
The Novel Antitumor Compound HCA Promotes Glioma Cell Death by Inducing Endoplasmic Reticulum Stress and Autophagy

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Animals, Tumor Xenografts, and Treatments
2.3. Cell Proliferation Assays
2.4. Cell Transfection, Gene Expression Silencing, and Commercial Inhibitors
2.5. Cell Lysis, Electrophoresis, and Immunoblotting and Protein Quantification
2.6. Immunofluorescence and Confocal Microscopy
2.7. Cathepsin Assay
2.8. Fatty Acid Analysis by Gas Chromatography (GC)
2.9. Data Analysis
3. Results
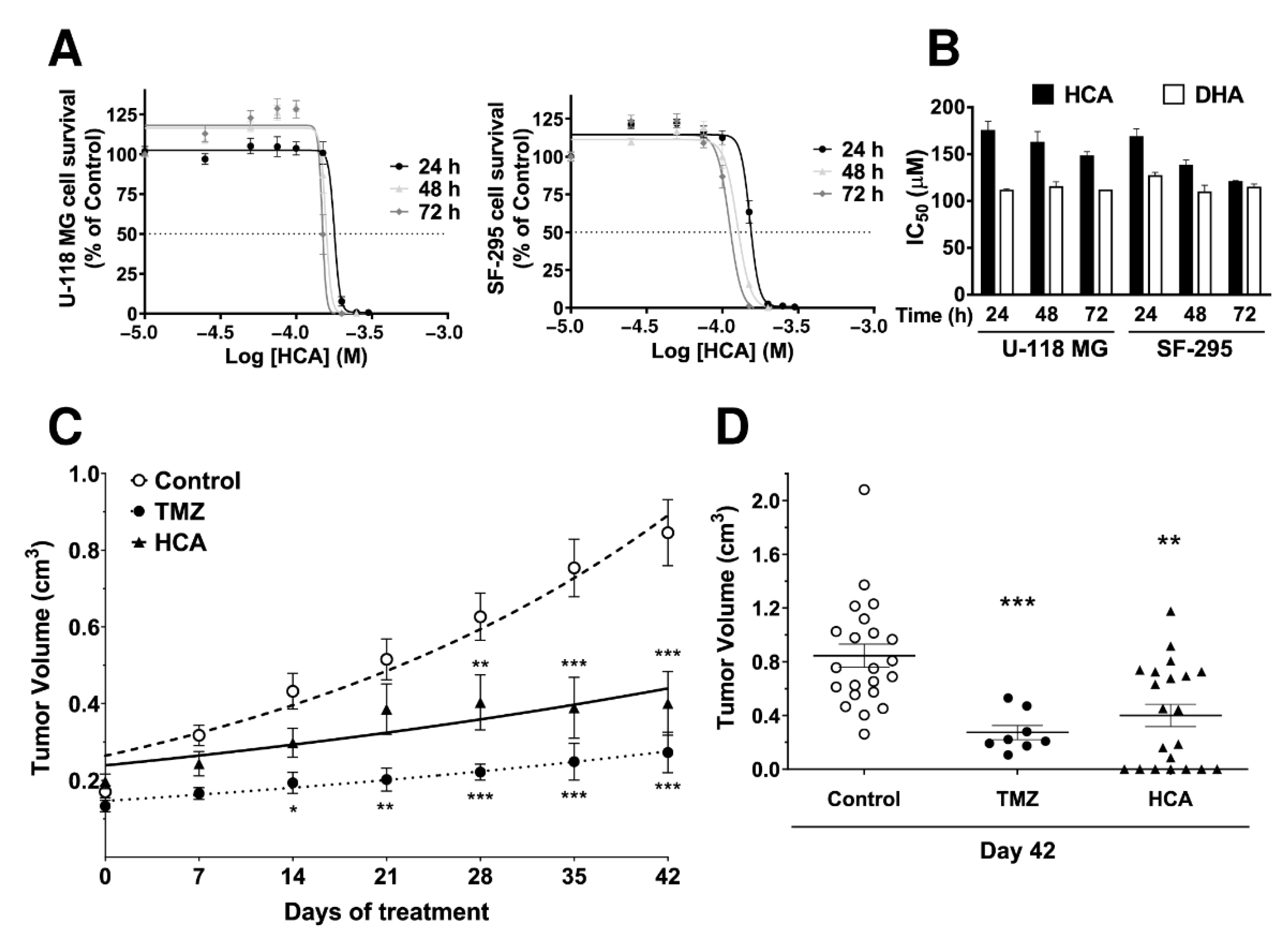
3.1. Efficacy of HCA against GBM
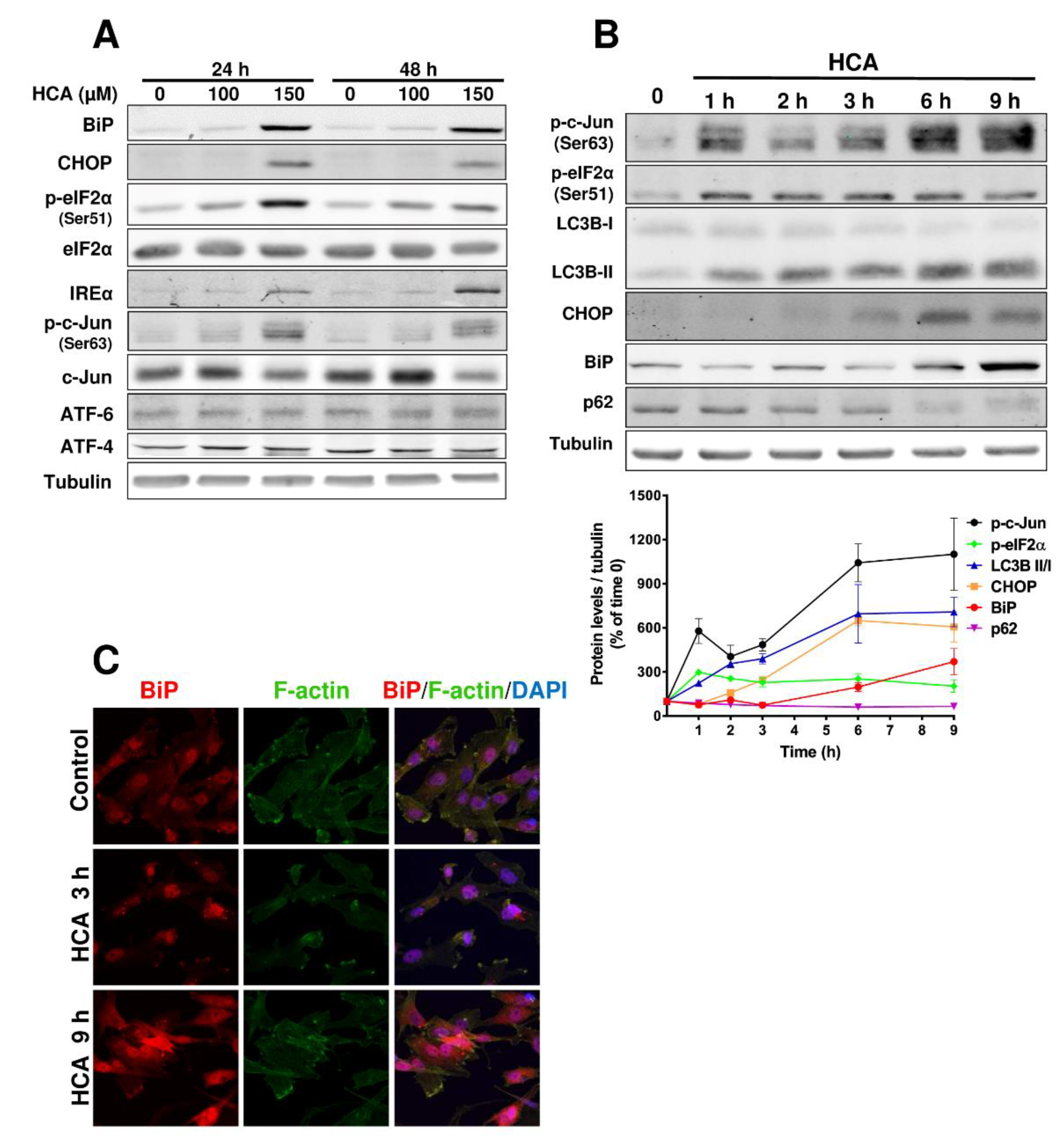
3.2. HCA Activates ER Stress/UPR Signaling Pathways in GBM Cells
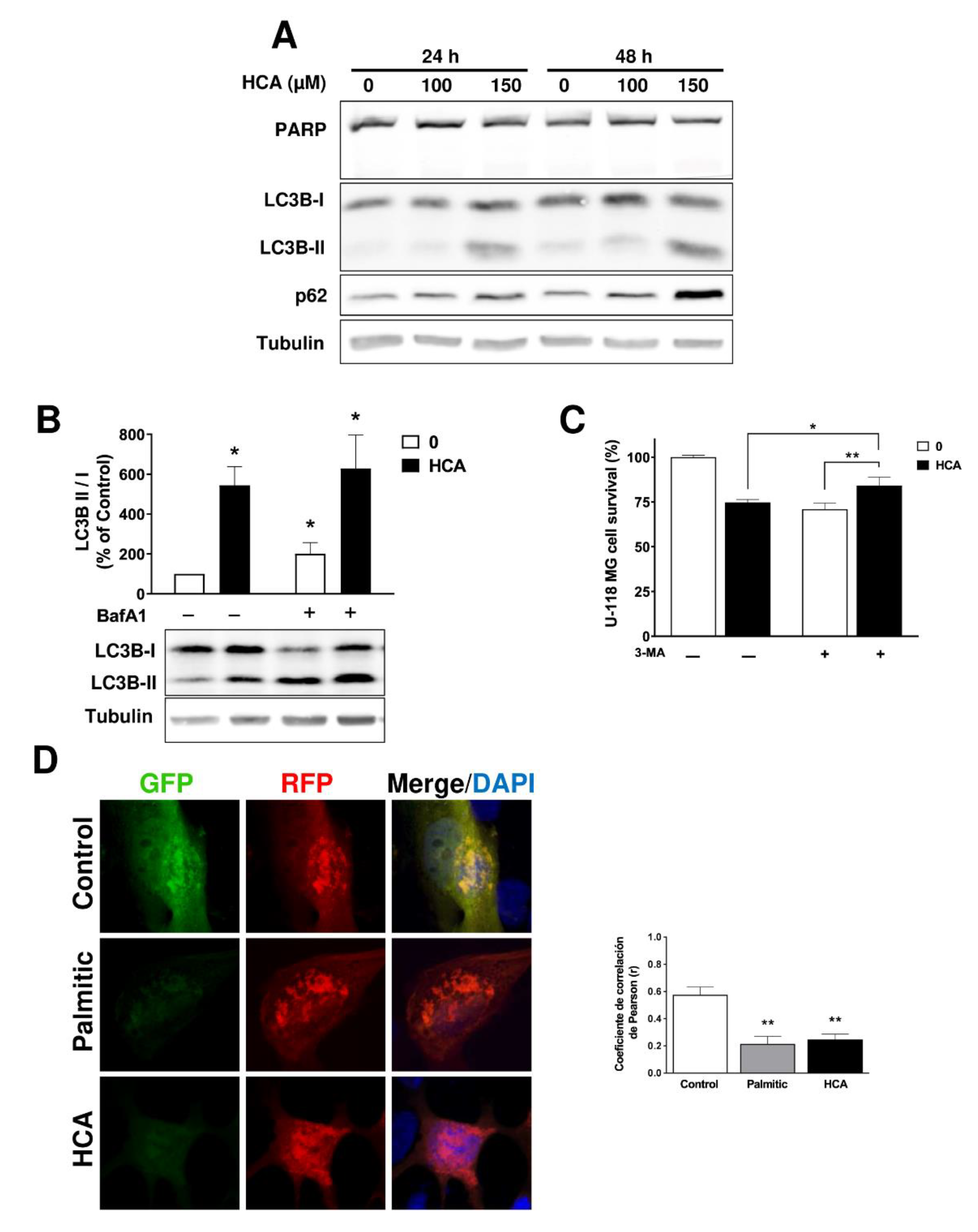
3.3. HCA Induces Autophagy in GBM Cells
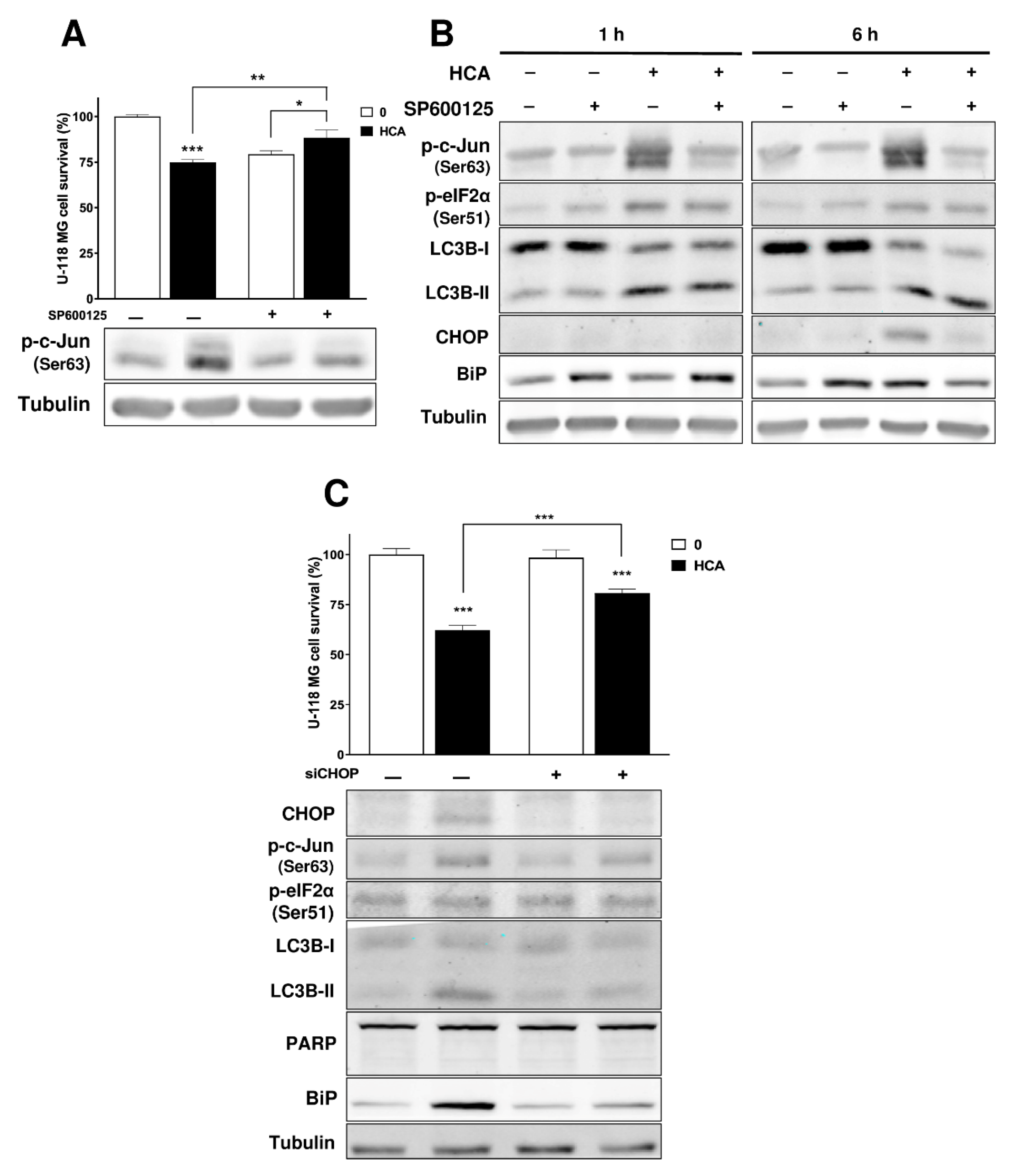
3.4. Activation of the JNK/c-Jun/CHOP Pathway Is Related to the Mechanism of Action of HCA
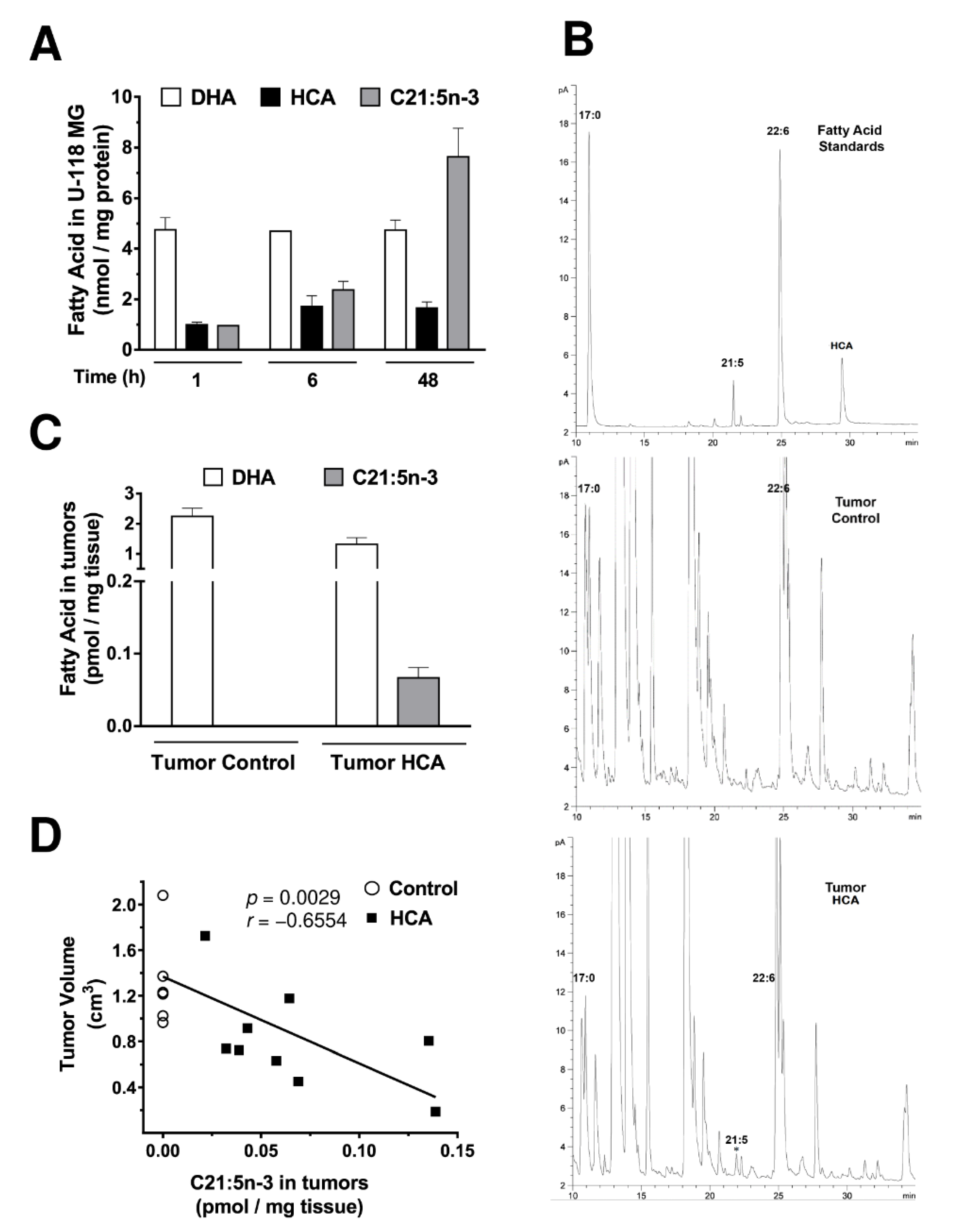
3.5. HCA Is Incorporated into GBM Cells and It Is Associated with the Appearance of Heneicosapentaenoic Acid (C21:5n-3)
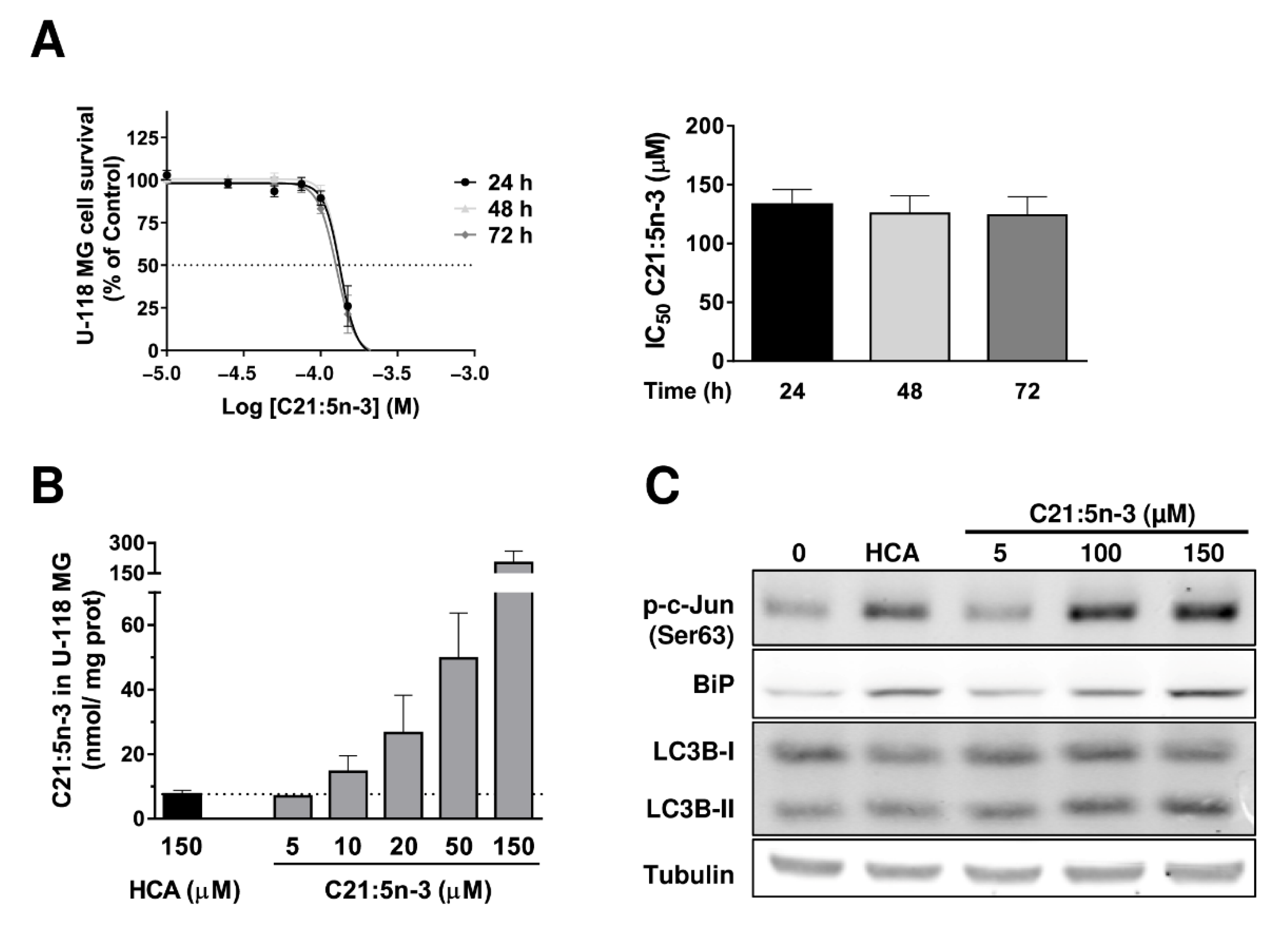
3.6. The Anti-Proliferative Effect of C21:5n-3
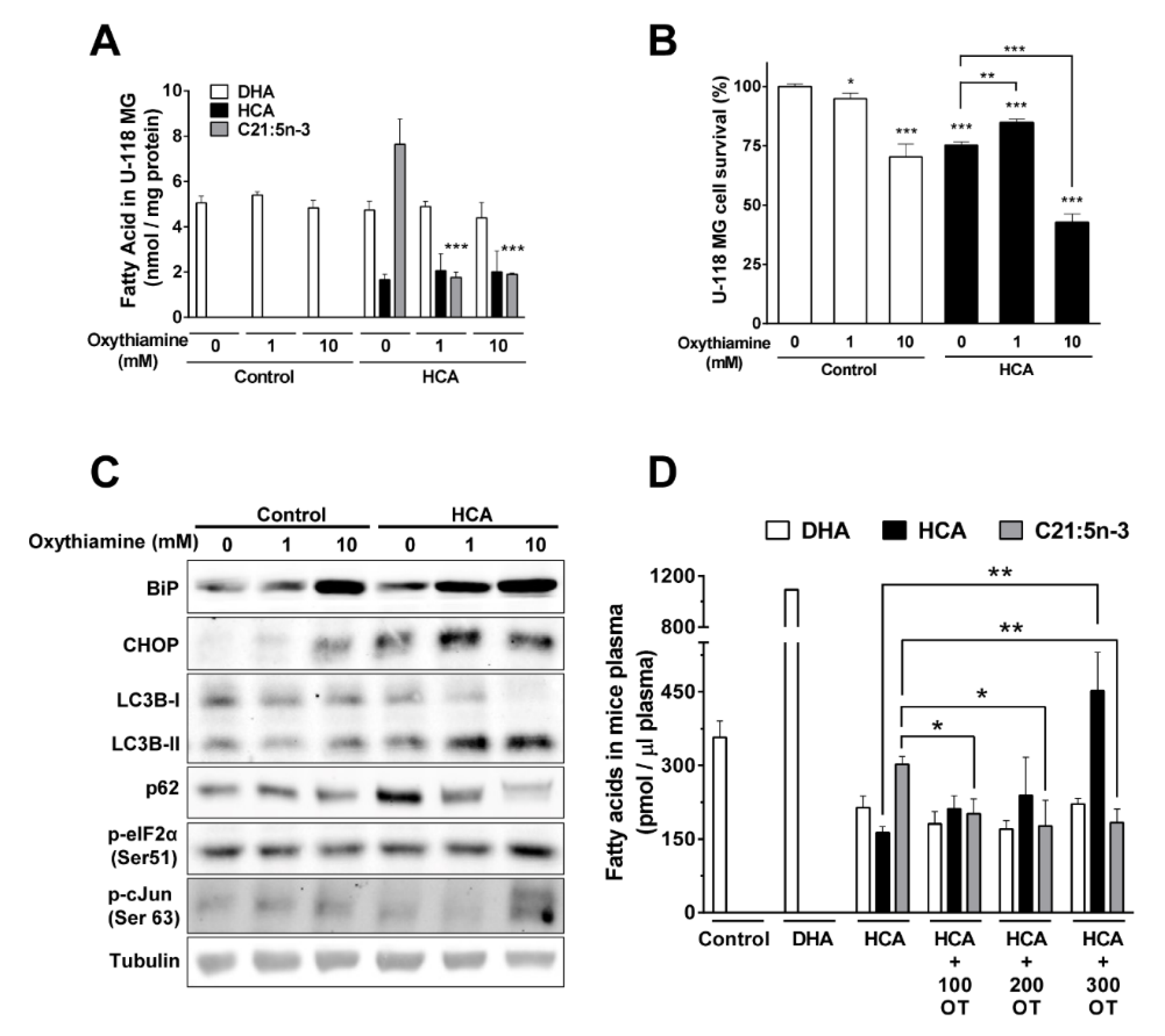
3.7. Metabolization of HCA to C21:5n-3 Is Produced through α-Oxidation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Batash, R.; Asna, N.; Schaffer, P.; Francis, N.; Schaffer, M. Glioblastoma Multiforme, Diagnosis and Treatment; Recent Literature Review. Curr. Med. Chem. 2017, 24, 3002–3009. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.; Gittleman, H.; Liao, P.; Rouse, C.; Chen, Y.; Dowling, J.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007-2011. Neuro-Oncology 2014, 16 (Suppl. 4), iv1–iv63. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escribá, P.V. Membrane-lipid therapy: A historical perspective of membrane-targeted therapies—From lipid bilayer structure to the pathophysiological regulation of cells. Biochim. Biophys. Acta (BBA) Biomembr. 2017, 1859, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Azaro, A.; Plummer, E.R.; Urruticoechea, A.; Rodon, J.; Haris, N.R.M.; Veal, G.; Perier, A.; Tur, V.; Escriba, P.V.; Busquets, X.; et al. Final report of a phase I study of 2-hydroxyoleic acid (2OHOA) a novel sphingomyelin synthase activator in patients (pt) with advanced solid tumors (AST) including recurrent high grade gliomas (rHGG). J. Clin. Oncol. 2017, 35, 2554. [Google Scholar] [CrossRef]
- Torres, M.; Price, S.L.; Fiol-deRoque, M.A.; Marcilla-Etxenike, A.; Ahyayauch, H.; Barceló-Coblijn, G.; Terés, S.; Katsouri, L.; Ordinas, M.; López, D.J.; et al. Membrane lipid modifications and therapeutic effects mediated by hydroxydocosahexaenoic acid on Alzheimer’s disease. Biochim. Biophys. Acta (BBA) Biomembr. 2014, 1838, 1680–1692. [Google Scholar] [CrossRef] [Green Version]
- Su, H.-M. Mechanisms of n-3 fatty acid-mediated development and maintenance of learning memory performance. J. Nutr. Biochem. 2010, 21, 364–373. [Google Scholar] [CrossRef]
- Martin, D.D.; Robbins, M.E.C.; Spector, A.A.; Wen, B.-C.; Hussey, D.H. The fatty acid composition of human gliomas differs from that found in nonmalignant brain tissue. Lipids 1996, 31, 1283–1288. [Google Scholar] [CrossRef]
- Rosselló, C.A.; Torres, M.; Busquets, X.; Escribá, P.V. Polyunsaturated Fatty Acids. In Encyclopedia of Cancer; Schwab, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–7. [Google Scholar]
- Elmesery, M.E.; Al-Gayyar, M.; Salem, H.; Darweish, M.M.; El-Mowafy, A. Chemopreventive and renal protective effects for docosahexaenoic acid (DHA): Implications of CRP and lipid peroxides. Cell Div. 2009, 4, 6. [Google Scholar] [CrossRef] [Green Version]
- Berquin, I.M.; Edwards, I.J.; Chen, Y.Q. Multi-targeted therapy of cancer by omega-3 fatty acids. Cancer Lett. 2008, 269, 363–377. [Google Scholar] [CrossRef] [Green Version]
- Newell, M.; Mackey, J.R.; Bigras, G.; Alvarez-Camacho, M.; Goruk, S.; Ghosh, S.; Schmidt, A.; Miede, D.; Chisotti, A.; Postovit, L.; et al. Comparing docosahexaenoic acid (DHA) concomitant with neoadjuvant chemotherapy versus neoadjuvant chemotherapy alone in the treatment of breast cancer (DHA WIN): Protocol of a double-blind, phase II, randomised controlled trial. BMJ Open 2019, 9, e030502. [Google Scholar] [CrossRef] [Green Version]
- Fiol-deRoque, M.A.; Lanza, R.G.; Terés, S.; Torres, M.; Barceló, P.; Rial, R.V.; Verkhratsky, A.; Escribá, P.V.; Busquets, X.; Rodríguez, J.J. Cognitive recovery and restoration of cell proliferation in the dentate gyrus in the 5XFAD transgenic mice model of Alzheimer’s disease following 2-hydroxy-DHA treatment. Biogerontology 2013, 14, 763–775. [Google Scholar] [CrossRef]
- Parets, S.; Irigoyen, Á.; Ordinas, M.; Cabot, J.; Miralles, M.; Arbona, L.; Péter, M.; Balogh, G.; Fernández-García, P.; Busquets, X.; et al. 2-Hydroxy-Docosahexaenoic Acid Is Converted Into Heneicosapentaenoic Acid via α-Oxidation: Implications for Alzheimer’s Disease Therapy. Front. Cell Dev. Biol. 2020, 8, 164. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Marcilla-Etxenike, A.; Fiol-deRoque, M.A.; Escribá, P.V.; Busquets, X. The unfolded protein response in the therapeutic effect of hydroxy-DHA against Alzheimer’s disease. Apoptosis 2015, 20, 712–724. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Kaufman, R.J. THE MAMMALIAN UNFOLDED PROTEIN RESPONSE. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [Green Version]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between Autophagy Receptors and Ubiquitin-like Proteins Form the Molecular Basis for Selective Autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Levine, B.L. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Fernández-García, P.; Rosselló, C.A.; Rodríguez-Lorca, R.; Beteta-Göbel, R.; Fernández-Díaz, J.; Lladó, V.; Busquets, X.; Escribá, P.V. The Opposing Contribution of SMS1 and SMS2 to Glioma Progression and Their Value in the Therapeutic Response to 2OHOA. Cancers 2019, 11, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guardiola-Serrano, F.; Beteta-Göbel, R.; Rodríguez-Lorca, R.; Ibarguren, M.; López, D.J.; Terés, S.; Alvarez, R.; Alonso-Sande, M.; Busquets, X.; Escribá, P.V. The Novel Anticancer Drug Hydroxytriolein Inhibits Lung Cancer Cell Proliferation via a Protein Kinase Cα– and Extracellular Signal-Regulated Kinase 1/2–Dependent Mechanism. J. Pharmacol. Exp. Ther. 2015, 354, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the Autophagosome Maturation Process by a Novel Reporter Protein, Tandem Fluorescent-Tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guardiola-Serrano, F.; Beteta-Göbel, R.; Rodríguez-Lorca, R.; Ibarguren, M.; López, D.J.; Terés, S.; Alonso-Sande, M.; Higuera, M.; Torres, M.; Busquets, X.; et al. The triacylglycerol, hydroxytriolein, inhibits triple negative mammary breast cancer cell proliferation through a mechanism dependent on dihydroceramide and Akt. Oncotarget 2019, 10, 2486–2507. [Google Scholar] [CrossRef] [Green Version]
- Ashizawa, T.; Miyata, H.; Iizuka, A.; Komiyama, M.; Oshita, C.; Kume, A.; Nogami, M.; Yagoto, M.; Ito, I.; Oishi, T.; et al. Effect of the STAT3 inhibitor STX-0119 on the proliferation of cancer stem-like cells derived from recurrent glioblastoma. Int. J. Oncol. 2013, 43, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Garros-Regulez, L.; Aldaz, P.; Arrizabalaga, O.; Moncho-Amor, V.; Carrasco-Garcia, E.; Manterola, L.; Moreno-Cugnon, L.; Barrena, C.; Villanua, J.; Ruiz, I.; et al. mTOR inhibition decreases SOX2-SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert Opin. Ther. Targets 2016, 20, 393–405. [Google Scholar] [CrossRef]
- Mohaibes, R.J.; Fiol-deRoque, M.A.; Torres, M.; Ordinas, M.; López, D.J.; Castro, J.A.; Escribá, P.V.; Busquets, X. The hydroxylated form of docosahexaenoic acid (DHA-H) modifies the brain lipid composition in a model of Alzheimer’s disease, improving behavioral motor function and survival. Biochim. Biophys. Acta (BBA) Biomembr. 2017, 1859, 1596–1603. [Google Scholar] [CrossRef]
- Kaufmann, S.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-T.; Tan, H.-L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.-M. Dual Role of 3-Methyladenine in Modulation of Autophagy via Different Temporal Patterns of Inhibition on Class I and III Phosphoinositide 3-Kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [Green Version]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic Palmitate But Not Oleate Exposure Induces Endoplasmic Reticulum Stress, Which May Contribute to INS-1 Pancreatic β-Cell Apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foulon, V.; Sniekers, M.; Huysmans, E.; Asselberghs, S.; Mahieu, V.; Mannaerts, G.P.; Van Veldhoven, P.P.; Casteels, M. Breakdown of 2-Hydroxylated Straight Chain Fatty Acids via Peroxisomal 2-Hydroxyphytanoyl-CoA Lyase. J. Biol. Chem. 2005, 280, 9802–9812. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Lan, W.; Bo, L.; Niu, C.; Zhou, J.; Zhu, H. Metabolic response of LLC xenografted mice to oxythiamine, as measured by [1H] NMR spectroscopy. Genet. Mol. Res. 2015, 14, 11043–11051. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Busquets, X.; Escribá, P.V. Brain Lipids in the Pathophysiology and Treatment of Alzheimer’s Disease. In Update on Dementia; Elsevier-Harvard: Chichago, IL, USA, 2016. [Google Scholar]
- Sulciner, M.L.; Serhan, C.N.; Gilligan, M.M.; Mudge, D.K.; Chang, J.; Gartung, A.; Lehner, K.A.; Bielenberg, D.R.; Schmidt, B.; Dalli, J.; et al. Resolvins suppress tumor growth and enhance cancer therapy. J. Exp. Med. 2017, 215, 115–140. [Google Scholar] [CrossRef] [PubMed]
- Paterson, I. CHEMISTRY: The Renaissance of Natural Products as Drug Candidates. Science 2005, 310, 451–453. [Google Scholar] [CrossRef]
- Torres, M.; Rosselló, C.A.; Fernández-García, P.; Lladó, V.; Kakhlon, O.; Escriba, P. The Implications for Cells of the Lipid Switches Driven by Protein–Membrane Interactions and the Development of Membrane Lipid Therapy. Int. J. Mol. Sci. 2020, 21, 2322. [Google Scholar] [CrossRef] [Green Version]
- Lladó, V.; López, D.J.; Ibarguren, M.; Alonso, M.; Soriano, J.B.; Escriba, P.; Busquets, X. Regulation of the cancer cell membrane lipid composition by NaCHOleate. Biochim. Biophys. Acta (BBA) Biomembr. 2014, 1838, 1619–1627. [Google Scholar] [CrossRef] [Green Version]
- Martínez, J.; Vögler, O.; Casas, J.; Barceló, F.; Alemany, R.; Prades, J.; Nagy, T.; Baamonde, C.; Kasprzyk, P.G.; Terés, S.; et al. Membrane Structure Modulation, Protein Kinase Cα Activation, and Anticancer Activity of Minerval. Mol. Pharmacol. 2004, 67, 531–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmo, A.; Carvalheiro, H.; Crespo, I.; Nunes, I.; Lopes, M.C. Effect of temozolomide on the U-118 glioma cell line. Oncol. Lett. 2011, 2, 1165–1170. [Google Scholar] [CrossRef] [Green Version]
- Mutter, N.; Stupp, R. Temozolomide: A milestone in neuro-oncology and beyond? Expert Rev. Anticancer. Ther. 2006, 6, 1187–1204. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Salazar, M.; Velasco, G.; Agostinis, P. Linking ER Stress to Autophagy: Potential Implications for Cancer Therapy. Int. J. Cell Biol. 2010, 2010, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Jawhari, S.; Ratinaud, M.-H.; Verdier, M. Glioblastoma, hypoxia and autophagy: A survival-prone ‘ménage-à-trois’. Cell Death Dis. 2016, 7, e2434. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer 2017, 8, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; An, G.; Kuo, M.T. C-Jun N-terminal kinase signalling pathway in response to cisplatin. J. Cell. Mol. Med. 2016, 20, 2013–2019. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Liu, L.; Naik, I.; Braunstein, Z.; Zhong, J.; Ren, B. Transcription Factor C/EBP Homologous Protein in Health and Diseases. Front. Immunol. 2017, 8, 1612. [Google Scholar] [CrossRef]
- Tsai, S.-F.; Tao, M.; Ho, L.-I.; Chiou, T.-W.; Lin, S.-Z.; Su, H.-L.; Harn, H.-J. Isochaihulactone-induced DDIT3 causes ER stress-PERK independent apoptosis in glioblastoma multiforme cells. Oncotarget 2016, 8, 4051–4061. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, X.; Ma, D.; Lee, W.-N.P.; Xiao, J.; Zhao, Y.; Go, V.L.; Wang, Q.; Yen, Y.; Recker, R.; et al. Inhibition of transketolase by oxythiamine altered dynamics of protein signals in pancreatic cancer cells. Exp. Hematol. Oncol. 2013, 2, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, S.D.; Wirth, M.; Statkevich, P.; Reidenberg, P.; Alton, K.; Sartorius, S.E.; Dugan, M.; Cutler, D.; Batra, V.; Grochow, L.B.; et al. Absorption, metabolism, and excretion of 14C-temozolomide following oral administration to patients with advanced cancer. Clin. Cancer Res. 1999, 5, 309–317. [Google Scholar]
- Larsen, L.N.; Høvik, K.; Bremer, J.; Holm, K.H.; Myhren, F.; Børretzen, B. Heneicosapentaenoate (21:5n-3): Its incorporation into lipids and its effects on arachidonic acid and eicosanoid synthesis. Lipids 1997, 32, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Masquio, D.C.L.; de Piano, A.; Oyama, L.M.; Campos, R.; Santamarina, A.B.; Souza, G.I.D.M.H.D.; Gomes, A.D.; Moreira, R.; Corgosinho, F.C.; Nascimento, C.M.O.D.; et al. The role of free fatty acids in the inflammatory and cardiometabolic profile in adolescents with metabolic syndrome engaged in interdisciplinary therapy. J. Nutr. Biochem. 2016, 33, 136–144. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beteta-Göbel, R.; Fernández-Díaz, J.; Arbona-González, L.; Rodríguez-Lorca, R.; Torres, M.; Busquets, X.; Fernández-García, P.; Escribá, P.V.; Lladó, V. The Novel Antitumor Compound HCA Promotes Glioma Cell Death by Inducing Endoplasmic Reticulum Stress and Autophagy. Cancers 2021, 13, 4290. https://doi.org/10.3390/cancers13174290
Beteta-Göbel R, Fernández-Díaz J, Arbona-González L, Rodríguez-Lorca R, Torres M, Busquets X, Fernández-García P, Escribá PV, Lladó V. The Novel Antitumor Compound HCA Promotes Glioma Cell Death by Inducing Endoplasmic Reticulum Stress and Autophagy. Cancers. 2021; 13(17):4290. https://doi.org/10.3390/cancers13174290
Chicago/Turabian StyleBeteta-Göbel, Roberto, Javier Fernández-Díaz, Laura Arbona-González, Raquel Rodríguez-Lorca, Manuel Torres, Xavier Busquets, Paula Fernández-García, Pablo V. Escribá, and Victoria Lladó. 2021. "The Novel Antitumor Compound HCA Promotes Glioma Cell Death by Inducing Endoplasmic Reticulum Stress and Autophagy" Cancers 13, no. 17: 4290. https://doi.org/10.3390/cancers13174290
APA StyleBeteta-Göbel, R., Fernández-Díaz, J., Arbona-González, L., Rodríguez-Lorca, R., Torres, M., Busquets, X., Fernández-García, P., Escribá, P. V., & Lladó, V. (2021). The Novel Antitumor Compound HCA Promotes Glioma Cell Death by Inducing Endoplasmic Reticulum Stress and Autophagy. Cancers, 13(17), 4290. https://doi.org/10.3390/cancers13174290