
Targeting PI3K Pathway in Pancreatic Ductal Adenocarcinoma: Rationale and Progress


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
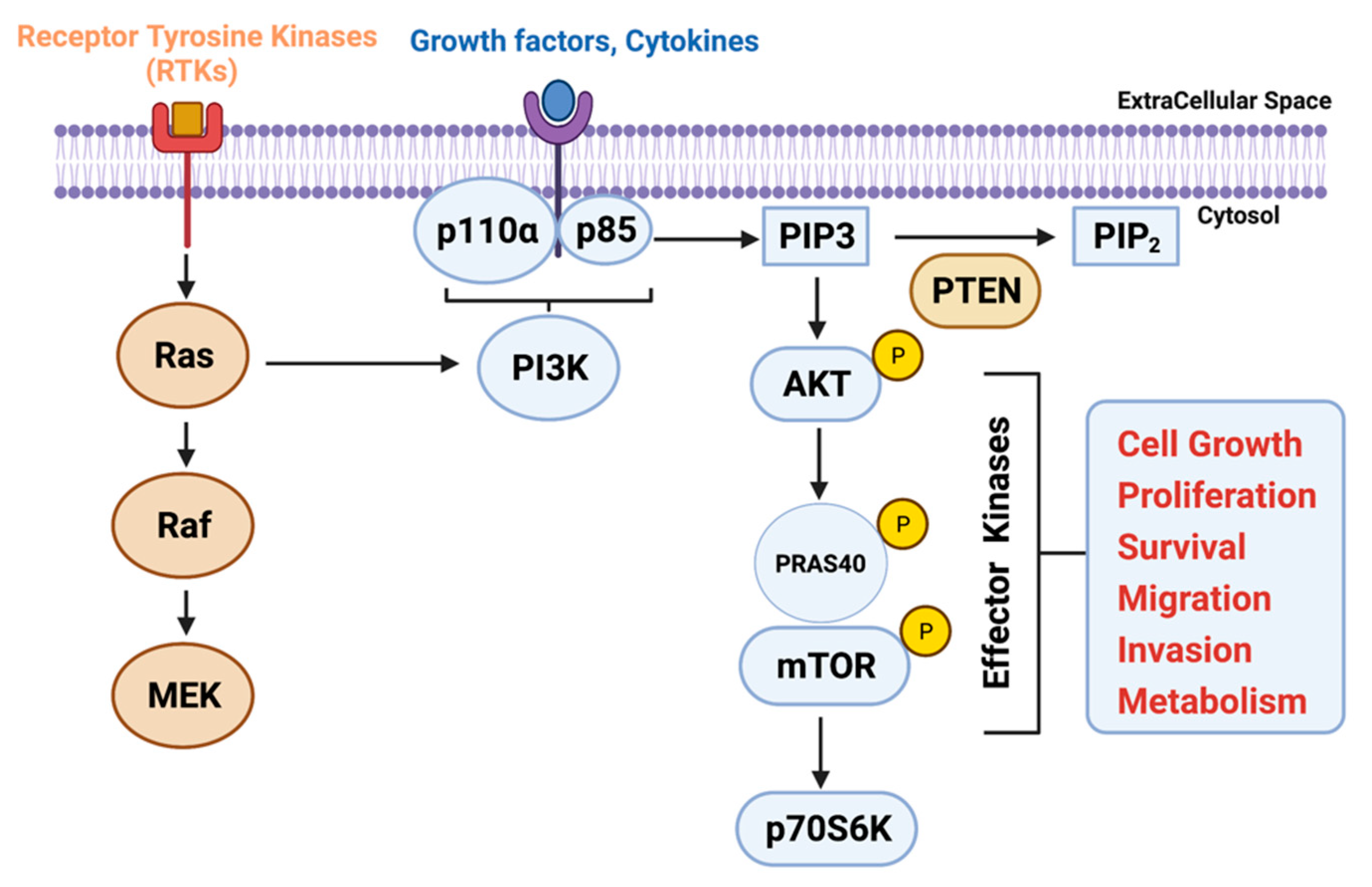
2. PI3K Signaling Activation in PDAC
3. Inhibition of PI3K Signaling in PDAC
4. Reciprocal Crosstalk Involving PI3K Signaling in PDAC
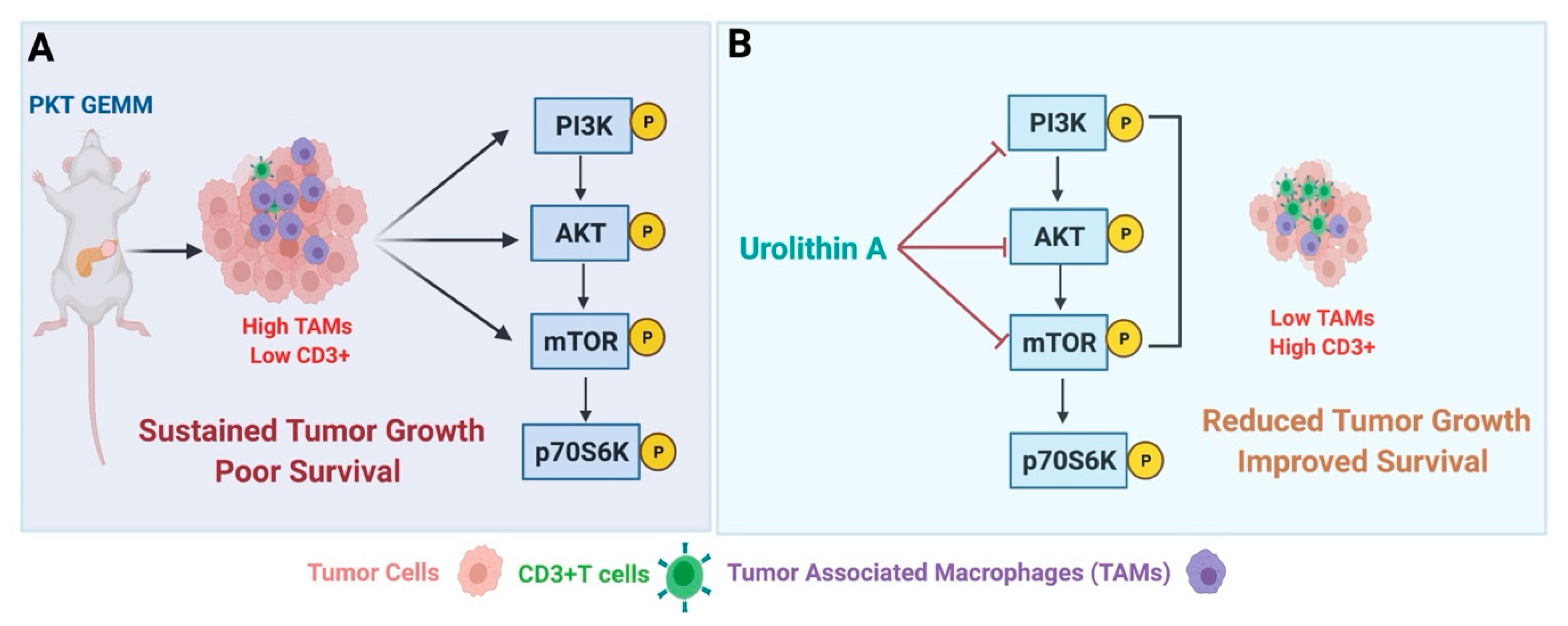
5. Impact of PI3K Inhibition on Tumor–Stromal Immune Crosstalk in PDAC
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ghadirian, P.; Lynch, H.T.; Krewski, D. Epidemiology of Pancreatic Cancer: An Overview. Cancer Detect. Prev. 2003, 27, 87–93. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Stocken, D.D.; Friess, H.; Bassi, C.; Dunn, J.A.; Hickey, H.; Beger, H.; Fernandez-Cruz, L.; Dervenis, C.; Lacaine, F.; et al. A Randomized Trial of Chemoradiotherapy and Chemotherapy after Resection of Pancreatic Cancer. N. Engl. J. Med. 2004, 350, 1200–1210. [Google Scholar] [CrossRef] [Green Version]
- Oettle, H.; Neuhaus, P.; Hochhaus, A.; Hartmann, J.T.; Gellert, K.; Ridwelski, K.; Niedergethmann, M.; Zülke, C.; Fahlke, J.; Arning, M.B.; et al. Adjuvant Chemotherapy with Gemcitabine and Long-Term Outcomes among Patients with Resected Pancreatic Cancer: The CONKO-001 Randomized Trial. JAMA 2013, 310, 1473–1481. [Google Scholar] [CrossRef] [Green Version]
- Winter, J.M.; Brennan, M.F.; Tang, L.H.; D’Angelica, M.I.; Dematteo, R.P.; Fong, Y.; Klimstra, D.S.; Jarnagin, W.R.; Allen, P.J. Survival after Resection of Pancreatic Adenocarcinoma: Results from a Single Institution over Three Decades. Ann. Surg. Oncol. 2012, 19, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Dergham, S.T.; Dugan, M.C.; Kucway, R.; Du, W.; Kamarauskiene, D.S.; Vaitkevicius, V.K.; Crissman, J.D.; Sarkar, F.H. Prevalence and Clinical Significance of Combined K-Ras Mutation and P53 Aberration in Pancreatic Adenocarcinoma. Int. J. Pancreatol. 1997, 21, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Shibata, D.; Almoguera, C.; Forrester, K.; Dunitz, J.; Martin, S.E.; Cosgrove, M.M.; Perucho, M.; Arnheim, N. Detection of C-K-Ras Mutations in Fine Needle Aspirates from Human Pancreatic Adenocarcinomas. Cancer Res. 1990, 50, 1279–1283. [Google Scholar]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT Signaling Pathway in Human Cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kwok-Shing, P.; Kucherlapati, M.; Chen, F.; Liu, Y.; Tsang, Y.H.; de Velasco, G.; Jeong, K.J.; Akbani, R.; Hadjipanayis, A.; et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/MTOR Pathway Alterations. Cancer Cell 2017, 31, 820–832.e3. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.Q.; Ruggeri, B.; Klein, W.M.; Sonoda, G.; Altomare, D.A.; Watson, D.K.; Testa, J.R. Amplification of AKT2 in Human Pancreatic Cancer Cells and Inhibition of AKT2 Expression and Tumorigenicity by Antisense RNA. Proc. Natl. Acad. Sci. USA 1996, 93, 3636–3641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantley, L.C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Jazirehi, A.R.; Wenn, P.B.; Damavand, M. Therapeutic Implications of Targeting the PI3Kinase/AKT/MTOR Signaling Module in Melanoma Therapy. Am. J. Cancer Res. 2012, 2, 178–191. [Google Scholar] [PubMed]
- Totiger, T.M.; Srinivasan, S.; Jala, V.R.; Lamichhane, P.; Dosch, A.R.; Gaidarski, A.A.; Joshi, C.; Rangappa, S.; Castellanos, J.; Vemula, P.K.; et al. Urolithin A, a Novel Natural Compound to Target PI3K/AKT/MTOR Pathway in Pancreatic Cancer. Mol. Cancer Ther. 2019, 18, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, A.L.; Morton, J.P.; Manoharan, I.; Nelson, D.M.; Jamieson, N.B.; Pawlikowski, J.S.; McBryan, T.; Doyle, B.; McKay, C.; Oien, K.A.; et al. Activation of the PIK3CA/AKT Pathway Suppresses Senescence Induced by an Activated RAS Oncogene to Promote Tumorigenesis. Mol. Cell 2011, 42, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibault, B.; Ramos-Delgado, F.; Pons-Tostivint, E.; Therville, N.; Cintas, C.; Arcucci, S.; Cassant-Sourdy, S.; Reyes-Castellanos, G.; Tosolini, M.; Villard, A.V.; et al. Pancreatic Cancer Intrinsic PI3Kα Activity Accelerates Metastasis and Rewires Macrophage Component. EMBO Mol. Med. 2021, 13, e13502. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.N.; Maher, M.E.; Tran, N.H.; Van De Hey, D.R.; Foley, T.M.; Yueh, A.E.; Leystra, A.A.; Pasch, C.A.; Jeffrey, J.J.; Clipson, L.; et al. PIK3CA Mutations Can Initiate Pancreatic Tumorigenesis and Are Targetable with PI3K Inhibitors. Oncogenesis 2015, 4, e169. [Google Scholar] [CrossRef] [Green Version]
- Eser, S.; Reiff, N.; Messer, M.; Seidler, B.; Gottschalk, K.; Dobler, M.; Hieber, M.; Arbeiter, A.; Klein, S.; Kong, B.; et al. Selective Requirement of PI3K/PDK1 Signaling for Kras Oncogene-Driven Pancreatic Cell Plasticity and Cancer. Cancer Cell 2013, 23, 406–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimeno, A.; Tan, A.C.; Coffa, J.; Rajeshkumar, N.V.; Kulesza, P.; Rubio-Viqueira, B.; Wheelhouse, J.; Diosdado, B.; Messersmith, W.A.; Iacobuzio-Donahue, C.; et al. Coordinated Epidermal Growth Factor Receptor Pathway Gene Overexpression Predicts Epidermal Growth Factor Receptor Inhibitor Sensitivity in Pancreatic Cancer. Cancer Res. 2008, 68, 2841–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baer, R.; Cintas, C.; Dufresne, M.; Cassant-Sourdy, S.; Schönhuber, N.; Planque, L.; Lulka, H.; Couderc, B.; Bousquet, C.; Garmy-Susini, B.; et al. Pancreatic Cell Plasticity and Cancer Initiation Induced by Oncogenic Kras Is Completely Dependent on Wild-Type PI 3-Kinase P110α. Genes Dev. 2014, 28, 2621–2635. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Tomita, Y.; Hoshida, Y.; Morooka, T.; Nagano, H.; Dono, K.; Umeshita, K.; Sakon, M.; Ishikawa, O.; Ohigashi, H.; et al. Prognostic Significance of Activated Akt Expression in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2004, 10, 2846–2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, S.R.; Downes, C.P.; Gigg, R.; Grove, S.J.; Holmes, A.B.; Alessi, D.R. Specific Binding of the Akt-1 Protein Kinase to Phosphatidylinositol 3,4,5-Trisphosphate without Subsequent Activation. Biochem. J. 1996, 315 Pt 3, 709–713. [Google Scholar] [CrossRef]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.J.; Reese, C.B.; Cohen, P. Characterization of a 3-Phosphoinositide-Dependent Protein Kinase Which Phosphorylates and Activates Protein Kinase Bα. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-MTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggeri, B.A.; Huang, L.; Wood, M.; Cheng, J.Q.; Testa, J.R. Amplification and Overexpression of the AKT2 Oncogene in a Subset of Human Pancreatic Ductal Adenocarcinomas. Mol. Carcinog. 1998, 21, 81–86. [Google Scholar] [CrossRef]
- Schlieman, M.G.; Fahy, B.N.; Ramsamooj, R.; Beckett, L.; Bold, R.J. Incidence, Mechanism and Prognostic Value of Activated AKT in Pancreas Cancer. Br. J. Cancer 2003, 89, 2110–2115. [Google Scholar] [CrossRef] [PubMed]
- Memmott, R.M.; Dennis, P.A. Akt-Dependent and -Independent Mechanisms of MTOR Regulation in Cancer. Cell. Signal. 2009, 21, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-Dependent Activation of the MTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef] [Green Version]
- Faivre, S.; Kroemer, G.; Raymond, E. Current Development of MTOR Inhibitors as Anticancer Agents. Nat. Rev. Drug Discov. 2006, 5, 671–688. [Google Scholar] [CrossRef]
- Pópulo, H.; Lopes, J.M.; Soares, P. The MTOR Signalling Pathway in Human Cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed]
- Bellizzi, A.M.; Bloomston, M.; Zhou, X.-P.; Iwenofu, O.H.; Frankel, W.L. The MTOR Pathway Is Frequently Activated in Pancreatic Ductal Adenocarcinoma and Chronic Pancreatitis. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.M.; Shroff, R.T.; Xiong, H.; Varadhachary, G.A.; Fogelman, D.; Reddy, S.A.; Davis, D.; Zhang, Y.; Wolff, R.A.; Abbruzzese, J.L. Inhibition of the Mammalian Target of Rapamycin (MTOR) in Advanced Pancreatic Cancer: Results of Two Phase II Studies. BMC Cancer 2010, 10, 368. [Google Scholar] [CrossRef] [Green Version]
- Utomo, W.K.; Narayanan, V.; Biermann, K.; van Eijck, C.H.J.; Bruno, M.J.; Peppelenbosch, M.P.; Braat, H. MTOR Is a Promising Therapeutical Target in a Subpopulation of Pancreatic Adenocarcinoma. Cancer Lett. 2014, 346, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.; Calvopina, J.H.; Kim, C.; Wang, Y.; Dawson, D.W.; Donahue, T.R.; Dry, S.; Wu, H. PTEN Loss Accelerates KrasG12D-Induced Pancreatic Cancer Development. Cancer Res. 2010, 70, 7114–7124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanger, B.Z.; Stiles, B.; Lauwers, G.Y.; Bardeesy, N.; Mendoza, M.; Wang, Y.; Greenwood, A.; Cheng, K.; McLaughlin, M.; Brown, D.; et al. Pten Constrains Centroacinar Cell Expansion and Malignant Transformation in the Pancreas. Cancer Cell 2005, 8, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Bondar, V.M.; Sweeney-Gotsch, B.; Andreeff, M.; Mills, G.B.; McConkey, D.J. Inhibition of the Phosphatidylinositol 3′-Kinase-AKT Pathway Induces Apoptosis in Pancreatic Carcinoma Cells in Vitro and in Vivo. Mol. Cancer Ther. 2002, 1, 989–997. [Google Scholar]
- Yip-Schneider, M.T.; Wiesenauer, C.A.; Schmidt, C.M. Inhibition of the Phosphatidylinositol 3′-Kinase Signaling Pathway Increases the Responsiveness of Pancreatic Carcinoma Cells to Sulindac. J. Gastrointest. Surg. 2003, 7, 354–363. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Yan, L.; Patnaik, A.; Fearen, I.; Olmos, D.; Papadopoulos, K.; Baird, R.D.; Delgado, L.; Taylor, A.; Lupinacci, L.; et al. First-in-Man Clinical Trial of the Oral Pan-AKT Inhibitor MK-2206 in Patients With Advanced Solid Tumors. JCO 2011, 29, 4688–4695. [Google Scholar] [CrossRef]
- Hu, C.; Dadon, T.; Chenna, V.; Yabuuchi, S.; Bannerji, R.; Booher, R.; Strack, P.; Azad, N.; Nelkin, B.D.; Maitra, A. Combined Inhibition of Cyclin-Dependent Kinases (Dinaciclib) and AKT (MK-2206) Blocks Pancreatic Tumor Growth and Metastases in Patient-Derived Xenograft Models. Mol. Cancer Ther. 2015, 14, 1532–1539. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of Insulin Feedback Enhances the Efficacy of PI3K Inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, K.E.; Rojo, F.; She, Q.-B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. MTOR Inhibition Induces Upstream Receptor Tyrosine Kinase Signaling and Activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Cao, P.; Maira, S.-M.; García-Echeverría, C.; Hedley, D.W. Activity of a Novel, Dual PI3-Kinase/MTor Inhibitor NVP-BEZ235 against Primary Human Pancreatic Cancers Grown as Orthotopic Xenografts. Br. J. Cancer 2009, 100, 1267–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahreddine, H.; Borden, K.L.B. Mechanisms and Insights into Drug Resistance in Cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.H.; Xue, A.; Baxter, R.C.; Pavlakis, N.; Smith, R.C. Upstream and Downstream Co-Inhibition of Mitogen-Activated Protein Kinase and PI3K/Akt/MTOR Pathways in Pancreatic Ductal Adenocarcinoma. Neoplasia 2016, 18, 425–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alagesan, B.; Contino, G.; Guimaraes, A.R.; Corcoran, R.B.; Deshpande, V.; Wojtkiewicz, G.R.; Hezel, A.F.; Wong, K.-K.; Loda, M.; Weissleder, R.; et al. Combined MEK and PI3K Inhibition in a Mouse Model of Pancreatic Cancer. Clin. Cancer Res. 2015, 21, 396–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junttila, M.R.; Devasthali, V.; Cheng, J.H.; Castillo, J.; Metcalfe, C.; Clermont, A.C.; Otter, D.D.; Chan, E.; Bou-Reslan, H.; Cao, T.; et al. Modeling Targeted Inhibition of MEK and PI3 Kinase in Human Pancreatic Cancer. Mol. Cancer Ther. 2015, 14, 40–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, W.S.; McDonald, P.C.; Nemirovsky, O.; Awrey, S.; Chafe, S.C.; Schaeffer, D.F.; Li, J.; Renouf, D.J.; Stanger, B.Z.; Dedhar, S. Overcoming Adaptive Resistance to KRAS and MEK Inhibitors by Co-Targeting MTORC1/2 Complexes in Pancreatic Cancer. Cell Rep. Med. 2020, 1, 100131. [Google Scholar] [CrossRef] [PubMed]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175.e27. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Baghdadi, M.; Tsuchikawa, T.; Wada, H.; Nakamura, T.; Abe, H.; Nakanishi, S.; Usui, Y.; Higuchi, K.; Takahashi, M.; et al. Chemotherapy-Derived Inflammatory Responses Accelerate the Formation of Immunosuppressive Myeloid Cells in the Tissue Microenvironment of Human Pancreatic Cancer. Cancer Res. 2015, 75, 2629–2640. [Google Scholar] [CrossRef] [Green Version]
- Winograd, R.; Byrne, K.T.; Evans, R.A.; Odorizzi, P.M.; Meyer, A.R.L.; Bajor, D.L.; Clendenin, C.; Stanger, B.Z.; Furth, E.E.; Wherry, E.J.; et al. Induction of T-Cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol. Res. 2015, 3, 399–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The Pancreas Cancer Microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, T.; Cantley, L. PI3K Pathway Alterations in Cancer: Variations on a Theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.-B.; Bousquet-Dubouch, M.-P.; et al. Pharmacological Targeting of the Protein Synthesis MTOR/4E-BP1 Pathway in Cancer-Associated Fibroblasts Abrogates Pancreatic Tumour Chemoresistance. EMBO Mol. Med. 2015, 7, 735–753. [Google Scholar] [CrossRef]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Fujitani, Y.; Fujitani, S.; Wright, C.V.E.; Moses, H.L. Aggressive Pancreatic Ductal Adenocarcinoma in Mice Caused by Pancreas-Specific Blockade of Transforming Growth Factor-Beta Signaling in Cooperation with Active Kras Expression. Genes Dev. 2006, 20, 3147–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Li, M.; Yang, Y.; Liu, Y.; Xie, H.; Yu, Q.; Tian, L.; Tang, X.; Ren, K.; Li, J.; et al. Remodeling Tumor Immune Microenvironment via Targeted Blockade of PI3K-γ and CSF-1/CSF-1R Pathways in Tumor Associated Macrophages for Pancreatic Cancer Therapy. J. Control. Release 2020, 321, 23–35. [Google Scholar] [CrossRef]
- Ali, K.; Soond, D.R.; Piñeiro, R.; Hagemann, T.; Pearce, W.; Lim, E.L.; Bouabe, H.; Scudamore, C.L.; Hancox, T.; Maecker, H.; et al. Inactivation of PI(3)K P110δ Breaks Regulatory T-Cell-Mediated Immune Tolerance to Cancer. Nature 2014, 510, 407–411. [Google Scholar] [CrossRef] [Green Version]
- Kaneda, M.M.; Cappello, P.; Nguyen, A.V.; Ralainirina, N.; Hardamon, C.R.; Foubert, P.; Schmid, M.C.; Sun, P.; Mose, E.; Bouvet, M.; et al. Macrophage PI3Kγ Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov. 2016, 6, 870–885. [Google Scholar] [CrossRef] [Green Version]
- Lien, E.C.; Dibble, C.C.; Toker, A. PI3K Signaling in Cancer: Beyond AKT. Curr. Opin. Cell Biol. 2017, 45, 62–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truitt, M.; Olson, P.; et al. Bruton Tyrosine Kinase–Dependent Immune Cell Cross-Talk Drives Pancreas Cancer. Cancer Discov. 2016, 6, 270–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehra, S.; Deshpande, N.; Nagathihalli, N. Targeting PI3K Pathway in Pancreatic Ductal Adenocarcinoma: Rationale and Progress. Cancers 2021, 13, 4434. https://doi.org/10.3390/cancers13174434
Mehra S, Deshpande N, Nagathihalli N. Targeting PI3K Pathway in Pancreatic Ductal Adenocarcinoma: Rationale and Progress. Cancers. 2021; 13(17):4434. https://doi.org/10.3390/cancers13174434
Chicago/Turabian StyleMehra, Siddharth, Nilesh Deshpande, and Nagaraj Nagathihalli. 2021. "Targeting PI3K Pathway in Pancreatic Ductal Adenocarcinoma: Rationale and Progress" Cancers 13, no. 17: 4434. https://doi.org/10.3390/cancers13174434
APA StyleMehra, S., Deshpande, N., & Nagathihalli, N. (2021). Targeting PI3K Pathway in Pancreatic Ductal Adenocarcinoma: Rationale and Progress. Cancers, 13(17), 4434. https://doi.org/10.3390/cancers13174434