges and Future Perspectives of Immunotherapy in Pancreatic Cancer" Cancers 13, no. 16: 4235. https://doi.org/10.3390/cancers13164235
APA StyleWandmacher, A. M., Letsch, A., & Sebens, S. (2021). Challenges and Future Perspectives of Immunotherapy in Pancreatic Cancer. Cancers, 13(16), 4235. https://doi.org/10.3390/cancers13164235