
Crosstalk between Environmental Inflammatory Stimuli and Non-Coding RNA in Cancer Occurrence and Development


Abstract
:Simple Summary
Abstract
1. Introduction
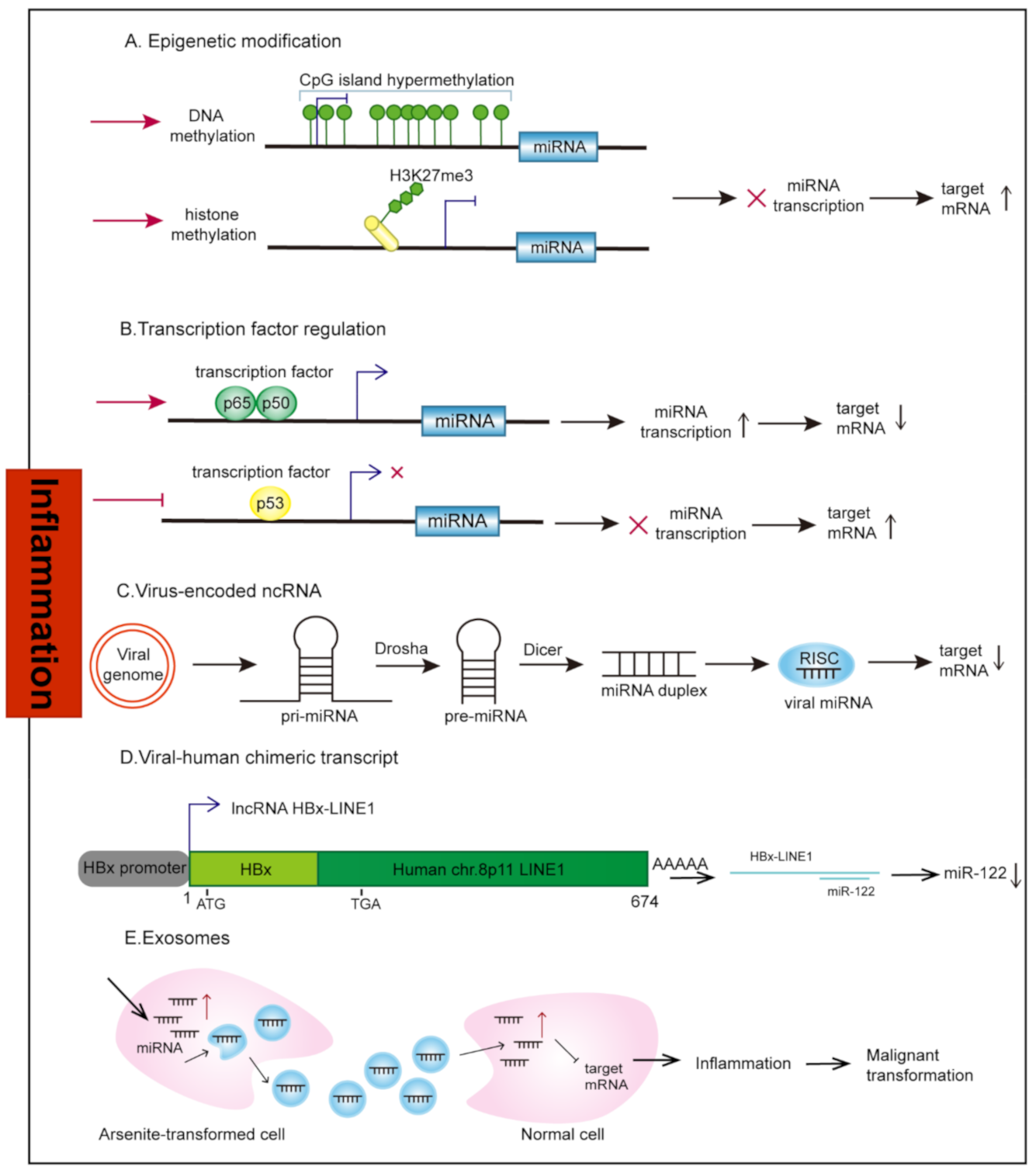
2. Aberrant Regulation of ncRNAs under Inflammation Stimuli
2.1. Transcriptional Regulation
2.1.1. Epigenetic Modification
2.1.2. Transcription Factors
2.2. Virus-Encoded ncRNAs
2.3. Human-Virus Fusion ncRNAs
2.4. Exosomes
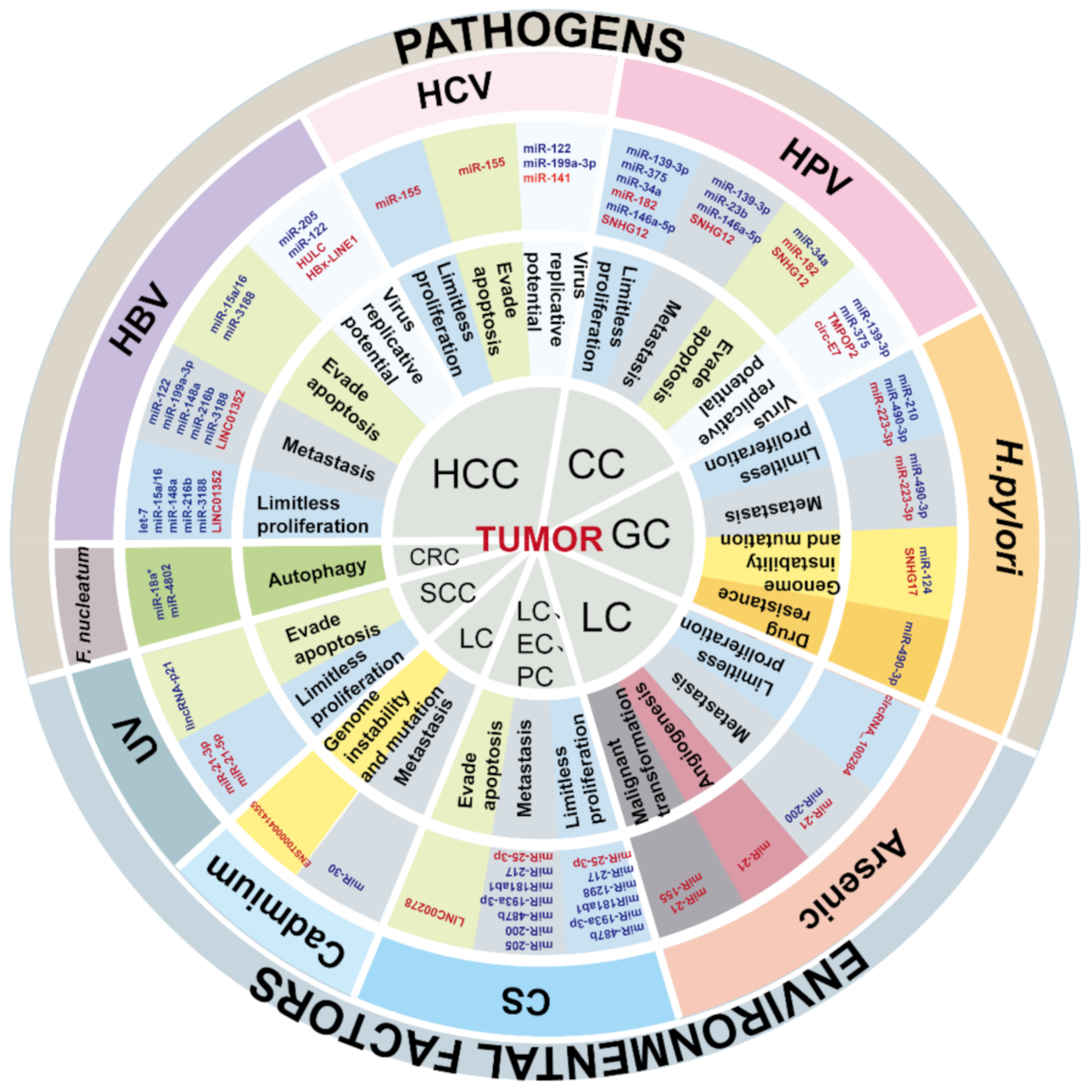
3. Cancers Caused by Pathogens
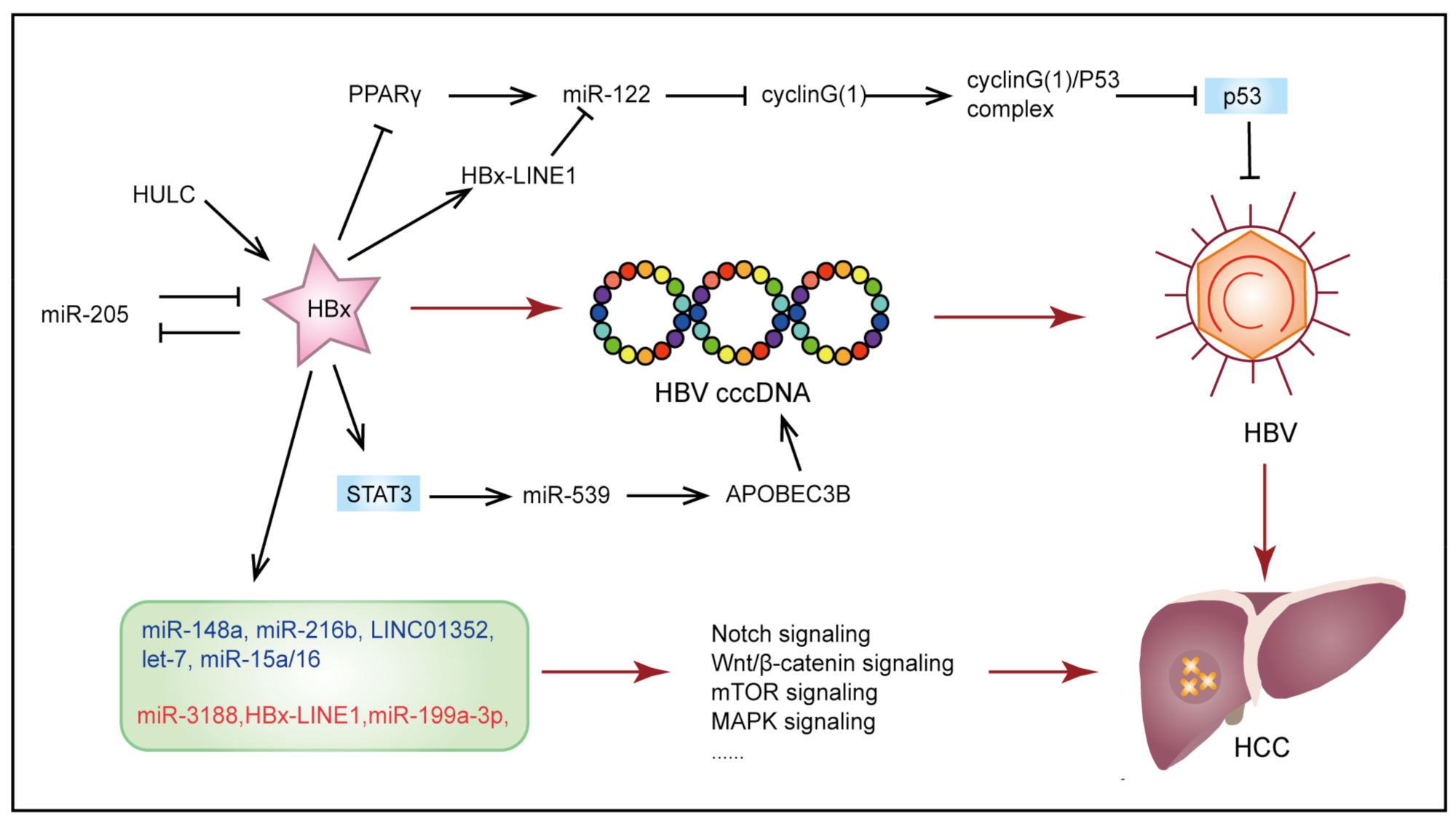
3.1. Hepatitis B Virus-Induced Hepatocellular Carcinoma
3.2. Human Papillomavirus-Induced Cervical Cancer
3.3. Helicobacter pylori-Induced Gastric Cancer
3.4. Cancers Associated with Other Pathogens
3.4.1. Intestinal Flora-Induced Colorectal Cancer
3.4.2. Hepatitis C Virus-Induced Hepatocellular Carcinoma
4. Cancers Associated with Environmental Factors
4.1. Inhalation Exposure-Induced Cancer
4.1.1. Cigarette Smoke
4.1.2. Lung Cancer
4.1.3. Esophageal Cancer
4.1.4. Pancreatic Cancer
4.2. Ingestion Exposure-Induced Cancers
4.2.1. Arsenic
4.2.2. Cadmium
4.3. Dermal Contact Exposure-Induced Cancers
Ultraviolet
5. ncRNAs as Therapeutic Targets
6. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Balkwill, F.; Mantovani, A. Inflammation and Cancer: Back to Virchow? Lancet Lond. Engl. 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Correa, P. Human Gastric Carcinogenesis: A Multistep and Multifactorial Process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar] [PubMed]
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and Colon Cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Crick, F. Central Dogma of Molecular Biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. Elegans Heterochronic Gene Lin-4 Encodes Small RNAs with Antisense Complementarity to Lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Alexander, R.P.; Fang, G.; Rozowsky, J.; Snyder, M.; Gerstein, M.B. Annotating Non-Coding Regions of the Genome. Nat. Rev. Genet. 2010, 11, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Parsons, C.; Walker, L.; Zhang, W.C.; Slack, F.J. Targeting Noncoding RNAs in Disease. J. Clin. Investig. 2017, 127, 761–771. [Google Scholar] [CrossRef]
- Quinn, J.J.; Chang, H.Y. Unique Features of Long Non-Coding RNA Biogenesis and Function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- O’Gorman, A.; Colleran, A.; Ryan, A.; Mann, J.; Egan, L.J. Regulation of NF-KappaB Responses by Epigenetic Suppression of IkappaBalpha Expression in HCT116 Intestinal Epithelial Cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G96–G105. [Google Scholar] [CrossRef]
- Hur, K.; Niwa, T.; Toyoda, T.; Tsukamoto, T.; Tatematsu, M.; Yang, H.-K.; Ushijima, T. Insufficient Role of Cell Proliferation in Aberrant DNA Methylation Induction and Involvement of Specific Types of Inflammation. Carcinogenesis 2011, 32, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Niwa, T.; Tsukamoto, T.; Toyoda, T.; Mori, A.; Tanaka, H.; Maekita, T.; Ichinose, M.; Tatematsu, M.; Ushijima, T. Inflammatory Processes Triggered by Helicobacter Pylori Infection Cause Aberrant DNA Methylation in Gastric Epithelial Cells. Cancer Res. 2010, 70, 1430–1440. [Google Scholar] [CrossRef] [Green Version]
- McDonald, O.G.; Wu, H.; Timp, W.; Doi, A.; Feinberg, A.P. Genome-Scale Epigenetic Reprogramming during Epithelial-to-Mesenchymal Transition. Nat. Struct. Mol. Biol. 2011, 18, 867–874. [Google Scholar] [CrossRef]
- Szulakowski, P.; Crowther, A.J.L.; Jiménez, L.A.; Donaldson, K.; Mayer, R.; Leonard, T.B.; MacNee, W.; Drost, E.M. The Effect of Smoking on the Transcriptional Regulation of Lung Inflammation in Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2006, 174, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Sundar, I.K.; Nevid, M.Z.; Friedman, A.E.; Rahman, I. Cigarette Smoke Induces Distinct Histone Modifications in Lung Cells: Implications for the Pathogenesis of COPD and Lung Cancer. J. Proteome Res. 2014, 13, 982–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, G.; Karin, M. NF-ΚB and STAT3—Key Players in Liver Inflammation and Cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodge, D.R.; Hurt, E.M.; Farrar, W.L. The Role of IL-6 and STAT3 in Inflammation and Cancer. Eur. J. Cancer 2005, 41, 2502–2512. [Google Scholar] [CrossRef] [PubMed]
- Sheedy, F.J.; Palsson-McDermott, E.; Hennessy, E.J.; Martin, C.; O’Leary, J.J.; Ruan, Q.; Johnson, D.S.; Chen, Y.; O’Neill, L.A.J. Negative Regulation of TLR4 via Targeting of the Proinflammatory Tumor Suppressor PDCD4 by the MicroRNA MiR-21. Nat. Immunol. 2010, 11, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Jaeger, S.A.; Hirsch, H.A.; Bulyk, M.L.; Struhl, K. STAT3 Activation of MiR-21 and MiR-181b-1 via PTEN and CYLD Are Part of the Epigenetic Switch Linking Inflammation to Cancer. Mol. Cell 2010, 39, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Rigby, C.M.; Roy, S.; Deep, G.; Guillermo-Lagae, R.; Jain, A.K.; Dhar, D.; Orlicky, D.J.; Agarwal, C.; Agarwal, R. Role of P53 in Silibinin-Mediated Inhibition of Ultraviolet B Radiation-Induced DNA Damage, Inflammation and Skin Carcinogenesis. Carcinogenesis 2017, 38, 40–50. [Google Scholar] [CrossRef]
- Cooks, T.; Harris, C.C.; Oren, M. Caught in the Cross Fire: P53 in Inflammation. Carcinogenesis 2014, 35, 1680–1690. [Google Scholar] [CrossRef]
- Levine, A.J.; Oren, M. The First 30 Years of P53: Growing Ever More Complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Rokavec, M.; Li, H.; Jiang, L.; Hermeking, H. The P53/MiR-34 Axis in Development and Disease. J. Mol. Cell Biol. 2014, 6, 214–230. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.; Zavolan, M.; Grässer, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of Virus-Encoded MicroRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef]
- Umbach, J.L.; Nagel, M.A.; Cohrs, R.J.; Gilden, D.H.; Cullen, B.R. Analysis of Human Alphaherpesvirus MicroRNA Expression in Latently Infected Human Trigeminal Ganglia. J. Virol. 2009, 83, 10677–10683. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Cullen, B.R. Analysis of the Interaction of Primate Retroviruses with the Human RNA Interference Machinery. J. Virol. 2007, 81, 12218–12226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, B.R. Five Questions about Viruses and MicroRNAs. PLoS Pathog. 2010, 6, e1000787. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grässer, F.A.; van Dyk, L.F.; Ho, C.K.; Shuman, S.; Chien, M.; et al. Identification of MicroRNAs of the Herpesvirus Family. Nat. Methods 2005, 2, 269–276. [Google Scholar] [CrossRef]
- Rossetto, C.C.; Tarrant-Elorza, M.; Verma, S.; Purushothaman, P.; Pari, G.S. Regulation of Viral and Cellular Gene Expression by Kaposi’s Sarcoma-Associated Herpesvirus Polyadenylated Nuclear RNA. J. Virol. 2013, 87, 5540–5553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortes, P.; Morris, K.V. Long Noncoding RNAs in Viral Infections. Virus Res. 2016, 212, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cullen, B.R. Viruses and MicroRNAs. Nat. Genet. 2006, 38, S25–S30. [Google Scholar] [CrossRef]
- Lau, C.-C.; Sun, T.; Ching, A.K.K.; He, M.; Li, J.-W.; Wong, A.M.; Co, N.N.; Chan, A.W.H.; Li, P.-S.; Lung, R.W.M.; et al. Viral-Human Chimeric Transcript Predisposes Risk to Liver Cancer Development and Progression. Cancer Cell 2014, 25, 335–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bello-Morales, R.; Praena, B.; de la Nuez, C.; Rejas, M.T.; Guerra, M.; Galán-Ganga, M.; Izquierdo, M.; Calvo, V.; Krummenacher, C.; López-Guerrero, J.A. Role of Microvesicles in the Spread of Herpes Simplex Virus 1 in Oligodendrocytic Cells. J. Virol. 2018, 92, e00088-18. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Han, Q.; Hou, Z.; Zhang, C.; Tian, Z.; Zhang, J. Exosomes Mediate Hepatitis B Virus (HBV) Transmission and NK-Cell Dysfunction. Cell. Mol. Immunol. 2017, 14, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Bukong, T.N.; Momen-Heravi, F.; Kodys, K.; Bala, S.; Szabo, G. Exosomes from Hepatitis C Infected Patients Transmit HCV Infection and Contain Replication Competent Viral RNA in Complex with Ago2-MiR122-HSP90. PLoS Pathog. 2014, 10, e1004424. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnaiah, V.; Thumann, C.; Fofana, I.; Habersetzer, F.; Pan, Q.; de Ruiter, P.E.; Willemsen, R.; Demmers, J.A.A.; Stalin Raj, V.; Jenster, G.; et al. Exosome-Mediated Transmission of Hepatitis C Virus between Human Hepatoma Huh7.5 Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13109–13113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenzie, A.J.; Hoshino, D.; Hong, N.H.; Cha, D.J.; Franklin, J.L.; Coffey, R.J.; Patton, J.G.; Weaver, A.M. KRAS-MEK Signaling Controls Ago2 Sorting into Exosomes. Cell Rep. 2016, 15, 978–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarroya-Beltri, C.; Gutiérrez-Vázquez, C.; Sánchez-Cabo, F.; Pérez-Hernández, D.; Vázquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sánchez-Madrid, F. Sumoylated HnRNPA2B1 Controls the Sorting of MiRNAs into Exosomes through Binding to Specific Motifs. Nat. Commun. 2013, 4, 2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shurtleff, M.J.; Temoche-Diaz, M.M.; Karfilis, K.V.; Ri, S.; Schekman, R. Y-Box Protein 1 Is Required to Sort MicroRNAs into Exosomes in Cells and in a Cell-Free Reaction. eLife 2016, 5, e19276. [Google Scholar] [CrossRef]
- Armstrong, H.; Bording-Jorgensen, M.; Dijk, S.; Wine, E. The Complex Interplay between Chronic Inflammation, the Microbiome, and Cancer: Understanding Disease Progression and What We Can Do to Prevent It. Cancers 2018, 10, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuper, H.; Adami, H.O.; Trichopoulos, D. Infections as a Major Preventable Cause of Human Cancer. J. Intern. Med. 2000, 248, 171–183. [Google Scholar] [CrossRef]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of Fusobacterium Persistence and Antibiotic Response in Colorectal Cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A Review of Human Carcinogens--Part B: Biological Agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Hoshida, Y.; Nijman, S.M.B.; Kobayashi, M.; Chan, J.A.; Brunet, J.-P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative Transcriptome Analysis Reveals Common Molecular Subclasses of Human Hepatocellular Carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef] [Green Version]
- Levrero, M.; Pollicino, T.; Petersen, J.; Belloni, L.; Raimondo, G.; Dandri, M. Control of CccDNA Function in Hepatitis B Virus Infection. J. Hepatol. 2009, 51, 581–592. [Google Scholar] [CrossRef] [Green Version]
- Pollicino, T.; Belloni, L.; Raffa, G.; Pediconi, N.; Squadrito, G.; Raimondo, G.; Levrero, M. Hepatitis B Virus Replication Is Regulated by the Acetylation Status of Hepatitis B Virus CccDNA-Bound H3 and H4 Histones. Gastroenterology 2006, 130, 823–837. [Google Scholar] [CrossRef]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B Virus X Protein Is Essential to Initiate and Maintain Virus Replication after Infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx Binds the HBV Minichromosome and Modifies the Epigenetic Regulation of CccDNA Function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrieri, F.; Belloni, L.; D’Andrea, D.; Pediconi, N.; Le Pera, L.; Testoni, B.; Scisciani, C.; Floriot, O.; Zoulim, F.; Tramontano, A.; et al. Genome-Wide Identification of Direct HBx Genomic Targets. BMC Genom. 2017, 18, 184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, J.; Cui, M.; Liu, F.; You, X.; Du, Y.; Gao, Y.; Zhang, S.; Lu, Z.; Ye, L.; et al. Hepatitis B Virus X Protein Inhibits Tumor Suppressor MiR-205 through Inducing Hypermethylation of MiR-205 Promoter to Enhance Carcinogenesis. Neoplasia 2013, 15, 1282–1291. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Feng, J.; Sun, M.; Yang, G.; Yuan, H.; Wang, Y.; Bu, Y.; Zhao, M.; Zhang, S.; Zhang, X. Long Non-Coding RNA HULC Activates HBV by Modulating HBx/STAT3/MiR-539/APOBEC3B Signaling in HBV-Related Hepatocellular Carcinoma. Cancer Lett. 2019, 454, 158–170. [Google Scholar] [CrossRef]
- Song, K.; Han, C.; Zhang, J.; Lu, D.; Dash, S.; Feitelson, M.; Lim, K.; Wu, T. Epigenetic Regulation of MiR-122 by PPARγ and Hepatitis B Virus X Protein in Hepatocellular Carcinoma Cells. Hepatology 2013, 58, 1681–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.-W.; Wang, N.; Wang, Y.; Wang, F.; Fu, Z.; Yan, X.; Zhu, H.; Diao, W.; Ding, Y.; Chen, X.; et al. Hepatitis B Virus-Human Chimeric Transcript HBx-LINE1 Promotes Hepatic Injury via Sequestering Cellular MicroRNA-122. J. Hepatol. 2016, 64, 278–291. [Google Scholar] [CrossRef]
- Wang, S.; Qiu, L.; Yan, X.; Jin, W.; Wang, Y.; Chen, L.; Wu, E.; Ye, X.; Gao, G.F.; Wang, F.; et al. Loss of MicroRNA 122 Expression in Patients with Hepatitis B Enhances Hepatitis B Virus Replication through Cyclin G(1)-Modulated P53 Activity. Hepatology 2012, 55, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.-S.; Chen, Y.; Yang, G.; Liao, Z.-B.; Zhang, H.-W.; Liang, H.-F.; Chen, X.-P.; Dong, H.-H. TGF-Β1 Accelerates the Hepatitis B Virus X-Induced Malignant Transformation of Hepatic Progenitor Cells by Upregulating MiR-199a-3p. Oncogene 2020, 39, 1807–1820. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Yu, F.; Xiao, Z.; Xu, K.; Xu, J.; Tang, W.; Wang, J.; Song, E. Hepatitis B Virus X Protein Downregulates Expression of the MiR-16 Family in Malignant Hepatocytes in Vitro. Br. J. Cancer 2011, 105, 146–153. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, Y.; Toh, S.T.; Sung, W.-K.; Tan, P.; Chow, P.; Chung, A.Y.F.; Jooi, L.L.P.; Lee, C.G.L. Lethal-7 Is down-Regulated by the Hepatitis B Virus x Protein and Targets Signal Transducer and Activator of Transcription 3. J. Hepatol. 2010, 53, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Fan, Z.; Kang, L.; Han, J.; Jiang, C.; Zheng, X.; Zhu, Z.; Jiao, H.; Lin, J.; Jiang, K.; et al. Hepatitis B Virus X Protein Represses MiRNA-148a to Enhance Tumorigenesis. J. Clin. Investig. 2013, 123, 630–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.-Y.; Zhou, S.-J.; Deng, Y.-L.; Zhang, Z.-Y.; Zhang, E.-L.; Wu, Z.-B.; Huang, Z.-Y.; Chen, X.-P. MiR-216b Is Involved in Pathogenesis and Progression of Hepatocellular Carcinoma through HBx-MiR-216b-IGF2BP2 Signaling Pathway. Cell Death Dis. 2015, 6, e1670. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.-J.; Deng, Y.-L.; Liang, H.-F.; Jaoude, J.C.; Liu, F.-Y. Hepatitis B Virus X Protein Promotes CREB-Mediated Activation of MiR-3188 and Notch Signaling in Hepatocellular Carcinoma. Cell Death Differ. 2017, 24, 1577–1587. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Xu, Q.; Yan, Y.; Lu, Y.; Hu, Z.; Ou, B.; Zhang, H.; Mao, K.; Zhang, J.; Wang, J.; et al. HBx/ERα Complex-Mediated LINC01352 Downregulation Promotes HBV-Related Hepatocellular Carcinoma via the MiR-135b-APC Axis. Oncogene 2020, 39, 3774–3789. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, N.; Bosch, F.X.; de Sanjosé, S.; Herrero, R.; Castellsagué, X.; Shah, K.V.; Snijders, P.J.F.; Meijer, C.J.L.M.; International Agency for Research on Cancer Multicenter Cervical Cancer Study Group. Epidemiologic Classification of Human Papillomavirus Types Associated with Cervical Cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Mazibrada, J.; Rittà, M.; Mondini, M.; De Andrea, M.; Azzimonti, B.; Borgogna, C.; Ciotti, M.; Orlando, A.; Surico, N.; Chiusa, L.; et al. Interaction between Inflammation and Angiogenesis during Different Stages of Cervical Carcinogenesis. Gynecol. Oncol. 2008, 108, 112–120. [Google Scholar] [CrossRef]
- Punt, S.; Houwing-Duistermaat, J.J.; Schulkens, I.A.; Thijssen, V.L.; Osse, E.M.; de Kroon, C.D.; Griffioen, A.W.; Fleuren, G.J.; Gorter, A.; Jordanova, E.S. Correlations between Immune Response and Vascularization QRT-PCR Gene Expression Clusters in Squamous Cervical Cancer. Mol. Cancer 2015, 14, 71. [Google Scholar] [CrossRef] [PubMed]
- Walch-Rückheim, B.; Ströder, R.; Theobald, L.; Pahne-Zeppenfeld, J.; Hegde, S.; Kim, Y.-J.; Bohle, R.M.; Juhasz-Böss, I.; Solomayer, E.-F.; Smola, S. Cervical Cancer-Instructed Stromal Fibroblasts Enhance IL23 Expression in Dendritic Cells to Support Expansion of Th17 Cells. Cancer Res. 2019, 79, 1573–1586. [Google Scholar] [CrossRef] [Green Version]
- Schröer, N.; Pahne, J.; Walch, B.; Wickenhauser, C.; Smola, S. Molecular Pathobiology of Human Cervical High-Grade Lesions: Paracrine STAT3 Activation in Tumor-Instructed Myeloid Cells Drives Local MMP-9 Expression. Cancer Res. 2011, 71, 87–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forman, D.; de Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M.; et al. Global Burden of Human Papillomavirus and Related Diseases. Vaccine 2012, 30 (Suppl. 5), F12–F23. [Google Scholar] [CrossRef] [Green Version]
- Hoppe-Seyler, K.; Bossler, F.; Braun, J.A.; Herrmann, A.L.; Hoppe-Seyler, F. The HPV E6/E7 Oncogenes: Key Factors for Viral Carcinogenesis and Therapeutic Targets. Trends Microbiol. 2018, 26, 158–168. [Google Scholar] [CrossRef]
- Roman, A.; Munger, K. The Papillomavirus E7 Proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vande Pol, S.B.; Klingelhutz, A.J. Papillomavirus E6 Oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sannigrahi, M.K.; Sharma, R.; Singh, V.; Panda, N.K.; Rattan, V.; Khullar, M. Role of Host MiRNA Hsa-MiR-139-3p in HPV-16-Induced Carcinomas. Clin. Cancer Res. 2017, 23, 3884–3895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.M.; Phillips, B.L.; Chan, E.K. MiR-375 Activates P21 and Suppresses Telomerase Activity by Coordinately Regulating HPV E6/E7, E6AP, CIP2A, and 14-3-3ζ. Mol. Cancer 2014, 13, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP Complex Functions as a Ubiquitin-Protein Ligase in the Ubiquitination of P53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- He, H.; Liu, X.; Liu, Y.; Zhang, M.; Lai, Y.; Hao, Y.; Wang, Q.; Shi, D.; Wang, N.; Luo, X.-G.; et al. Human Papillomavirus E6/E7 and Long Noncoding RNA TMPOP2 Mutually Upregulated Gene Expression in Cervical Cancer Cells. J. Virol. 2019, 93, e01808-18. [Google Scholar] [CrossRef] [Green Version]
- Qian, K.; Pietilä, T.; Rönty, M.; Michon, F.; Frilander, M.J.; Ritari, J.; Tarkkanen, J.; Paulín, L.; Auvinen, P.; Auvinen, E. Identification and Validation of Human Papillomavirus Encoded MicroRNAs. PLoS ONE 2013, 8, e70202. [Google Scholar] [CrossRef]
- Zhao, J.; Lee, E.E.; Kim, J.; Yang, R.; Chamseddin, B.; Ni, C.; Gusho, E.; Xie, Y.; Chiang, C.-M.; Buszczak, M.; et al. Transforming Activity of an Oncoprotein-Encoding Circular RNA from Human Papillomavirus. Nat. Commun. 2019, 10, 2300. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, H.-K.; McCoy, J.P.; Banerjee, N.S.; Rader, J.S.; Broker, T.R.; Meyers, C.; Chow, L.T.; Zheng, Z.-M. Oncogenic HPV Infection Interrupts the Expression of Tumor-Suppressive MiR-34a through Viral Oncoprotein E6. RNA 2009, 15, 637–647. [Google Scholar] [CrossRef] [Green Version]
- Au Yeung, C.L.; Tsang, T.Y.; Yau, P.L.; Kwok, T.T. Human Papillomavirus Type 16 E6 Induces Cervical Cancer Cell Migration through the P53/MicroRNA-23b/Urokinase-Type Plasminogen Activator Pathway. Oncogene 2011, 30, 2401–2410. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Deng, Y.; Ao, L.; Song, Y.; Xu, Y.; Wang, C.C.; Choy, K.W.; Tony Chung, K.H.; Du, Q.; Sui, Y.; et al. The High-Risk HPV Oncogene E7 Upregulates MiR-182 Expression through the TGF-β/Smad Pathway in Cervical Cancer. Cancer Lett. 2019, 460, 75–85. [Google Scholar] [CrossRef]
- Peta, E.; Sinigaglia, A.; Masi, G.; Di Camillo, B.; Grassi, A.; Trevisan, M.; Messa, L.; Loregian, A.; Manfrin, E.; Brunelli, M.; et al. HPV16 E6 and E7 Upregulate the Histone Lysine Demethylase KDM2B through the C-MYC/MiR-146a-5p Axys. Oncogene 2018, 37, 1654–1668. [Google Scholar] [CrossRef]
- Lai, S.-Y.; Guan, H.-M.; Liu, J.; Huang, L.-J.; Hu, X.-L.; Chen, Y.-H.; Wu, Y.-H.; Wang, Y.; Yang, Q.; Zhou, J.-Y. Long Noncoding RNA SNHG12 Modulated by Human Papillomavirus 16 E6/E7 Promotes Cervical Cancer Progression via ERK/Slug Pathway. J. Cell. Physiol. 2020, 235, 7911–7922. [Google Scholar] [CrossRef]
- Fischbach, W.; Malfertheiner, P. Helicobacter Pylori Infection. Dtsch. Arzteblatt Int. 2018, 115, 429–436. [Google Scholar] [CrossRef]
- Schistosomes, Liver Flukes and Helicobacter Pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7–14 June 1994. IARC Monogr. Eval. Carcinog. Risks Hum. 1994, 61, 1–241. [Google Scholar]
- Shen, J.; Xiao, Z.; Wu, W.K.K.; Wang, M.H.; To, K.F.; Chen, Y.; Yang, W.; Li, M.S.M.; Shin, V.Y.; Tong, J.H.; et al. Epigenetic Silencing of MiR-490-3p Reactivates the Chromatin Remodeler SMARCD1 to Promote Helicobacter Pylori-Induced Gastric Carcinogenesis. Cancer Res. 2015, 75, 754–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Khalafi, S.; Chen, Z.; Poveda, J.; Peng, D.; Lu, H.; Soutto, M.; Que, J.; Garcia-Buitrago, M.; Zaika, A.; et al. Silencing of MiR490-3p by H. Pylori Activates DARPP-32 and Induces Resistance to Gefitinib. Cancer Lett. 2020, 491, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Kiga, K.; Mimuro, H.; Suzuki, M.; Shinozaki-Ushiku, A.; Kobayashi, T.; Sanada, T.; Kim, M.; Ogawa, M.; Iwasaki, Y.W.; Kayo, H.; et al. Epigenetic Silencing of MiR-210 Increases the Proliferation of Gastric Epithelium during Chronic Helicobacter Pylori Infection. Nat. Commun. 2014, 5, 4497. [Google Scholar] [CrossRef] [PubMed]
- Murray-Stewart, T.; Sierra, J.C.; Piazuelo, M.B.; Mera, R.M.; Chaturvedi, R.; Bravo, L.E.; Correa, P.; Schneider, B.G.; Wilson, K.T.; Casero, R.A. Epigenetic Silencing of MiR-124 Prevents Spermine Oxidase Regulation: Implications for Helicobacter Pylori-Induced Gastric Cancer. Oncogene 2016, 35, 5480–5488. [Google Scholar] [CrossRef] [Green Version]
- Crabtree, J.E.; Taylor, J.D.; Wyatt, J.I.; Heatley, R.V.; Shallcross, T.M.; Tompkins, D.S.; Rathbone, B.J. Mucosal IgA Recognition of Helicobacter Pylori 120 KDa Protein, Peptic Ulceration, and Gastric Pathology. Lancet 1991, 338, 332–335. [Google Scholar] [CrossRef]
- Backert, S.; Selbach, M. Role of Type IV Secretion in Helicobacter Pylori Pathogenesis. Cell. Microbiol. 2008, 10, 1573–1581. [Google Scholar] [CrossRef]
- Hatakeyama, M. Helicobacter Pylori CagA and Gastric Cancer: A Paradigm for Hit-and-Run Carcinogenesis. Cell Host Microbe 2014, 15, 306–316. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Xu, Y.; Liu, C.; Ma, C.; Zou, S.; Xu, X.; Jia, J.; Liu, Z. NF-ΚB/MiR-223-3p/ARID1A Axis Is Involved in Helicobacter Pylori CagA-Induced Gastric Carcinogenesis and Progression. Cell Death Dis. 2018, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, T.; Jing, X.; Bao, J.; Zhao, L.; Zhang, A.; Miao, R.; Guo, H.; Zhou, B.; Zhang, S.; Sun, J.; et al. H. Pylori Infection Alters Repair of DNA Double-Strand Breaks via SNHG17. J. Clin. Investig. 2020, 130, 3901–3918. [Google Scholar] [CrossRef]
- Li, Y.; Kundu, P.; Seow, S.W.; de Matos, C.T.; Aronsson, L.; Chin, K.C.; Kärre, K.; Pettersson, S.; Greicius, G. Gut Microbiota Accelerate Tumor Growth via C-Jun and STAT3 Phosphorylation in APCMin/+ Mice. Carcinogenesis 2012, 33, 1231–1238. [Google Scholar] [CrossRef] [Green Version]
- Vannucci, L.; Stepankova, R.; Kozakova, H.; Fiserova, A.; Rossmann, P.; Tlaskalova-Hogenova, H. Colorectal Carcinogenesis in Germ-Free and Conventionally Reared Rats: Different Intestinal Environments Affect the Systemic Immunity. Int. J. Oncol. 2008, 32, 609–617. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Rhee, K.-J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.-R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A Human Colonic Commensal Promotes Colon Tumorigenesis via Activation of T Helper Type 17 T Cell Responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium Nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via Its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium Nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563.e16. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; da Cunha, A.P.; Rezende, R.M.; Cialic, R.; Wei, Z.; Bry, L.; Comstock, L.E.; Gandhi, R.; Weiner, H.L. The Host Shapes the Gut Microbiota via Fecal MicroRNA. Cell Host Microbe 2016, 19, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global Cancer Statistics, 2012. CA. Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Neufeldt, C.J.; Cortese, M.; Acosta, E.G.; Bartenschlager, R. Rewiring Cellular Networks by Members of the Flaviviridae Family. Nat. Rev. Microbiol. 2018, 16, 125–142. [Google Scholar] [CrossRef] [PubMed]
- Niepmann, M.; Shalamova, L.A.; Gerresheim, G.K.; Rossbach, O. Signals Involved in Regulation of Hepatitis C Virus RNA Genome Translation and Replication. Front. Microbiol. 2018, 9, 395. [Google Scholar] [CrossRef] [PubMed]
- Niepmann, M.; Gerresheim, G.K. Hepatitis C Virus Translation Regulation. Int. J. Mol. Sci. 2020, 21, 2328. [Google Scholar] [CrossRef] [Green Version]
- Luna, J.M.; Scheel, T.K.H.; Danino, T.; Shaw, K.S.; Mele, A.; Fak, J.J.; Nishiuchi, E.; Takacs, C.N.; Catanese, M.T.; de Jong, Y.P.; et al. Hepatitis C Virus RNA Functionally Sequesters MiR-122. Cell 2015, 160, 1099–1110. [Google Scholar] [CrossRef] [Green Version]
- Banaudha, K.; Kaliszewski, M.; Korolnek, T.; Florea, L.; Yeung, M.L.; Jeang, K.-T.; Kumar, A. MicroRNA Silencing of Tumor Suppressor DLC-1 Promotes Efficient Hepatitis C Virus Replication in Primary Human Hepatocytes. Hepatology 2011, 53, 53–61. [Google Scholar] [CrossRef]
- Murakami, Y.; Aly, H.H.; Tajima, A.; Inoue, I.; Shimotohno, K. Regulation of the Hepatitis C Virus Genome Replication by MiR-199a. J. Hepatol. 2009, 50, 453–460. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, W.; Cheng, N.; Wang, K.; Li, B.; Jiang, X.; Sun, S. Hepatitis C Virus-Induced up-Regulation of MicroRNA-155 Promotes Hepatocarcinogenesis by Activating Wnt Signaling. Hepatology 2012, 56, 1631–1640. [Google Scholar] [CrossRef]
- Stämpfli, M.R.; Anderson, G.P. How Cigarette Smoke Skews Immune Responses to Promote Infection, Lung Disease and Cancer. Nat. Rev. Immunol. 2009, 9, 377–384. [Google Scholar] [CrossRef]
- Shopland, D.R. Tobacco Use and Its Contribution to Early Cancer Mortality with a Special Emphasis on Cigarette Smoking. Environ. Health Perspect. 1995, 103 (Suppl. 8), 131–142. [Google Scholar] [CrossRef]
- Bracke, K.R.; D’hulst, A.I.; Maes, T.; Moerloose, K.B.; Demedts, I.K.; Lebecque, S.; Joos, G.F.; Brusselle, G.G. Cigarette Smoke-Induced Pulmonary Inflammation and Emphysema Are Attenuated in CCR6-Deficient Mice. J. Immunol. 2006, 177, 4350–4359. [Google Scholar] [CrossRef] [Green Version]
- Sopori, M. Effects of Cigarette Smoke on the Immune System. Nat. Rev. Immunol. 2002, 2, 372–377. [Google Scholar] [CrossRef]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils Sense Microbe Size and Selectively Release Neutrophil Extracellular Traps in Response to Large Pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Hosseinzadeh, A.; Thompson, P.R.; Segal, B.H.; Urban, C.F. Nicotine Induces Neutrophil Extracellular Traps. J. Leukoc. Biol. 2016, 100, 1105–1112. [Google Scholar] [CrossRef]
- Huang, W.; Li, M.D. Differential Allelic Expression of Dopamine D1 Receptor Gene (DRD1) Is Modulated by MicroRNA MiR-504. Biol. Psychiatry 2009, 65, 702–705. [Google Scholar] [CrossRef] [Green Version]
- Leng, S.; Stidley, C.A.; Bernauer, A.M.; Picchi, M.A.; Sheng, X.; Frasco, M.A.; Van Den Berg, D.; Gilliland, F.D.; Crowell, R.E.; Belinsky, S.A. Haplotypes of DNMT1 and DNMT3B Are Associated with Mutagen Sensitivity Induced by Benzo[a]Pyrene Diol Epoxide among Smokers. Carcinogenesis 2008, 29, 1380–1385. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.-R.; Chida, A.S.; Bauter, M.R.; Shafiq, N.; Seweryniak, K.; Maggirwar, S.B.; Kilty, I.; Rahman, I. Cigarette Smoke Induces Proinflammatory Cytokine Release by Activation of NF-KappaB and Posttranslational Modifications of Histone Deacetylase in Macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L46–L57. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhou, Y.; Boggs, S.E.; Belinsky, S.A.; Liu, J. Cigarette Smoke Induces Demethylation of Prometastatic Oncogene Synuclein-Gamma in Lung Cancer Cells by Downregulation of DNMT3B. Oncogene 2007, 26, 5900–5910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Jia, M.; Zhang, Y.; Breitling, L.P.; Brenner, H. DNA Methylation Changes of Whole Blood Cells in Response to Active Smoking Exposure in Adults: A Systematic Review of DNA Methylation Studies. Clin. Epigenetics 2015, 7, 113. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Cui, S.; Ma, J.; Lu, Q.; Kong, C.; Liu, T.; Sun, Z. Cigarette Smoking Extract Causes Hypermethylation and Inactivation of WWOX Gene in T-24 Human Bladder Cancer Cells. Neoplasma 2012, 59, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Tellez, C.S.; Juri, D.E.; Do, K.; Bernauer, A.M.; Thomas, C.L.; Damiani, L.A.; Tessema, M.; Leng, S.; Belinsky, S.A. EMT and Stem Cell-like Properties Associated with MiR-205 and MiR-200 Epigenetic Silencing Are Early Manifestations during Carcinogen-Induced Transformation of Human Lung Epithelial Cells. Cancer Res. 2011, 71, 3087–3097. [Google Scholar] [CrossRef] [Green Version]
- Xi, S.; Xu, H.; Shan, J.; Tao, Y.; Hong, J.A.; Inchauste, S.; Zhang, M.; Kunst, T.F.; Mercedes, L.; Schrump, D.S. Cigarette Smoke Mediates Epigenetic Repression of MiR-487b during Pulmonary Carcinogenesis. J. Clin. Investig. 2013, 123, 1241–1261. [Google Scholar] [CrossRef] [PubMed]
- Vähäkangas, K.H.; Bennett, W.P.; Castrén, K.; Welsh, J.A.; Khan, M.A.; Blömeke, B.; Alavanja, M.C.; Harris, C.C. P53 and K-Ras Mutations in Lung Cancers from Former and Never-Smoking Women. Cancer Res. 2001, 61, 4350–4356. [Google Scholar]
- Chapman, A.M.; Sun, K.Y.; Ruestow, P.; Cowan, D.M.; Madl, A.K. Lung Cancer Mutation Profile of EGFR, ALK, and KRAS: Meta-Analysis and Comparison of Never and Ever Smokers. Lung Cancer 2016, 102, 122–134. [Google Scholar] [CrossRef]
- Sun, S.; Schiller, J.H.; Gazdar, A.F. Lung Cancer in Never Smokers—A Different Disease. Nat. Rev. Cancer 2007, 7, 778–790. [Google Scholar] [CrossRef]
- Chin, L.J.; Ratner, E.; Leng, S.; Zhai, R.; Nallur, S.; Babar, I.; Muller, R.-U.; Straka, E.; Su, L.; Burki, E.A.; et al. A SNP in a Let-7 MicroRNA Complementary Site in the KRAS 3’ Untranslated Region Increases Non-Small Cell Lung Cancer Risk. Cancer Res. 2008, 68, 8535–8540. [Google Scholar] [CrossRef] [Green Version]
- Seviour, E.G.; Sehgal, V.; Mishra, D.; Rupaimoole, R.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Lee, J.-S.; Sood, A.K.; Kim, M.P.; Mills, G.B.; et al. Targeting KRas-Dependent Tumour Growth, Circulating Tumour Cells and Metastasis in Vivo by Clinically Significant MiR-193a-3p. Oncogene 2017, 36, 1339–1350. [Google Scholar] [CrossRef] [Green Version]
- Valencia, K.; Erice, O.; Kostyrko, K.; Hausmann, S.; Guruceaga, E.; Tathireddy, A.; Flores, N.M.; Sayles, L.C.; Lee, A.G.; Fragoso, R.; et al. The Mir181ab1 Cluster Promotes KRAS-Driven Oncogenesis and Progression in Lung and Pancreas. J. Clin. Investig. 2020, 130, 1879–1895. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Dang, J.; Chang, K.-Y.; Yau, E.; Aza-Blanc, P.; Moscat, J.; Rana, T.M. MiR-1298 Inhibits Mutant KRAS-Driven Tumor Growth by Repressing FAK and LAMB3. Cancer Res. 2016, 76, 5777–5787. [Google Scholar] [CrossRef] [Green Version]
- Engel, L.S.; Chow, W.-H.; Vaughan, T.L.; Gammon, M.D.; Risch, H.A.; Stanford, J.L.; Schoenberg, J.B.; Mayne, S.T.; Dubrow, R.; Rotterdam, H.; et al. Population Attributable Risks of Esophageal and Gastric Cancers. J. Natl. Cancer Inst. 2003, 95, 1404–1413. [Google Scholar] [CrossRef]
- Colleypriest, B.J.; Ward, S.G.; Tosh, D. How Does Inflammation Cause Barrett’s Metaplasia? Curr. Opin. Pharmacol. 2009, 9, 721–726. [Google Scholar] [CrossRef]
- Hardikar, S.; Onstad, L.; Song, X.; Wilson, A.M.; Montine, T.J.; Kratz, M.; Anderson, G.L.; Blount, P.L.; Reid, B.J.; White, E.; et al. Inflammation and Oxidative Stress Markers and Esophageal Adenocarcinoma Incidence in a Barrett’s Esophagus Cohort. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2393–2403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, M.B.; Barnett, M.J.; Bock, C.H.; Cross, A.J.; Goodman, P.J.; Goodman, G.E.; Haiman, C.A.; Khaw, K.-T.; McCullough, M.L.; Newton, C.C.; et al. Prediagnostic Circulating Markers of Inflammation and Risk of Oesophageal Adenocarcinoma: A Study within the National Cancer Institute Cohort Consortium. Gut 2019, 68, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Xi, S.; Inchauste, S.; Guo, H.; Shan, J.; Xiao, Z.; Xu, H.; Miettenen, M.; Zhang, M.R.; Hong, J.A.; Raiji, M.T.; et al. Cigarette Smoke Mediates Epigenetic Repression of MiR-217 during Esophageal Adenocarcinogenesis. Oncogene 2015, 34, 5548–5559. [Google Scholar] [CrossRef]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer Statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Hsia, J.; Yang, G. Prevalence of Smoking in China in 2010. N. Engl. J. Med. 2011, 364, 2469–2470. [Google Scholar] [CrossRef]
- Pennathur, A.; Gibson, M.K.; Jobe, B.A.; Luketich, J.D. Oesophageal Carcinoma. Lancet 2013, 381, 400–412. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Zhang, L.; Deng, J.; Guo, B.; Li, F.; Wang, Y.; Wu, R.; Zhang, S.; Lu, J.; Zhou, Y. A Novel Micropeptide Encoded by Y-Linked LINC00278 Links Cigarette Smoking and AR Signaling in Male Esophageal Squamous Cell Carcinoma. Cancer Res. 2020, 80, 2790–2803. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, M. Pancreatic Cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, D.; Lowenfels, A.B. The Epidemiology of Pancreatitis and Pancreatic Cancer. Gastroenterology 2013, 144, 1252–1261. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Zhong, R.; Shen, N.; Chen, W.; Zhu, B.; Ke, J.; Lu, X.; Zhang, T.; Lou, J.; Wang, Z.; et al. Non-Linear Dose-Response Relationship between Cigarette Smoking and Pancreatic Cancer Risk: Evidence from a Meta-Analysis of 42 Observational Studies. Eur. J. Cancer 2014, 50, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Morales-Oyarvide, V.; Babic, A.; Clish, C.B.; Kraft, P.; Bao, Y.; Qian, Z.R.; Rubinson, D.A.; Ng, K.; Giovannucci, E.L.; et al. Cigarette Smoking and Pancreatic Cancer Survival. J. Clin. Oncol. 2017, 35, 1822–1828. [Google Scholar] [CrossRef]
- Zhang, J.; Bai, R.; Li, M.; Ye, H.; Wu, C.; Wang, C.; Li, S.; Tan, L.; Mai, D.; Li, G.; et al. Excessive MiR-25-3p Maturation via N6-Methyladenosine Stimulated by Cigarette Smoke Promotes Pancreatic Cancer Progression. Nat. Commun. 2019, 10, 1858. [Google Scholar] [CrossRef]
- Stohs, S.J.; Bagchi, D. Oxidative Mechanisms in the Toxicity of Metal Ions. Free Radic. Biol. Med. 1995, 18, 321–336. [Google Scholar] [CrossRef] [Green Version]
- Gebel, T.W. Arsenic and Drinking Water Contamination. Science 1999, 283, 1458–1459. [Google Scholar] [CrossRef]
- Gebel, T.W. Genotoxicity of Arsenical Compounds. Int. J. Hyg. Environ. Health 2001, 203, 249–262. [Google Scholar] [CrossRef]
- Mass, M.J.; Wang, L. Arsenic Alters Cytosine Methylation Patterns of the Promoter of the Tumor Suppressor Gene P53 in Human Lung Cells: A Model for a Mechanism of Carcinogenesis. Mutat. Res. 1997, 386, 263–277. [Google Scholar] [CrossRef]
- Treas, J.; Tyagi, T.; Singh, K.P. Chronic Exposure to Arsenic, Estrogen, and Their Combination Causes Increased Growth and Transformation in Human Prostate Epithelial Cells Potentially by Hypermethylation-Mediated Silencing of MLH1. Prostate 2013, 73, 1660–1672. [Google Scholar] [CrossRef]
- Zhou, X.; Li, Q.; Arita, A.; Sun, H.; Costa, M. Effects of Nickel, Chromate, and Arsenite on Histone 3 Lysine Methylation. Toxicol. Appl. Pharmacol. 2009, 236, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhao, Y.; Xu, W.; Luo, F.; Wang, B.; Li, Y.; Pang, Y.; Liu, Q. Involvement of HIF-2α-Mediated Inflammation in Arsenite-Induced Transformation of Human Bronchial Epithelial Cells. Toxicol. Appl. Pharmacol. 2013, 272, 542–550. [Google Scholar] [CrossRef]
- Chen, C.; Luo, F.; Liu, X.; Lu, L.; Xu, H.; Yang, Q.; Xue, J.; Shi, L.; Li, J.; Zhang, A.; et al. NF-KB-Regulated Exosomal MiR-155 Promotes the Inflammation Associated with Arsenite Carcinogenesis. Cancer Lett. 2017, 388, 21–33. [Google Scholar] [CrossRef]
- Chen, C.; Luo, F.; Yang, Q.; Wang, D.; Yang, P.; Xue, J.; Dai, X.; Liu, X.; Xu, H.; Lu, J.; et al. NF-ΚB-Regulated MiR-155, via Repression of QKI, Contributes to the Acquisition of CSC-like Phenotype during the Neoplastic Transformation of Hepatic Cells Induced by Arsenite. Mol. Carcinog. 2018, 57, 483–493. [Google Scholar] [CrossRef]
- Dai, X.; Chen, C.; Yang, Q.; Xue, J.; Chen, X.; Sun, B.; Luo, F.; Liu, X.; Xiao, T.; Xu, H.; et al. Exosomal CircRNA_100284 from Arsenite-Transformed Cells, via MicroRNA-217 Regulation of EZH2, Is Involved in the Malignant Transformation of Human Hepatic Cells by Accelerating the Cell Cycle and Promoting Cell Proliferation. Cell Death Dis. 2018, 9, 454. [Google Scholar] [CrossRef]
- Lu, X.; Luo, F.; Liu, Y.; Zhang, A.; Li, J.; Wang, B.; Xu, W.; Shi, L.; Liu, X.; Lu, L.; et al. The IL-6/STAT3 Pathway via MiR-21 Is Involved in the Neoplastic and Metastatic Properties of Arsenite-Transformed Human Keratinocytes. Toxicol. Lett. 2015, 237, 191–199. [Google Scholar] [CrossRef]
- Luo, F.; Ji, J.; Liu, Y.; Xu, Y.; Zheng, G.; Jing, J.; Wang, B.; Xu, W.; Shi, L.; Lu, X.; et al. MicroRNA-21, up-Regulated by Arsenite, Directs the Epithelial-Mesenchymal Transition and Enhances the Invasive Potential of Transformed Human Bronchial Epithelial Cells by Targeting PDCD4. Toxicol. Lett. 2015, 232, 301–309. [Google Scholar] [CrossRef]
- Luo, F.; Xu, Y.; Ling, M.; Zhao, Y.; Xu, W.; Liang, X.; Jiang, R.; Wang, B.; Bian, Q.; Liu, Q. Arsenite Evokes IL-6 Secretion, Autocrine Regulation of STAT3 Signaling, and MiR-21 Expression, Processes Involved in the EMT and Malignant Transformation of Human Bronchial Epithelial Cells. Toxicol. Appl. Pharmacol. 2013, 273, 27–34. [Google Scholar] [CrossRef]
- Zhao, Y.; Xu, Y.; Luo, F.; Xu, W.; Wang, B.; Pang, Y.; Zhou, J.; Wang, X.; Liu, Q. Angiogenesis, Mediated by MiR-21, Is Involved Arsenite-Induced Carcinogenesis. Toxicol. Lett. 2013, 223, 35–41. [Google Scholar] [CrossRef]
- Xu, Y.; Luo, F.; Liu, Y.; Shi, L.; Lu, X.; Xu, W.; Liu, Q. Exosomal MiR-21 Derived from Arsenite-Transformed Human Bronchial Epithelial Cells Promotes Cell Proliferation Associated with Arsenite Carcinogenesis. Arch. Toxicol. 2015, 89, 1071–1082. [Google Scholar] [CrossRef]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A Reciprocal Repression between ZEB1 and Members of the MiR-200 Family Promotes EMT and Invasion in Cancer Cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [Green Version]
- Bracken, C.P.; Gregory, P.A.; Kolesnikoff, N.; Bert, A.G.; Wang, J.; Shannon, M.F.; Goodall, G.J. A Double-Negative Feedback Loop between ZEB1-SIP1 and the MicroRNA-200 Family Regulates Epithelial-Mesenchymal Transition. Cancer Res. 2008, 68, 7846–7854. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhao, Y.; Smith, E.; Goodall, G.J.; Drew, P.A.; Brabletz, T.; Yang, C. Reversal and Prevention of Arsenic-Induced Human Bronchial Epithelial Cell Malignant Transformation by MicroRNA-200b. Toxicol. Sci. 2011, 121, 110–122. [Google Scholar] [CrossRef]
- Wang, Z.; Humphries, B.; Xiao, H.; Jiang, Y.; Yang, C. MicroRNA-200b Suppresses Arsenic-Transformed Cell Migration by Targeting Protein Kinase Cα and Wnt5b-Protein Kinase Cα Positive Feedback Loop and Inhibiting Rac1 Activation. J. Biol. Chem. 2014, 289, 18373–18386. [Google Scholar] [CrossRef] [Green Version]
- Filipic, M.; Fatur, T.; Vudrag, M. Molecular Mechanisms of Cadmium Induced Mutagenicity. Hum. Exp. Toxicol. 2006, 25, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Waalkes, M.P. Cadmium Carcinogenesis. Mutat. Res. 2003, 533, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, H.; Cai, D.; Li, P.; Jin, J.; Jiang, X.; Li, Z.; Tian, L.; Chen, G.; Sun, J.; et al. Chronic Oral Exposure to Cadmium Causes Liver Inflammation by NLRP3 Inflammasome Activation in Pubertal Mice. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2021, 148, 111944. [Google Scholar] [CrossRef] [PubMed]
- Neagu, M.; Constantin, C.; Cretoiu, S.M.; Zurac, S. MiRNAs in the Diagnosis and Prognosis of Skin Cancer. Front. Cell Dev. Biol. 2020, 8, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handa, H.; Murakami, Y.; Ishihara, R.; Kimura-Masuda, K.; Masuda, Y. The Role and Function of MicroRNA in the Pathogenesis of Multiple Myeloma. Cancers 2019, 11, 1738. [Google Scholar] [CrossRef] [Green Version]
- Weng, S.; Wang, W.; Li, Y.; Li, H.; Lu, X.; Xiao, S.; Wu, T.; Xie, M.; Zhang, W. Continuous Cadmium Exposure from Weaning to Maturity Induces Downregulation of Ovarian Follicle Development-Related SCF/c-Kit Gene Expression and the Corresponding Changes of DNA Methylation/MicroRNA Pattern. Toxicol. Lett. 2014, 225, 367–377. [Google Scholar] [CrossRef]
- Tanwar, V.S.; Zhang, X.; Jagannathan, L.; Jose, C.C.; Cuddapah, S. Cadmium Exposure Upregulates SNAIL through MiR-30 Repression in Human Lung Epithelial Cells. Toxicol. Appl. Pharmacol. 2019, 373, 1–9. [Google Scholar] [CrossRef]
- Tani, H.; Onuma, Y.; Ito, Y.; Torimura, M. Long Non-Coding RNAs as Surrogate Indicators for Chemical Stress Responses in Human-Induced Pluripotent Stem Cells. PLoS ONE 2014, 9, e106282. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, H.; Wang, C.; Lu, Q.; Huang, Q.; Zheng, C.; Lei, Y. Long Non-Coding RNAs as Novel Expression Signatures Modulate DNA Damage and Repair in Cadmium Toxicology. Sci. Rep. 2015, 5, 15293. [Google Scholar] [CrossRef] [Green Version]
- El Ghissassi, F.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A Review of Human Carcinogens—Part D: Radiation. Lancet Oncol. 2009, 10, 751–752. [Google Scholar] [CrossRef]
- Armstrong, B.K.; Kricker, A. The Epidemiology of UV Induced Skin Cancer. J. Photochem. Photobiol. B 2001, 63, 8–18. [Google Scholar] [CrossRef]
- Chen, H.; Weng, Q.Y.; Fisher, D.E. UV Signaling Pathways within the Skin. J. Investig. Dermatol. 2014, 134, 2080–2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, R.J.; Ray, A. Epigenetic Regulation of MiRNA Genes and Their Role in Human Melanomas. Epigenomics 2012, 4, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Bald, T.; Quast, T.; Landsberg, J.; Rogava, M.; Glodde, N.; Lopez-Ramos, D.; Kohlmeyer, J.; Riesenberg, S.; van den Boorn-Konijnenberg, D.; Hömig-Hölzel, C.; et al. Ultraviolet-Radiation-Induced Inflammation Promotes Angiotropism and Metastasis in Melanoma. Nature 2014, 507, 109–113. [Google Scholar] [CrossRef]
- Zaidi, M.R.; Davis, S.; Noonan, F.P.; Graff-Cherry, C.; Hawley, T.S.; Walker, R.L.; Feigenbaum, L.; Fuchs, E.; Lyakh, L.; Young, H.A.; et al. Interferon-γ Links Ultraviolet Radiation to Melanomagenesis in Mice. Nature 2011, 469, 548–553. [Google Scholar] [CrossRef] [Green Version]
- Bernard, J.J.; Cowing-Zitron, C.; Nakatsuji, T.; Muehleisen, B.; Muto, J.; Borkowski, A.W.; Martinez, L.; Greidinger, E.L.; Yu, B.D.; Gallo, R.L. Ultraviolet Radiation Damages Self Noncoding RNA and Is Detected by TLR3. Nat. Med. 2012, 18, 1286–1290. [Google Scholar] [CrossRef] [PubMed]
- Degueurce, G.; D’Errico, I.; Pich, C.; Ibberson, M.; Schütz, F.; Montagner, A.; Sgandurra, M.; Mury, L.; Jafari, P.; Boda, A.; et al. Identification of a Novel PPARβ/δ/MiR-21-3p Axis in UV-induced Skin Inflammation. EMBO Mol. Med. 2016, 8, 919–936. [Google Scholar] [CrossRef]
- Ge, Y.; Zhang, L.; Nikolova, M.; Reva, B.; Fuchs, E. Strand-Specific in Vivo Screen of Cancer-Associated MiRNAs Unveils a Role for MiR-21(∗) in SCC Progression. Nat. Cell Biol. 2016, 18, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Huang, Z.-X.; Chen, X.-W.; Deng, Q.-K.; Yan, W.; Zhou, M.-J.; Ou, C.-S.; Ding, Z.-H. Differential Expression Profiles of MicroRNAs in NIH3T3 Cells in Response to UVB Irradiation. Photochem. Photobiol. 2009, 85, 765–773. [Google Scholar] [CrossRef]
- Darido, C.; Georgy, S.R.; Wilanowski, T.; Dworkin, S.; Auden, A.; Zhao, Q.; Rank, G.; Srivastava, S.; Finlay, M.J.; Papenfuss, A.T.; et al. Targeting of the Tumor Suppressor GRHL3 by a MiR-21-Dependent Proto-Oncogenic Network Results in PTEN Loss and Tumorigenesis. Cancer Cell 2011, 20, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.R.; Messenger, Z.J.; Tam, H.W.; Phillips, S.L.; Recio, L.; Smart, R.C. Long Noncoding RNA LincRNA-P21 Is the Major Mediator of UVB-Induced and P53-Dependent Apoptosis in Keratinocytes. Cell Death Dis. 2015, 6, e1700. [Google Scholar] [CrossRef] [PubMed]
- Kramata, P.; Lu, Y.-P.; Lou, Y.-R.; Singh, R.N.; Kwon, S.M.; Conney, A.H. Patches of Mutant P53-Immunoreactive Epidermal Cells Induced by Chronic UVB Irradiation Harbor the Same P53 Mutations as Squamous Cell Carcinomas in the Skin of Hairless SKH-1 Mice. Cancer Res. 2005, 65, 3577–3585. [Google Scholar] [CrossRef] [Green Version]
- Matsui, M.; Corey, D.R. Non-Coding RNAs as Drug Targets. Nat. Rev. Drug Discov. 2017, 16, 167–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haussecker, D.; Kay, M.A. MiR-122 Continues to Blaze the Trail for MicroRNA Therapeutics. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 240–242. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA Sponges: Competitive Inhibitors of Small RNAs in Mammalian Cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Jost, I.; Shalamova, L.A.; Gerresheim, G.K.; Niepmann, M.; Bindereif, A.; Rossbach, O. Functional Sequestration of MicroRNA-122 from Hepatitis C Virus by Circular RNA Sponges. RNA Biol. 2018, 15, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G.; Shair, K.H.Y.; Marquitz, A.R.; Kung, C.-P.; Edwards, R.H.; Raab-Traub, N. Human Tumor Virus Utilizes Exosomes for Intercellular Communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar] [CrossRef] [Green Version]
- Pegtel, D.M.; Cosmopoulos, K.; Thorley-Lawson, D.A.; van Eijndhoven, M.A.J.; Hopmans, E.S.; Lindenberg, J.L.; de Gruijl, T.D.; Würdinger, T.; Middeldorp, J.M. Functional Delivery of Viral MiRNAs via Exosomes. Proc. Natl. Acad. Sci. USA 2010, 107, 6328–6333. [Google Scholar] [CrossRef] [Green Version]
- Wahlgren, J.; De L Karlson, T.; Brisslert, M.; Vaziri Sani, F.; Telemo, E.; Sunnerhagen, P.; Valadi, H. Plasma Exosomes Can Deliver Exogenous Short Interfering RNA to Monocytes and Lymphocytes. Nucleic Acids Res. 2012, 40, e130. [Google Scholar] [CrossRef] [Green Version]
- Shtam, T.A.; Kovalev, R.A.; Varfolomeeva, E.Y.; Makarov, E.M.; Kil, Y.V.; Filatov, M.V. Exosomes Are Natural Carriers of Exogenous SiRNA to Human Cells in Vitro. Cell Commun. Signal. CCS 2013, 11, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binenbaum, Y.; Na’ara, S.; Gil, Z. Gemcitabine Resistance in Pancreatic Ductal Adenocarcinoma. Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother. 2015, 23, 55–68. [Google Scholar] [CrossRef]
- Iqbal, M.A.; Arora, S.; Prakasam, G.; Calin, G.A.; Syed, M.A. MicroRNA in Lung Cancer: Role, Mechanisms, Pathways and Therapeutic Relevance. Mol. Aspects Med. 2019, 70, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varn, F.S.; Schaafsma, E.; Wang, Y.; Cheng, C. Genomic Characterization of Six Virus-Associated Cancers Identifies Changes in the Tumor Immune Microenvironment and Altered Genetic Programs. Cancer Res. 2018, 78, 6413–6423. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Farmer, E.; Wu, T.C.; Hung, C.-F. Perspectives for Therapeutic HPV Vaccine Development. J. Biomed. Sci. 2016, 23, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, K.; Nimura, K.; Ito, R.; Saga, K.; Inohara, H.; Kaneda, Y. Evaluation of HPV16 E7 Expression in Head and Neck Carcinoma Cell Lines and Clinical Specimens. Sci. Rep. 2020, 10, 22138. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| miRNA | Expression | Target | Related Cancers | Related Inflammation | Reference |
|---|---|---|---|---|---|
| miR-205 | ↓ 18 | HBx mRNA | HCC 1 | HBV 11 | [51] |
| miR-205 | ↓ | unknown | LC 2 | CS 12 | [120] |
| miR-122 | ↓ | cyclin G(1) | HCC | HBV | [53,54,55] |
| miR-122 | ↓ | HCV mRNA | HCC | HCV 13 | [102,103,104] |
| miR-199a-3p | ↑ 19 | unknown | HCC | HBV | [56] |
| miR-199a-3p | ↓ | HCV mRNA | unknown | HCV | [106] |
| let-7 | ↓ | STAT3 | HCC | HBV | [58] |
| let7 | ↓ | KRAS | LC | CS | [125] |
| miR-15a/16 | ↓ | cyclin D1 | HCC | HBV | [57] |
| miR-148a | ↓ | HPIP | HCC | HBV | [59] |
| miR-216b | ↓ | IGF2BP2 | HCC | HBV | [60] |
| miR-3188 | ↑ | ZHX2 | HCC | HBV | [61] |
| miR-139-3p | ↓ | E6/E7 | CC 3, HNC 4 | HPV 14 | [72] |
| miR-375 | ↓ | E6AP | CC | HPV | [73] |
| miR-34a | ↓ | unknown | CC | HPV | [78] |
| miR-23b | ↓ | uPA | CC | HPV | [79] |
| miR-182 | ↑ | unknown | CC | HPV | [80] |
| miR-146a-5p | ↓ | KDM2B | CC | HPV | [81] |
| miR-210 | ↓ | STMN1, DIMT1 | GC 5 | H. pylori15 | [87] |
| miR-490-3p | ↓ | SMARCD1, DARPP-32 | GC | H. pylori | [85,86] |
| miR-124 | ↓ | SMOX | GC | H. pylori | [88] |
| miR-223-3p | ↑ | ARID1A | GC | H. pylori | [92] |
| miR-18a* | ↓ | ULK1 | CRC 6 | F. nucleatum | [98] |
| miR-4802 | ↓ | ATG7 | CRC | F. nucleatum | [98] |
| miR-515-5p | unknown | unknown | unknown | Escherichia coli and F. nucleatum 16 | [99] |
| miR-1226-5p | unknown | unknown | unknown | Escherichia coli and F. nucleatum | [99] |
| miR-141 | ↑ | DLC-1 | unknown | HCV | [105] |
| miR-155 | ↑ | unknown | HCC | HCV | [107] |
| miR-155 | ↑ | QKI | unknown | Arsenic | [150,151] |
| miR-504 | unknown | DRD1 | unknown | CS | [114] |
| miR-200b, miR-200c | ↓ | unknown | LC | CS | [120] |
| miR-200b | ↓ | PKCα | unknown | Arsenic | [161] |
| miR-487b | ↓ | SUZ12, BMI1, WNT5A, MYC, and KRAS | LC | CS | [121] |
| miR-193a-3p | ↓ | KRAS | LC | CS | [126] |
| miR181ab1 | ↓ | unknown | LC, PDAC 7 | CS | [127] |
| miR-1298 | ↓ | FAK, LAMB3 | LC | CS | [128] |
| miR-217 | ↓ | KLK7 | EC 8 | CS | [133] |
| miR-25-3p | ↑ | PHLPP2 | PC 9 | CS | [142] |
| miR-221 | ↑ | unknown | unknown | Cadmium | [167] |
| miR-30 | ↓ | Snail | unknown | Cadmium | [168] |
| miR-21 | ↑ | PDCD4 | LC | Arsenic | [153,154,155,156,157] |
| miR-21-3p | ↑ | SMAD7 | unknown | UV 17 | [178] |
| miR-21-3p | ↑ | PHACTR4 | SCC 10 | unknown | [179] |
| miR-21-5p | ↑ | GRHL3, PTEN | SCC | UV | [180,181] |
| lncRNA | Expression | Target | Related Diseases | Related Inflammation | Reference |
|---|---|---|---|---|---|
| HULC | ↑ 10 | unknown | HCC 1 | HBV 5 | [52] |
| HBx-LINE1 | ↑ | miR-122 | HCC | HBV | [54] |
| LINC01352 | ↓ 11 | miR-135b | HCC | HBV | [62] |
| TMPOP2 | ↑ | miR-375, miR-139 | CC 2 | HPV 6 | [75] |
| SNHG12 | ↑ | unknown | CC | HPV | [82] |
| SNHG17 | ↑ | NONO, miR-3909 | GC 3 | H. pylori7 | [93] |
| LINC00278 | ↓ | unknown | EC 4 | CS 8 | [137] |
| GABPB1-AS1 | ↑ | unknown | unknown | Cadmium | [169] |
| LINC00152 | ↑ | unknown | unknown | Cadmium | [169] |
| ENST00000414355 | ↑ | unknown | unknown | Cadmium | [170] |
| lincRNA-p21 | ↓ | unknown | unknown | UV 9 | [182] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, T.; Xie, M.; Jing, X.; Cui, J.; Wu, X.; Shu, Y. Crosstalk between Environmental Inflammatory Stimuli and Non-Coding RNA in Cancer Occurrence and Development. Cancers 2021, 13, 4436. https://doi.org/10.3390/cancers13174436
Xu T, Xie M, Jing X, Cui J, Wu X, Shu Y. Crosstalk between Environmental Inflammatory Stimuli and Non-Coding RNA in Cancer Occurrence and Development. Cancers. 2021; 13(17):4436. https://doi.org/10.3390/cancers13174436
Chicago/Turabian StyleXu, Tingting, Mengyan Xie, Xinming Jing, Jiahua Cui, Xi Wu, and Yongqian Shu. 2021. "Crosstalk between Environmental Inflammatory Stimuli and Non-Coding RNA in Cancer Occurrence and Development" Cancers 13, no. 17: 4436. https://doi.org/10.3390/cancers13174436
APA StyleXu, T., Xie, M., Jing, X., Cui, J., Wu, X., & Shu, Y. (2021). Crosstalk between Environmental Inflammatory Stimuli and Non-Coding RNA in Cancer Occurrence and Development. Cancers, 13(17), 4436. https://doi.org/10.3390/cancers13174436