
Emerging Principles in the Transcriptional Control by YAP and TAZ

{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. YAP/TAZ Are Transcriptional Cofactors
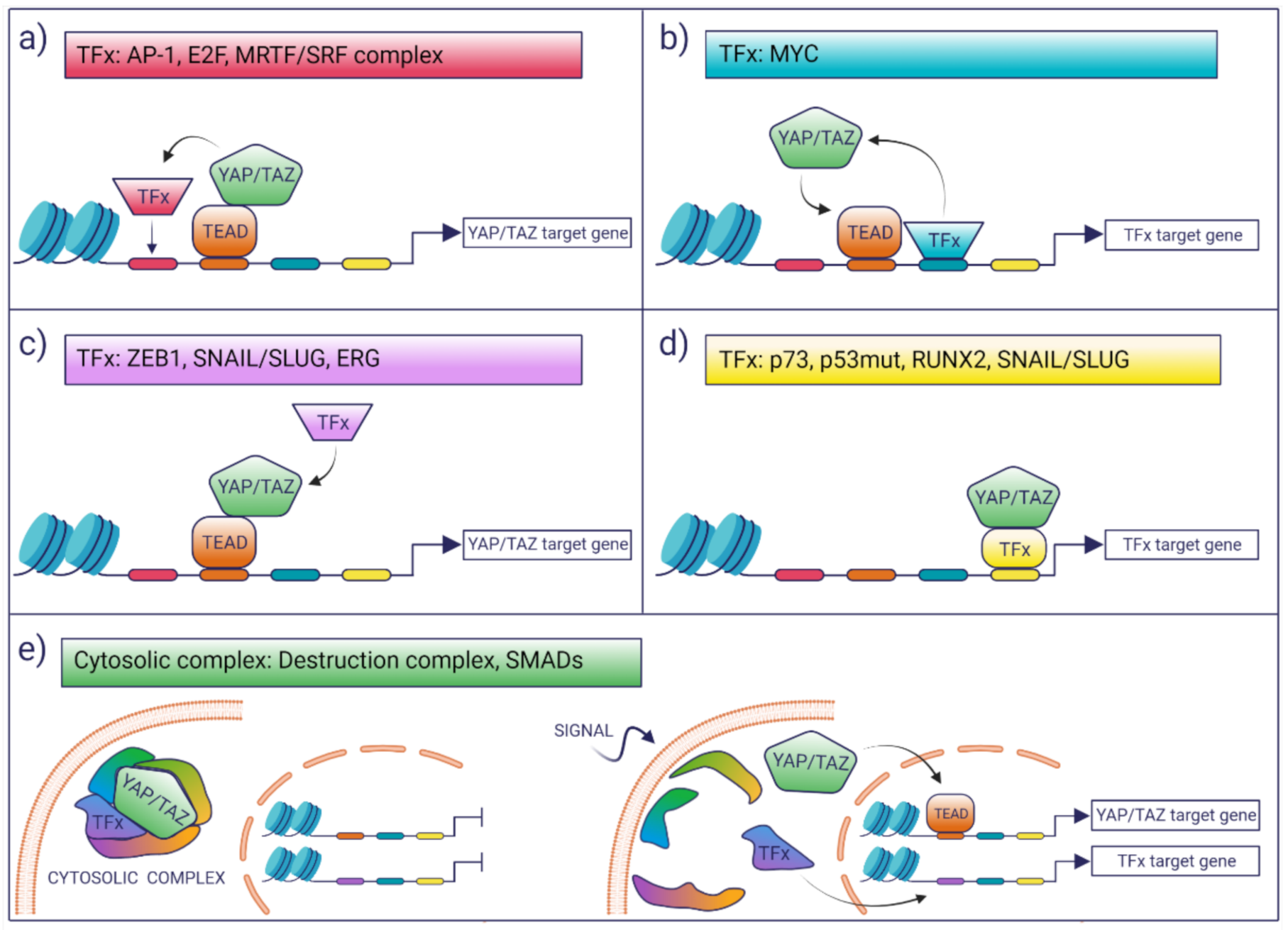
3. Interaction with Other TFs
- (i)
- TFs may modulate the YAP/TAZ-dependent transcriptional landscape by boosting the activation of a subset of potential YAP/TAZ targets. This selection may be dictated in cis by the presence of cognate TF-binding motifs which favor the colocalization of YAP/TAZ with other TFs on proximal and distal regulatory regions, as is the case for AP-1 or MRTF/SRF [43,44].
- (ii)
- TFs may enhance the binding of YAP/TAZ to TEAD loci that would otherwise be low-affinity sites for YAP/TAZ binding. This is the case of MYC target genes, whereby MYC promotes the recruitment of YAP/TAZ on constitutive TEAD-bound loci, which, in the absence of MYC expression, are not bound or regulated by YAP [45].
- (iii)
- Co-operating TFs might directly interact with YAP/TAZ-TEAD and can be recruited to YAP/TAZ-bound loci in a manner that is independent of the presence of their cognate DNA motif, as is the case of ZEB1 [46]. In these latter cases, the open question is what are the factors that account for the selective interaction of these TFs with a subset of YAP/TAZ–TEAD-bound loci. A possibility is that these are mediated only by protein–protein interaction and that the topology or the composition of the chromatin-associated complexes dictates selective association and transcriptional regulation.
- (iv)
- There are a number of examples where YAP/TAZ have been proposed to regulate transcription in a TEAD-independent manner, as is the case of p73, mutant p53, and the regulation of osteogenic programs by RUNX2 and SNAIL/SLUG [47,48,49,50]. In these instances, YAP/TAZ would function as transcriptional modulators of other transcriptional programs.
- (v)
4. YAP/TAZ Regulate Enhancers
5. Epigenetic Regulation
6. Transcriptional Condensates and Phase Separation
7. Hyper-Transcription and Transcriptional Addiction
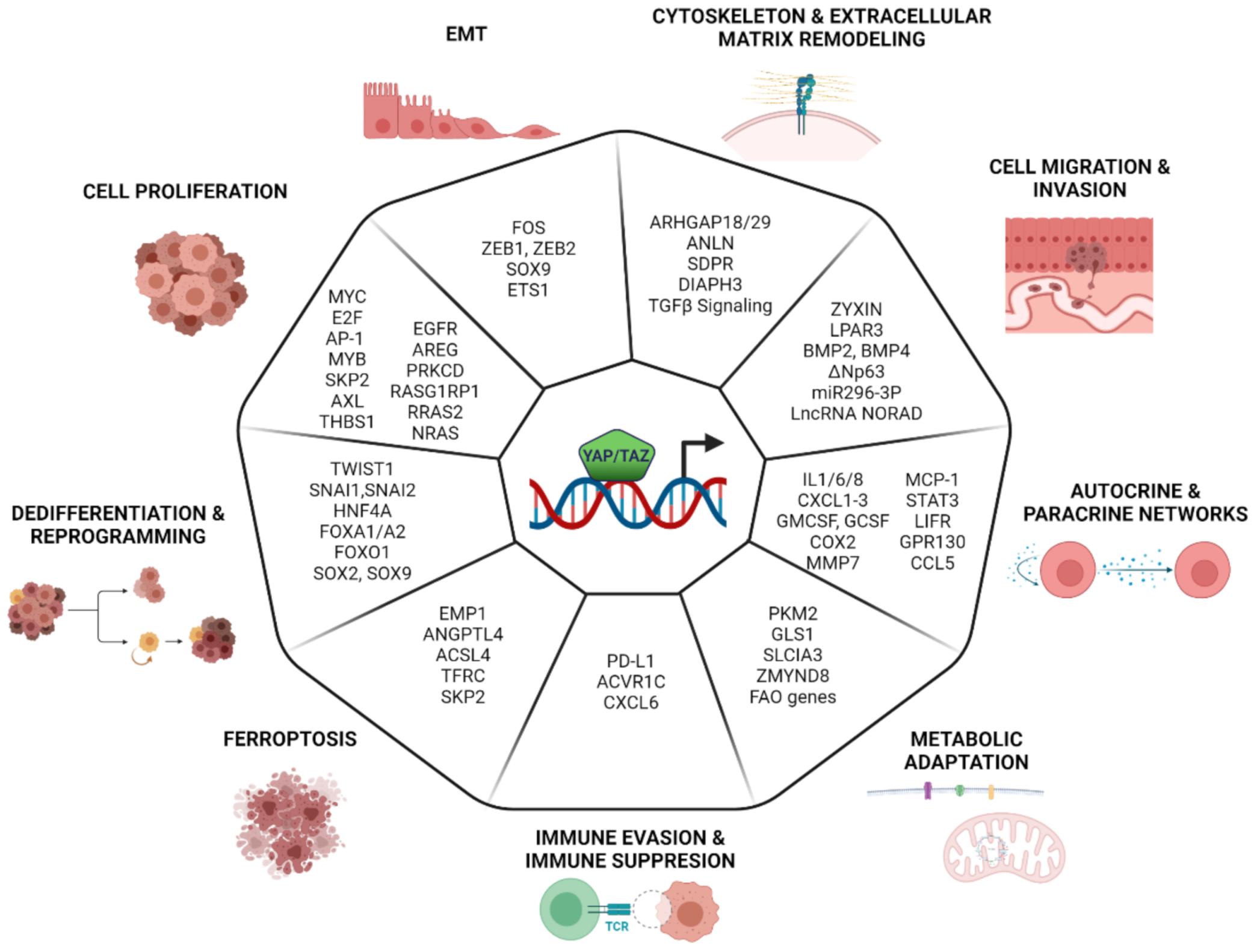
8. Transcriptional Program Controlled by YAP/TAZ in Cancer
8.1. Cell Proliferation
8.2. EMT
8.3. Cytoskeleton and ECM Remodeling
8.4. Cell Migration and Invasion
8.5. Metabolic Adaptation
8.6. Autocrine and Paracrine Signaling
8.7. Immune Evasion and Immune Suppression
8.8. Ferroptosis
8.9. De-Differentiation and Reprogramming
9. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Justice, R.W.; Zilian, O.; Woods, D.F.; Noll, M.; Bryant, P.J. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995, 9, 534–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [Green Version]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Moya, I.M.; Halder, G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol. 2019, 20, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Samavarchi-Tehrani, P.; Narimatsu, M.; Weiss, A.; Cockburn, K.; Larsen, B.G.; Rossant, J.; Wrana, J.L. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-beta-SMAD pathway. Dev. Cell 2010, 19, 831–844. [Google Scholar] [CrossRef]
- Grzeschik, N.A.; Parsons, L.M.; Allott, M.L.; Harvey, K.F.; Richardson, H.E. Lgl, aPKC, and Crumbs regulate the Salvador/Warts/Hippo pathway through two distinct mechanisms. Curr. Biol. 2010, 20, 573–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Huang, J.; Chen, J. Angiomotin-like proteins associate with and negatively regulate YAP1. J. Biol. Chem. 2011, 286, 4364–4370. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Li, L.; Lu, Q.; Wang, L.H.; Liu, C.Y.; Lei, Q.; Guan, K.L. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011, 25, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Yin, F.; Yu, J.; Zheng, Y.; Chen, Q.; Zhang, N.; Pan, D. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell 2013, 154, 1342–1355. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Zheng, Y.; Dong, J.; Klusza, S.; Deng, W.M.; Pan, D. Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Dev. Cell 2010, 18, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef]
- Martin-Belmonte, F.; Perez-Moreno, M. Epithelial cell polarity, stem cells and cancer. Nat. Rev. Cancer 2011, 12, 23–38. [Google Scholar] [CrossRef]
- Bennett, F.C.; Harvey, K.F. Fat cadherin modulates organ size in Drosophila via the Salvador/Warts/Hippo signaling pathway. Curr. Biol. 2006, 16, 2101–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matakatsu, H.; Blair, S.S. Separating planar cell polarity and Hippo pathway activities of the protocadherins Fat and Dachsous. Development 2012, 139, 1498–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.X.; Guan, K.L. The Hippo pathway: Regulators and regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [Green Version]
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A tense situation: Forcing tumour progression. Nat. Rev. Cancer 2009, 9, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Wang, L.; Luo, J.Y.; Li, B.; Tian, X.Y.; Chen, L.J.; Huang, Y.; Liu, J.; Deng, D.; Lau, C.W.; Wan, S.; et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 2016, 540, 579–582. [Google Scholar] [CrossRef]
- Sansores-Garcia, L.; Bossuyt, W.; Wada, K.; Yonemura, S.; Tao, C.; Sasaki, H.; Halder, G. Modulating F-actin organization induces organ growth by affecting the Hippo pathway. EMBO J. 2011, 30, 2325–2335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Maitra, A.; Anders, R.A.; Taketo, M.M.; Pan, D. beta-Catenin destruction complex-independent regulation of Hippo-YAP signaling by APC in intestinal tumorigenesis. Genes Dev. 2015, 29, 1493–1506. [Google Scholar] [CrossRef] [Green Version]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.X.; Alexander, C.M.; et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Liu, C.Y.; Zha, Z.Y.; Zhao, B.; Yao, J.; Zhao, S.; Xiong, Y.; Lei, Q.Y.; Guan, K.L. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J. Biol. Chem. 2009, 284, 13355–13362. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [Green Version]
- Vassilev, A.; Kaneko, K.J.; Shu, H.; Zhao, Y.; DePamphilis, M.L. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 2001, 15, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Mahoney, W.M., Jr.; Hong, J.H.; Yaffe, M.B.; Farrance, I.K. The transcriptional co-activator TAZ interacts differentially with transcriptional enhancer factor-1 (TEF-1) family members. Biochem. J. 2005, 388, 217–225. [Google Scholar] [CrossRef]
- Kim, M.K.; Jang, J.W.; Bae, S.C. DNA binding partners of YAP/TAZ. BMB Rep. 2018, 51, 126–133. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.C.; Park, H.W.; Guan, K.L. Regulation of the Hippo Pathway Transcription Factor TEAD. Trends Biochem. Sci. 2017, 42, 862–872. [Google Scholar] [CrossRef]
- Huh, H.D.; Kim, D.H.; Jeong, H.S.; Park, H.W. Regulation of TEAD Transcription Factors in Cancer Biology. Cells 2019, 8, 600. [Google Scholar] [CrossRef] [Green Version]
- Jacquemin, P.; Hwang, J.J.; Martial, J.A.; Dolle, P.; Davidson, I. A novel family of developmentally regulated mammalian transcription factors containing the TEA/ATTS DNA binding domain. J. Biol. Chem. 1996, 271, 21775–21785. [Google Scholar] [CrossRef] [Green Version]
- Koontz, L.M.; Liu-Chittenden, Y.; Yin, F.; Zheng, Y.; Yu, J.; Huang, B.; Chen, Q.; Wu, S.; Pan, D. The Hippo effector Yorkie controls normal tissue growth by antagonizing scalloped-mediated default repression. Dev. Cell 2013, 25, 388–401. [Google Scholar] [CrossRef] [Green Version]
- Stein, C.; Bardet, A.F.; Roma, G.; Bergling, S.; Clay, I.; Ruchti, A.; Agarinis, C.; Schmelzle, T.; Bouwmeester, T.; Schubeler, D.; et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet. 2015, 11, e1005465. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; Di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610. [Google Scholar] [CrossRef]
- Oh, H.; Slattery, M.; Ma, L.; Crofts, A.; White, K.P.; Mann, R.S.; Irvine, K.D. Genome-wide association of Yorkie with chromatin and chromatin-remodeling complexes. Cell Rep. 2013, 3, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Galli, G.G.; Carrara, M.; Yuan, W.C.; Valdes-Quezada, C.; Gurung, B.; Pepe-Mooney, B.; Zhang, T.; Geeven, G.; Gray, N.S.; de Laat, W.; et al. YAP Drives Growth by Controlling Transcriptional Pause Release from Dynamic Enhancers. Mol. Cell 2015, 60, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Tropberger, P.; Pott, S.; Keller, C.; Kamieniarz-Gdula, K.; Caron, M.; Richter, F.; Li, G.; Mittler, G.; Liu, E.T.; Buhler, M.; et al. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell 2013, 152, 859–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.S.; Li, S.; Wang, X.; Zhu, J.; Zhuo, S.; Han, Y.; Yue, T.; Yang, Y.; Jiang, J. CDK7 regulates organ size and tumor growth by safeguarding the Hippo pathway effector Yki/Yap/Taz in the nucleus. Genes Dev. 2020, 34, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Estaras, C.; Benner, C.; Jones, K.A. SMADs and YAP compete to control elongation of beta-catenin:LEF-1-recruited RNAPII during hESC differentiation. Mol. Cell 2015, 58, 780–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef]
- Liu, X.; Li, H.; Rajurkar, M.; Li, Q.; Cotton, J.L.; Ou, J.; Zhu, L.J.; Goel, H.L.; Mercurio, A.M.; Park, J.S.; et al. Tead and AP1 Coordinate Transcription and Motility. Cell Rep. 2016, 14, 1169–1180. [Google Scholar] [CrossRef] [Green Version]
- Croci, O.; De Fazio, S.; Biagioni, F.; Donato, E.; Caganova, M.; Curti, L.; Doni, M.; Sberna, S.; Aldeghi, D.; Biancotto, C.; et al. Transcriptional integration of mitogenic and mechanical signals by Myc and YAP. Genes Dev. 2017, 31, 2017–2022. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, W.; Mossmann, D.; Kleemann, J.; Mock, K.; Meisinger, C.; Brummer, T.; Herr, R.; Brabletz, S.; Stemmler, M.P.; Brabletz, T. ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat. Commun. 2016, 7, 10498. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Lin, S.J.; Chen, Y.; Voon, D.C.; Zhu, F.; Chuang, L.S.; Wang, T.; Tan, P.; Lee, S.C.; Yeoh, K.G.; et al. RUNX3 is a novel negative regulator of oncogenic TEAD-YAP complex in gastric cancer. Oncogene 2016, 35, 2664–2674. [Google Scholar] [CrossRef]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell Death Differ. 2007, 14, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Di Agostino, S.; Sorrentino, G.; Ingallina, E.; Valenti, F.; Ferraiuolo, M.; Bicciato, S.; Piazza, S.; Strano, S.; Del Sal, G.; Blandino, G. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. 2016, 17, 188–201. [Google Scholar] [CrossRef]
- Tang, Y.; Feinberg, T.; Keller, E.T.; Li, X.Y.; Weiss, S.J. Snail/Slug binding interactions with YAP/TAZ control skeletal stem cell self-renewal and differentiation. Nat. Cell Biol. 2016, 18, 917–929. [Google Scholar] [CrossRef]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, J.H.; Plouffe, S.W.; Meng, Z.; Lee, D.H.; Yang, D.; Lim, D.S.; Wang, C.Y.; Guan, K.L. Induction of AP-1 by YAP/TAZ contributes to cell proliferation and organ growth. Genes Dev. 2020, 34, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Maglic, D.; Schlegelmilch, K.; Dost, A.F.; Panero, R.; Dill, M.T.; Calogero, R.A.; Camargo, F.D. YAP-TEAD signaling promotes basal cell carcinoma development via a c-JUN/AP1 axis. EMBO J. 2018, 37, e98642. [Google Scholar] [CrossRef]
- Vierbuchen, T.; Ling, E.; Cowley, C.J.; Couch, C.H.; Wang, X.; Harmin, D.A.; Roberts, C.W.M.; Greenberg, M.E. AP-1 Transcription Factors and the BAF Complex Mediate Signal-Dependent Enhancer Selection. Mol. Cell 2017, 68, 1067–1082.e12. [Google Scholar] [CrossRef] [Green Version]
- Nicolay, B.N.; Bayarmagnai, B.; Islam, A.B.; Lopez-Bigas, N.; Frolov, M.V. Cooperation between dE2F1 and Yki/Sd defines a distinct transcriptional program necessary to bypass cell cycle exit. Genes Dev. 2011, 25, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Sabo, A.; Kress, T.R.; Pelizzola, M.; de Pretis, S.; Gorski, M.M.; Tesi, A.; Morelli, M.J.; Bora, P.; Doni, M.; Verrecchia, A.; et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014, 511, 488–492. [Google Scholar] [CrossRef]
- Walz, S.; Lorenzin, F.; Morton, J.; Wiese, K.E.; von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014, 511, 483–487. [Google Scholar] [CrossRef]
- Pattschull, G.; Walz, S.; Grundl, M.; Schwab, M.; Ruhl, E.; Baluapuri, A.; Cindric-Vranesic, A.; Kneitz, S.; Wolf, E.; Ade, C.P.; et al. The Myb-MuvB Complex Is Required for YAP-Dependent Transcription of Mitotic Genes. Cell Rep. 2019, 27, 3533–3546.e3537. [Google Scholar] [CrossRef] [Green Version]
- Foster, C.T.; Gualdrini, F.; Treisman, R. Mutual dependence of the MRTF-SRF and YAP-TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev. 2017, 31, 2361–2375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.; Hwang, D.; Lee, D.; Kim, J.H.; Kim, S.Y.; Lim, D.S. MRTF potentiates TEAD-YAP transcriptional activity causing metastasis. EMBO J. 2017, 36, 520–535. [Google Scholar] [CrossRef] [Green Version]
- Passaniti, A.; Brusgard, J.L.; Qiao, Y.; Sudol, M.; Finch-Edmondson, M. Roles of RUNX in Hippo Pathway Signaling. Adv. Exp. Med. Biol. 2017, 962, 435–448. [Google Scholar] [CrossRef]
- Hong, J.H.; Hwang, E.S.; McManus, M.T.; Amsterdam, A.; Tian, Y.; Kalmukova, R.; Mueller, E.; Benjamin, T.; Spiegelman, B.M.; Sharp, P.A.; et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 2005, 309, 1074–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitolo, M.I.; Anglin, I.E.; Mahoney, W.M., Jr.; Renoud, K.J.; Gartenhaus, R.B.; Bachman, K.E.; Passaniti, A. The RUNX2 transcription factor cooperates with the YES-associated protein, YAP65, to promote cell transformation. Cancer Biol. Ther. 2007, 6, 856–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elster, D.; Tollot, M.; Schlegelmilch, K.; Ori, A.; Rosenwald, A.; Sahai, E.; von Eyss, B. TRPS1 shapes YAP/TEAD-dependent transcription in breast cancer cells. Nat. Commun. 2018, 9, 3115. [Google Scholar] [CrossRef] [Green Version]
- Feldker, N.; Ferrazzi, F.; Schuhwerk, H.; Widholz, S.A.; Guenther, K.; Frisch, I.; Jakob, K.; Kleemann, J.; Riegel, D.; Bonisch, U.; et al. Genome-wide cooperation of EMT transcription factor ZEB1 with YAP and AP-1 in breast cancer. EMBO J. 2020, 39, e103209. [Google Scholar] [CrossRef] [PubMed]
- Rayon, T.; Menchero, S.; Nieto, A.; Xenopoulos, P.; Crespo, M.; Cockburn, K.; Canon, S.; Sasaki, H.; Hadjantonakis, A.K.; de la Pompa, J.L.; et al. Notch and hippo converge on Cdx2 to specify the trophectoderm lineage in the mouse blastocyst. Dev. Cell 2014, 30, 410–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manderfield, L.J.; Aghajanian, H.; Engleka, K.A.; Lim, L.Y.; Liu, F.; Jain, R.; Li, L.; Olson, E.N.; Epstein, J.A. Hippo signaling is required for Notch-dependent smooth muscle differentiation of neural crest. Development 2015, 142, 2962–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komuro, A.; Nagai, M.; Navin, N.E.; Sudol, M. WW domain-containing protein YAP associates with ErbB-4 and acts as a co-transcriptional activator for the carboxyl-terminal fragment of ErbB-4 that translocates to the nucleus. J. Biol. Chem. 2003, 278, 33334–33341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haskins, J.W.; Nguyen, D.X.; Stern, D.F. Neuregulin 1-activated ERBB4 interacts with YAP to induce Hippo pathway target genes and promote cell migration. Sci. Signal. 2014, 7, ra116. [Google Scholar] [CrossRef] [Green Version]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, M.; Nakagawa, M.; Olson, E.N.; Nakagawa, O. A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt-Oram syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 18034–18039. [Google Scholar] [CrossRef] [Green Version]
- Beyer, T.A.; Weiss, A.; Khomchuk, Y.; Huang, K.; Ogunjimi, A.A.; Varelas, X.; Wrana, J.L. Switch enhancers interpret TGF-beta and Hippo signaling to control cell fate in human embryonic stem cells. Cell Rep. 2013, 5, 1611–1624. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Holle, A.; Li, J.; Posa, F.; Biagioni, F.; Croci, O.; Benk, A.S.; Young, J.; Noureddine, F.; Deng, J.; et al. BMP-2 Signaling and Mechanotransduction Synergize to Drive Osteogenic Differentiation via YAP/TAZ. Adv. Sci. 2020, 7, 1902931. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Tretiakova, M.S.; Silvis, M.R.; Lucas, J.; Klezovitch, O.; Coleman, I.; Bolouri, H.; Kutyavin, V.I.; Morrissey, C.; True, L.D.; et al. ERG Activates the YAP1 Transcriptional Program and Induces the Development of Age-Related Prostate Tumors. Cancer Cell 2015, 27, 797–808. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Zhai, P.; Del Re, D.P.; Sciarretta, S.; Yabuta, N.; Nojima, H.; Lim, D.S.; Pan, D.; Sadoshima, J. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nat. Commun. 2014, 5, 3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellan, M.; Guarnieri, A.; Fujimura, A.; Zanconato, F.; Battilana, G.; Panciera, T.; Sladitschek, H.L.; Contessotto, P.; Citron, A.; Grilli, A.; et al. Single-cell analyses reveal YAP/TAZ as regulators of stemness and cell plasticity in Glioblastoma. Nat. Cancer 2021, 2, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Tao, G.; Kahr, P.C.; Morikawa, Y.; Zhang, M.; Rahmani, M.; Heallen, T.R.; Li, L.; Sun, Z.; Olson, E.N.; Amendt, B.A.; et al. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature 2016, 534, 119–123. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, Y.; Yuan, F.; Huang, J.; Xu, W.; Mao, B.; Yuan, Z.; Bi, W. TNFalpha-YAP/p65-HK2 axis mediates breast cancer cell migration. Oncogenesis 2017, 6, e383. [Google Scholar] [CrossRef] [Green Version]
- Chokas, A.L.; Bickford, J.S.; Barilovits, S.J.; Rogers, R.J.; Qiu, X.; Newsom, K.J.; Beachy, D.E.; Nick, H.S. A TEAD1/p65 complex regulates the eutherian-conserved MnSOD intronic enhancer, eRNA transcription and the innate immune response. Biochim. Biophys. Acta 2014, 1839, 1205–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottini, F.; Hideshima, T.; Xu, C.; Sattler, M.; Dori, M.; Agnelli, L.; ten Hacken, E.; Bertilaccio, M.T.; Antonini, E.; Neri, A.; et al. Rescue of Hippo coactivator YAP1 triggers DNA damage-induced apoptosis in hematological cancers. Nat. Med. 2014, 20, 599–606. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol. Cell 2008, 29, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Lapi, E.; Di Agostino, S.; Donzelli, S.; Gal, H.; Domany, E.; Rechavi, G.; Pandolfi, P.P.; Givol, D.; Strano, S.; Lu, X.; et al. PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Mol. Cell 2008, 32, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Lian, I.; Kim, J.; Okazawa, H.; Zhao, J.; Zhao, B.; Yu, J.; Chinnaiyan, A.; Israel, M.A.; Goldstein, L.S.; Abujarour, R.; et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010, 24, 1106–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Ugalde, A.P.; Scheele, C.; Dieter, S.M.; Nagel, R.; Ma, J.; Pataskar, A.; Korkmaz, G.; Elkon, R.; Chien, M.P.; et al. A comprehensive enhancer screen identifies TRAM2 as a key and novel mediator of YAP oncogenesis. Genome Biol. 2021, 22, 54. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Azambuja, A.P.; Simoes-Costa, M. Metabolic Reprogramming Promotes Neural Crest Migration via Yap/Tead Signaling. Dev. Cell 2020, 53, 199–211.e196. [Google Scholar] [CrossRef]
- Long, H.K.; Prescott, S.L.; Wysocka, J. Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 2016, 167, 1170–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinz, S.; Romanoski, C.E.; Benner, C.; Glass, C.K. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 2015, 16, 144–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cebola, I.; Rodriguez-Segui, S.A.; Cho, C.H.; Bessa, J.; Rovira, M.; Luengo, M.; Chhatriwala, M.; Berry, A.; Ponsa-Cobas, J.; Maestro, M.A.; et al. TEAD and YAP regulate the enhancer network of human embryonic pancreatic progenitors. Nat. Cell Biol. 2015, 17, 615–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alder, O.; Cullum, R.; Lee, S.; Kan, A.C.; Wei, W.; Yi, Y.; Garside, V.C.; Bilenky, M.; Griffith, M.; Morrissy, A.S.; et al. Hippo signaling influences HNF4A and FOXA2 enhancer switching during hepatocyte differentiation. Cell Rep. 2014, 9, 261–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monroe, T.O.; Hill, M.C.; Morikawa, Y.; Leach, J.P.; Heallen, T.; Cao, S.; Krijger, P.H.L.; de Laat, W.; Wehrens, X.H.T.; Rodney, G.G.; et al. YAP Partially Reprograms Chromatin Accessibility to Directly Induce Adult Cardiogenesis In Vivo. Dev. Cell 2019, 48, 765–779.e767. [Google Scholar] [CrossRef] [Green Version]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef] [Green Version]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Chapuy, B.; McKeown, M.R.; Lin, C.Y.; Monti, S.; Roemer, M.G.; Qi, J.; Rahl, P.B.; Sun, H.H.; Yeda, K.T.; Doench, J.G.; et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 2013, 24, 777–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [Green Version]
- Della Chiara, G.; Gervasoni, F.; Fakiola, M.; Godano, C.; D’Oria, C.; Azzolin, L.; Bonnal, R.J.P.; Moreni, G.; Drufuca, L.; Rossetti, G.; et al. Epigenomic landscape of human colorectal cancer unveils an aberrant core of pan-cancer enhancers orchestrated by YAP/TAZ. Nat. Commun. 2021, 12, 2340. [Google Scholar] [CrossRef]
- Hillmer, R.E.; Link, B.A. The Roles of Hippo Signaling Transducers Yap and Taz in Chromatin Remodeling. Cells 2019, 8, 502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadoch, C.; Crabtree, G.R. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 2015, 1, e1500447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Xu, J.; Yin, M.X.; Lu, Y.; Hu, L.; Li, P.; Zhang, P.; Yuan, Z.; Ho, M.S.; Ji, H.; et al. Brahma is essential for Drosophila intestinal stem cell proliferation and regulated by Hippo signaling. eLife 2013, 2, e00999. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, D.; Wang, Y.; Pei, C.; Liu, S.; Zhang, L.; Yuan, Z.; Zhang, P. Brahma regulates the Hippo pathway activity through forming complex with Yki-Sd and regulating the transcription of Crumbs. Cell Signal. 2015, 27, 606–613. [Google Scholar] [CrossRef] [Green Version]
- Skibinski, A.; Breindel, J.L.; Prat, A.; Galvan, P.; Smith, E.; Rolfs, A.; Gupta, P.B.; LaBaer, J.; Kuperwasser, C. The Hippo transducer TAZ interacts with the SWI/SNF complex to regulate breast epithelial lineage commitment. Cell Rep. 2014, 6, 1059–1072. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Azzolin, L.; Di Biagio, D.; Zanconato, F.; Battilana, G.; Lucon Xiccato, R.; Aragona, M.; Giulitti, S.; Panciera, T.; Gandin, A.; et al. The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature 2018, 563, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Saladi, S.V.; Ross, K.; Karaayvaz, M.; Tata, P.R.; Mou, H.; Rajagopal, J.; Ramaswamy, S.; Ellisen, L.W. ACTL6A Is Co-Amplified with p63 in Squamous Cell Carcinoma to Drive YAP Activation, Regenerative Proliferation, and Poor Prognosis. Cancer Cell 2017, 31, 35–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strano, S.; Monti, O.; Pediconi, N.; Baccarini, A.; Fontemaggi, G.; Lapi, E.; Mantovani, F.; Damalas, A.; Citro, G.; Sacchi, A.; et al. The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA Damage. Mol. Cell 2005, 18, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tu, K.; Liu, D.; Guo, L.; Chen, Y.; Li, Q.; Maiers, J.L.; Liu, Z.; Shah, V.H.; Dou, C.; et al. p300 Acetyltransferase Is a Cytoplasm-to-Nucleus Shuttle for SMAD2/3 and TAZ Nuclear Transport in Transforming Growth Factor beta-Stimulated Hepatic Stellate Cells. Hepatology 2019, 70, 1409–1423. [Google Scholar] [CrossRef]
- Song, S.; Li, Y.; Xu, Y.; Ma, L.; Pool Pizzi, M.; Jin, J.; Scott, A.W.; Huo, L.; Wang, Y.; Lee, J.H.; et al. Targeting Hippo coactivator YAP1 through BET bromodomain inhibition in esophageal adenocarcinoma. Mol. Oncol. 2020, 14, 1410–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devaiah, B.N.; Case-Borden, C.; Gegonne, A.; Hsu, C.H.; Chen, Q.; Meerzaman, D.; Dey, A.; Ozato, K.; Singer, D.S. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat. Struct. Mol. Biol. 2016, 23, 540–548. [Google Scholar] [CrossRef] [Green Version]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Shilatifard, A. The COMPASS family of histone H3K4 methylases: Mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012, 81, 65–95. [Google Scholar] [CrossRef] [Green Version]
- Ardehali, M.B.; Mei, A.; Zobeck, K.L.; Caron, M.; Lis, J.T.; Kusch, T. Drosophila Set1 is the major histone H3 lysine 4 trimethyltransferase with role in transcription. EMBO J. 2011, 30, 2817–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herz, H.M.; Mohan, M.; Garruss, A.S.; Liang, K.; Takahashi, Y.H.; Mickey, K.; Voets, O.; Verrijzer, C.P.; Shilatifard, A. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev. 2012, 26, 2604–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.; Slattery, M.; Ma, L.; White, K.P.; Mann, R.S.; Irvine, K.D. Yorkie promotes transcription by recruiting a histone methyltransferase complex. Cell Rep. 2014, 8, 449–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qing, Y.; Yin, F.; Wang, W.; Zheng, Y.; Guo, P.; Schozer, F.; Deng, H.; Pan, D. The Hippo effector Yorkie activates transcription by interacting with a histone methyltransferase complex through Ncoa6. eLife 2014, 3, e02564. [Google Scholar] [CrossRef]
- Kim, M.; Kim, T.; Johnson, R.L.; Lim, D.S. Transcriptional co-repressor function of the hippo pathway transducers YAP and TAZ. Cell Rep. 2015, 11, 270–282. [Google Scholar] [CrossRef] [Green Version]
- Torchy, M.P.; Hamiche, A.; Klaholz, B.P. Structure and function insights into the NuRD chromatin remodeling complex. Cell Mol. Life Sci. 2015, 72, 2491–2507. [Google Scholar] [CrossRef]
- Xue, Y.; Wong, J.; Moreno, G.T.; Young, M.K.; Côté, J.; Wang, W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol. Cell 1998, 2, 851–861. [Google Scholar] [CrossRef]
- Cai, D.; Feliciano, D.; Dong, P.; Flores, E.; Gruebele, M.; Porat-Shliom, N.; Sukenik, S.; Liu, Z.; Lippincott-Schwartz, J. Phase separation of YAP reorganizes genome topology for long-term YAP target gene expression. Nat. Cell Biol. 2019, 21, 1578–1589. [Google Scholar] [CrossRef]
- Lu, Y.; Wu, T.; Gutman, O.; Lu, H.; Zhou, Q.; Henis, Y.I.; Luo, K. Phase separation of TAZ compartmentalizes the transcription machinery to promote gene expression. Nat. Cell Biol. 2020, 22, 453–464. [Google Scholar] [CrossRef]
- Guzman-Ayala, M.; Sachs, M.; Koh, F.M.; Onodera, C.; Bulut-Karslioglu, A.; Lin, C.J.; Wong, P.; Nitta, R.; Song, J.S.; Ramalho-Santos, M. Chd1 is essential for the high transcriptional output and rapid growth of the mouse epiblast. Development 2015, 142, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Koh, F.M.; Lizama, C.O.; Wong, P.; Hawkins, J.S.; Zovein, A.C.; Ramalho-Santos, M. Emergence of hematopoietic stem and progenitor cells involves a Chd1-dependent increase in total nascent transcription. Proc. Natl. Acad. Sci. USA 2015, 112, E1734–E1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Percharde, M.; Bulut-Karslioglu, A.; Ramalho-Santos, M. Hypertranscription in Development, Stem Cells, and Regeneration. Dev. Cell 2017, 40, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorthi, A.; Romero, J.C.; Loranc, E.; Cao, L.; Lawrence, L.A.; Goodale, E.; Iniguez, A.B.; Bernard, X.; Masamsetti, V.P.; Roston, S.; et al. EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature 2018, 555, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Bowry, A.; Kelly, R.D.W.; Petermann, E. Hypertranscription and replication stress in cancer. Trends Cancer 2021, 7, 863–877. [Google Scholar] [CrossRef]
- Sun, C.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.; Vellano, C.P.; Ju, Z.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018, 33, 401–416.e408. [Google Scholar] [CrossRef] [Green Version]
- Lavado, A.; Park, J.Y.; Pare, J.; Finkelstein, D.; Pan, H.; Xu, B.; Fan, Y.; Kumar, R.P.; Neale, G.; Kwak, Y.D.; et al. The Hippo Pathway Prevents YAP/TAZ-Driven Hypertranscription and Controls Neural Progenitor Number. Dev. Cell 2018, 47, 576–591.e578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.H.; Park, J.O.; Kim, T.S.; Kim, S.K.; Kim, T.H.; Kim, M.C.; Park, G.S.; Kim, J.H.; Kuninaka, S.; Olson, E.N.; et al. LATS-YAP/TAZ controls lineage specification by regulating TGFbeta signaling and Hnf4alpha expression during liver development. Nat. Commun. 2016, 7, 11961. [Google Scholar] [CrossRef]
- Halder, G.; Camargo, F.D. The hippo tumor suppressor network: From organ size control to stem cells and cancer. Cancer Res. 2013, 73, 6389–6392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, W.; Guan, K.L. The YAP and TAZ transcription co-activators: Key downstream effectors of the mammalian Hippo pathway. Semin. Cell Dev. Biol. 2012, 23, 785–793. [Google Scholar] [CrossRef] [Green Version]
- Moroishi, T.; Hansen, C.G.; Guan, K.L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [Green Version]
- Grieve, S.; Wajnberg, G.; Lees, M.; Chacko, S.; Weir, J.; Crapoulet, N.; Reiman, T. TAZ functions as a tumor suppressor in multiple myeloma by downregulating MYC. Blood Adv. 2019, 3, 3613–3625. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.; Xiol, J.; Dill, M.T.; Yuan, W.C.; Panero, R.; Roper, J.; Osorio, F.G.; Maglic, D.; Li, Q.; Gurung, B.; et al. Regenerative Reprogramming of the Intestinal Stem Cell State via Hippo Signaling Suppresses Metastatic Colorectal Cancer. Cell Stem Cell 2020, 27, 590–604.e599. [Google Scholar] [CrossRef] [PubMed]
- Ehmer, U.; Zmoos, A.F.; Auerbach, R.K.; Vaka, D.; Butte, A.J.; Kay, M.A.; Sage, J. Organ size control is dominant over Rb family inactivation to restrict proliferation in vivo. Cell Rep. 2014, 8, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Fitamant, J.; Kottakis, F.; Benhamouche, S.; Tian, H.S.; Chuvin, N.; Parachoniak, C.A.; Nagle, J.M.; Perera, R.M.; Lapouge, M.; Deshpande, V.; et al. YAP Inhibition Restores Hepatocyte Differentiation in Advanced HCC, Leading to Tumor Regression. Cell Rep. 2015, 10, 1692–1707. [Google Scholar] [CrossRef] [Green Version]
- Choi, W.; Kim, J.; Park, J.; Lee, D.H.; Hwang, D.; Kim, J.H.; Ashktorab, H.; Smoot, D.; Kim, S.Y.; Choi, C.; et al. YAP/TAZ Initiates Gastric Tumorigenesis via Upregulation of MYC. Cancer Res. 2018, 78, 3306–3320. [Google Scholar] [CrossRef] [Green Version]
- Jang, W.; Kim, T.; Koo, J.S.; Kim, S.K.; Lim, D.S. Mechanical cue-induced YAP instructs Skp2-dependent cell cycle exit and oncogenic signaling. EMBO J. 2017, 36, 2510–2528. [Google Scholar] [CrossRef]
- Zhang, J.; Ji, J.Y.; Yu, M.; Overholtzer, M.; Smolen, G.A.; Wang, R.; Brugge, J.S.; Dyson, N.J.; Haber, D.A. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat. Cell Biol. 2009, 11, 1444–1450. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Z.; Chan, S.W.; Liu, A.M.; Wong, K.F.; Fan, S.T.; Chen, J.; Poon, R.T.; Zender, L.; Lowe, S.W.; Hong, W.; et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene 2011, 30, 1229–1240. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yang, L.; Szeto, P.; Abali, G.K.; Zhang, Y.; Kulkarni, A.; Amarasinghe, K.; Li, J.; Vergara, I.A.; Molania, R.; et al. The Hippo pathway oncoprotein YAP promotes melanoma cell invasion and spontaneous metastasis. Oncogene 2020, 39, 5267–5281. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rendueles, M.E.; Ricarte-Filho, J.C.; Untch, B.R.; Landa, I.; Knauf, J.A.; Voza, F.; Smith, V.E.; Ganly, I.; Taylor, B.S.; Persaud, Y.; et al. NF2 Loss Promotes Oncogenic RAS-Induced Thyroid Cancers via YAP-Dependent Transactivation of RAS Proteins and Sensitizes Them to MEK Inhibition. Cancer Discov. 2015, 5, 1178–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Li, Q.; Dang, K.; Ma, S.; Cotton, J.L.; Yang, S.; Zhu, L.J.; Deng, A.C.; Ip, Y.T.; Johnson, R.L.; et al. YAP/TAZ Activation Drives Uveal Melanoma Initiation and Progression. Cell Rep. 2019, 29, 3200–3211.e3204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overholtzer, M.; Zhang, J.; Smolen, G.A.; Muir, B.; Li, W.; Sgroi, D.C.; Deng, C.X.; Brugge, J.S.; Haber, D.A. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. USA 2006, 103, 12405–12410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Q.Y.; Zhang, H.; Zhao, B.; Zha, Z.Y.; Bai, F.; Pei, X.H.; Zhao, S.; Xiong, Y.; Guan, K.L. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol. Cell Biol. 2008, 28, 2426–2436. [Google Scholar] [CrossRef] [Green Version]
- Xie, D.; Cui, J.; Xia, T.; Jia, Z.; Wang, L.; Wei, W.; Zhu, A.; Gao, Y.; Xie, K.; Quan, M. Hippo transducer TAZ promotes epithelial mesenchymal transition and supports pancreatic cancer progression. Oncotarget 2015, 6, 35949–35963. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Mauri, F.; Song, Y.; de Cock, F.; Meeusen, B.; Swedlund, B.; Impens, F.; Van Haver, D.; Opitz, M.; Thery, M.; et al. Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature 2021, 589, 448–455. [Google Scholar] [CrossRef]
- Dasgupta, I.; McCollum, D. Control of cellular responses to mechanical cues through YAP/TAZ regulation. J. Biol. Chem. 2019, 294, 17693–17706. [Google Scholar] [CrossRef] [Green Version]
- Oria, R.; Wiegand, T.; Escribano, J.; Elosegui-Artola, A.; Uriarte, J.J.; Moreno-Pulido, C.; Platzman, I.; Delcanale, P.; Albertazzi, L.; Navajas, D.; et al. Force loading explains spatial sensing of ligands by cells. Nature 2017, 552, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Iskratsch, T.; Wolfenson, H.; Sheetz, M.P. Appreciating force and shape-the rise of mechanotransduction in cell biology. Nat. Rev. Mol. Cell Biol. 2014, 15, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Chen, J.; Lim, Y.B.; Finch-Edmondson, M.L.; Seshachalam, V.P.; Qin, L.; Jiang, T.; Low, B.C.; Singh, H.; Lim, C.T.; et al. YAP Regulates Actin Dynamics through ARHGAP29 and Promotes Metastasis. Cell Rep. 2017, 19, 1495–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porazinski, S.; Wang, H.; Asaoka, Y.; Behrndt, M.; Miyamoto, T.; Morita, H.; Hata, S.; Sasaki, T.; Krens, S.F.G.; Osada, Y.; et al. YAP is essential for tissue tension to ensure vertebrate 3D body shape. Nature 2015, 521, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.G.; Hynes, R.O. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc. Natl. Acad. Sci. USA 2012, 109, E2441–E2450. [Google Scholar] [CrossRef] [Green Version]
- Diepenbruck, M.; Waldmeier, L.; Ivanek, R.; Berninger, P.; Arnold, P.; van Nimwegen, E.; Christofori, G. Tead2 expression levels control the subcellular distribution of Yap and Taz, zyxin expression and epithelial-mesenchymal transition. J. Cell Sci. 2014, 127, 1523–1536. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhang, L.; Purohit, V.; Shukla, S.K.; Chen, X.; Yu, F.; Fu, K.; Chen, Y.; Solheim, J.; Singh, P.K.; et al. Active YAP promotes pancreatic cancer cell motility, invasion and tumorigenesis in a mitotic phosphorylation-dependent manner through LPAR3. Oncotarget 2015, 6, 36019–36031. [Google Scholar] [CrossRef] [Green Version]
- Lai, D.; Yang, X. BMP4 is a novel transcriptional target and mediator of mammary cell migration downstream of the Hippo pathway component TAZ. Cell Signal. 2013, 25, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Valencia-Sama, I.; Zhao, Y.; Lai, D.; Janse van Rensburg, H.J.; Hao, Y.; Yang, X. Hippo Component TAZ Functions as a Co-repressor and Negatively Regulates DeltaNp63 Transcription through TEA Domain (TEAD) Transcription Factor. J. Biol. Chem. 2015, 290, 16906–16917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, B.S.; Yang, M.C.; Singh, S.; Chou, Y.C.; Chen, H.Y.; Wang, M.Y.; Wang, Y.C.; Chen, R.H. LncRNA NORAD is repressed by the YAP pathway and suppresses lung and breast cancer metastasis by sequestering S100P. Oncogene 2019, 38, 5612–5626. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Hwang, S.J.; Kim, H.R.; Shin, C.H.; Choi, K.H.; Joung, J.G.; Kim, H.H. Neurofibromatosis 2 (NF2) controls the invasiveness of glioblastoma through YAP-dependent expression of CYR61/CCN1 and miR-296-3p. Biochim. Biophys. Acta 2016, 1859, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Enzo, E.; Santinon, G.; Pocaterra, A.; Aragona, M.; Bresolin, S.; Forcato, M.; Grifoni, D.; Pession, A.; Zanconato, F.; Guzzo, G.; et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. 2015, 34, 1349–1370. [Google Scholar] [CrossRef]
- Wang, W.; Xiao, Z.D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.C.; Ling, H.H.; Chiang, M.C.; Chung, C.H.; Lee, W.Y.; Chu, C.Y.; Wu, Y.C.; Chen, C.H.; Lai, Y.W.; Tsai, I.L.; et al. Metastatic colorectal cancer rewrites metabolic program through a Glut3-YAP-dependent signaling circuit. Theranostics 2019, 9, 2526–2540. [Google Scholar] [CrossRef]
- Bertero, T.; Oldham, W.M.; Grasset, E.M.; Bourget, I.; Boulter, E.; Pisano, S.; Hofman, P.; Bellvert, F.; Meneguzzi, G.; Bulavin, D.V.; et al. Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab 2019, 29, 124–140.e110. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.K.; Jeong, S.H.; Jang, C.; Bae, H.; Kim, Y.H.; Park, I.; Kim, S.K.; Koh, G.Y. Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science 2019, 363, 644–649. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Zhong, S.; Wang, H.; Wang, X.; Li, N.; Li, Y.; Zhang, G.; Yuan, H.; Lian, Y.; Chen, Q.; et al. The ZMYND8-regulated mevalonate pathway endows YAP-high intestinal cancer with metabolic vulnerability. Mol. Cell 2021, 81, 2736–2751.e2738. [Google Scholar] [CrossRef]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Sharif, G.M.; Schmidt, M.O.; Yi, C.; Hu, Z.; Haddad, B.R.; Glasgow, E.; Riegel, A.T.; Wellstein, A. Cell growth density modulates cancer cell vascular invasion via Hippo pathway activity and CXCR2 signaling. Oncogene 2015, 34, 5879–5889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Sun, Y.; Wei, Y.; Zhang, P.; Rezaeian, A.H.; Teruya-Feldstein, J.; Gupta, S.; Liang, H.; Lin, H.K.; Hung, M.C.; et al. LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat. Med. 2012, 18, 1511–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Nandakumar, N.; Shi, Y.; Manzano, M.; Smith, A.; Graham, G.; Gupta, S.; Vietsch, E.E.; Laughlin, S.Z.; Wadhwa, M.; et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci. Signal. 2014, 7, ra42. [Google Scholar] [CrossRef] [Green Version]
- Gruber, R.; Panayiotou, R.; Nye, E.; Spencer-Dene, B.; Stamp, G.; Behrens, A. YAP1 and TAZ Control Pancreatic Cancer Initiation in Mice by Direct Up-regulation of JAK-STAT3 Signaling. Gastroenterology 2016, 151, 526–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, K.; Wu, L.W.; Grivennikov, S.I.; de Jong, P.R.; Lian, I.; Yu, F.X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 2015, 519, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Rouse, C.; Jasper, J.S.; Pendergast, A.M. ABL kinases promote breast cancer osteolytic metastasis by modulating tumor-bone interactions through TAZ and STAT5 signaling. Sci. Signal. 2016, 9, ra12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Fan, Y.; Jing, X.; Zhao, L.; Liu, T.; Wang, L.; Zhang, L.; Gu, S.; Zhao, X.; Teng, Y. OTUD5-mediated deubiquitination of YAP in macrophage promotes M2 phenotype polarization and favors triple-negative breast cancer progression. Cancer Lett. 2021, 504, 104–115. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Y.; Hong, T.; Ye, J.; Chu, C.; Zuo, L.; Zhang, J.; Cui, X. Targeting a positive regulatory loop in the tumor-macrophage interaction impairs the progression of clear cell renal cell carcinoma. Cell Death Differ. 2021, 28, 932–951. [Google Scholar] [CrossRef]
- Miao, J.; Hsu, P.C.; Yang, Y.L.; Xu, Z.; Dai, Y.; Wang, Y.; Chan, G.; Huang, Z.; Hu, B.; Li, H.; et al. YAP regulates PD-L1 expression in human NSCLC cells. Oncotarget 2017, 8, 114576–114587. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Yang, H.; Zhang, Y.; Wei, H.; Zhu, Z.; Zhu, B.; Yang, M.; Cao, W.; Wang, L.; Wu, Z. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene 2017, 36, 5829–5839. [Google Scholar] [CrossRef]
- Janse van Rensburg, H.J.; Azad, T.; Ling, M.; Hao, Y.; Snetsinger, B.; Khanal, P.; Minassian, L.M.; Graham, C.H.; Rauh, M.J.; Yang, X. The Hippo Pathway Component TAZ Promotes Immune Evasion in Human Cancer through PD-L1. Cancer Res. 2018, 78, 1457–1470. [Google Scholar] [CrossRef] [Green Version]
- Ni, X.; Tao, J.; Barbi, J.; Chen, Q.; Park, B.V.; Li, Z.; Zhang, N.; Lebid, A.; Ramaswamy, A.; Wei, P.; et al. YAP Is Essential for Treg-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2018, 8, 1026–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef]
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.H.; Ding, C.C.; Sun, T.; Rupprecht, G.; Lin, C.C.; Hsu, D.; Chi, J.T. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep. 2019, 28, 2501–2508. [Google Scholar] [CrossRef]
- Yang, W.H.; Huang, Z.; Wu, J.; Ding, C.C.; Murphy, S.K.; Chi, J.T. A TAZ-ANGPTL4-NOX2 Axis Regulates Ferroptotic Cell Death and Chemoresistance in Epithelial Ovarian Cancer. Mol. Cancer Res. 2020, 18, 79–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.H.; Lin, C.C.; Wu, J.; Chao, P.Y.; Chen, K.; Chen, P.H.; Chi, J.T. The Hippo Pathway Effector YAP Promotes Ferroptosis via the E3 Ligase SKP2. Mol. Cancer Res. 2021, 19, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Boumahdi, S.; de Sauvage, F.J. The great escape: Tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 2020, 19, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Friedmann-Morvinski, D.; Verma, I.M. Dedifferentiation and reprogramming: Origins of cancer stem cells. EMBO Rep. 2014, 15, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Norgard, R.J.; Stanger, B.Z. Cellular Plasticity in Cancer. Cancer Discov. 2019, 9, 837–851. [Google Scholar] [CrossRef] [Green Version]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Zhang, N.; Zheng, Y.; de Wilde, R.F.; Maitra, A.; Pan, D. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev. 2010, 24, 2383–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yimlamai, D.; Christodoulou, C.; Galli, G.G.; Yanger, K.; Pepe-Mooney, B.; Gurung, B.; Shrestha, K.; Cahan, P.; Stanger, B.Z.; Camargo, F.D. Hippo pathway activity influences liver cell fate. Cell 2014, 157, 1324–1338. [Google Scholar] [CrossRef] [Green Version]
- Hong, A.W.; Meng, Z.; Guan, K.L. The Hippo pathway in intestinal regeneration and disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 324–337. [Google Scholar] [CrossRef]
- Hageman, J.H.; Heinz, M.C.; Kretzschmar, K.; van der Vaart, J.; Clevers, H.; Snippert, H.J.G. Intestinal Regeneration: Regulation by the Microenvironment. Dev. Cell 2020, 54, 435–446. [Google Scholar] [CrossRef]
- Gregorieff, A.; Liu, Y.; Inanlou, M.R.; Khomchuk, Y.; Wrana, J.L. Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature 2015, 526, 715–718. [Google Scholar] [CrossRef]
- Ayyaz, A.; Kumar, S.; Sangiorgi, B.; Ghoshal, B.; Gosio, J.; Ouladan, S.; Fink, M.; Barutcu, S.; Trcka, D.; Shen, J.; et al. Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 2019, 569, 121–125. [Google Scholar] [CrossRef]
- Panciera, T.; Azzolin, L.; Fujimura, A.; Di Biagio, D.; Frasson, C.; Bresolin, S.; Soligo, S.; Basso, G.; Bicciato, S.; Rosato, A.; et al. Induction of Expandable Tissue-Specific Stem/Progenitor Cells through Transient Expression of YAP/TAZ. Cell Stem Cell 2016, 19, 725–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartucci, M.; Dattilo, R.; Moriconi, C.; Pagliuca, A.; Mottolese, M.; Federici, G.; Benedetto, A.D.; Todaro, M.; Stassi, G.; Sperati, F.; et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene 2015, 34, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Higashi, T.; Yokoyama, N.; Kaida, T.; Sakamoto, K.; Fukushima, Y.; Ishimoto, T.; Kuroki, H.; Nitta, H.; Hashimoto, D.; et al. An Imbalance in TAZ and YAP Expression in Hepatocellular Carcinoma Confers Cancer Stem Cell-like Behaviors Contributing to Disease Progression. Cancer Res. 2015, 75, 4985–4997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu-Roy, U.; Bayin, N.S.; Rattanakorn, K.; Han, E.; Placantonakis, D.G.; Mansukhani, A.; Basilico, C. Sox2 antagonizes the Hippo pathway to maintain stemness in cancer cells. Nat. Commun. 2015, 6, 6411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, A.M.; Missiaglia, E.; Galli, G.G.; Hettmer, S.; Urcia, R.; Carrara, M.; Judson, R.N.; Thway, K.; Nadal, G.; Selfe, J.L.; et al. The Hippo transducer YAP1 transforms activated satellite cells and is a potent effector of embryonal rhabdomyosarcoma formation. Cancer Cell 2014, 26, 273–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bora-Singhal, N.; Nguyen, J.; Schaal, C.; Perumal, D.; Singh, S.; Coppola, D.; Chellappan, S. YAP1 Regulates OCT4 Activity and SOX2 Expression to Facilitate Self-Renewal and Vascular Mimicry of Stem-Like Cells. Stem Cells 2015, 33, 1705–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhang, Z.; Yu, X.; Huang, X.; Liu, Z.; Chai, Y.; Yang, L.; Wang, Q.; Li, M.; Zhao, J.; et al. Unbalanced YAP-SOX9 circuit drives stemness and malignant progression in esophageal squamous cell carcinoma. Oncogene 2019, 38, 2042–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopez-Hernandez, A.; Sberna, S.; Campaner, S. Emerging Principles in the Transcriptional Control by YAP and TAZ. Cancers 2021, 13, 4242. https://doi.org/10.3390/cancers13164242
Lopez-Hernandez A, Sberna S, Campaner S. Emerging Principles in the Transcriptional Control by YAP and TAZ. Cancers. 2021; 13(16):4242. https://doi.org/10.3390/cancers13164242
Chicago/Turabian StyleLopez-Hernandez, Alejandro, Silvia Sberna, and Stefano Campaner. 2021. "Emerging Principles in the Transcriptional Control by YAP and TAZ" Cancers 13, no. 16: 4242. https://doi.org/10.3390/cancers13164242
APA StyleLopez-Hernandez, A., Sberna, S., & Campaner, S. (2021). Emerging Principles in the Transcriptional Control by YAP and TAZ. Cancers, 13(16), 4242. https://doi.org/10.3390/cancers13164242