
Insights into Mechanisms of Pheochromocytomas and Paragangliomas Driven by Known or New Genetic Drivers

 , and
, and 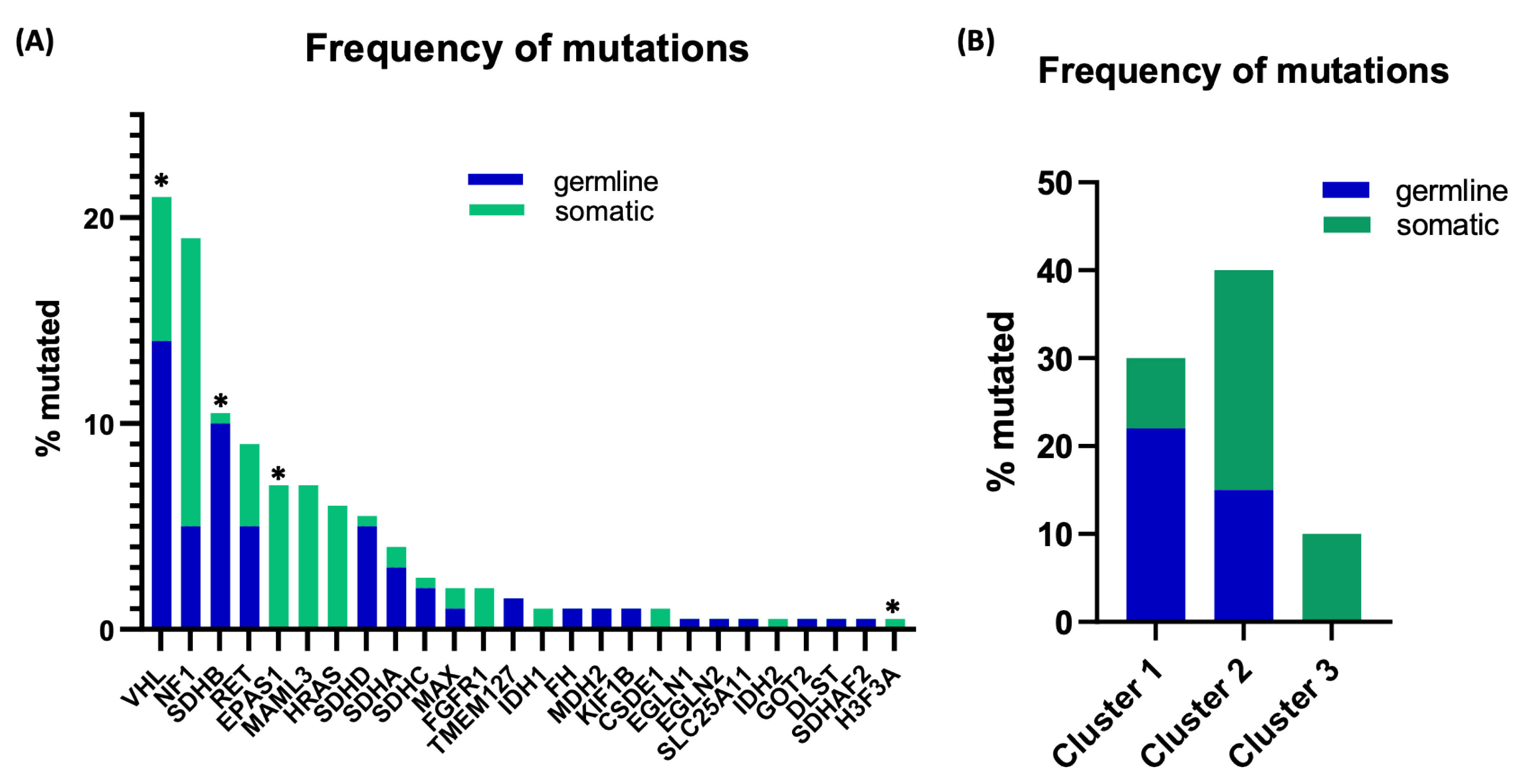{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Overview and Current Status of Genetic Drivers
2. Leveraging Clinical and Genetic Data for Classification and Patient Management
3. Detecting and Interpreting Variants: Protocols and Challenges
4. A Workflow to Identify a Driver Mutation in PPGLs
5. Lessons Learned from Atypical/Novel/Unsuspected Genetic Disruptions
6. Epistatic Interactions between Genetics and the Environment
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr.; Endocrine, S. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.P.H.; Young, W.F., Jr.; Eng, C. Pheochromocytoma and Paraganglioma. N. Engl. J. Med. 2019, 381, 552–565. [Google Scholar] [CrossRef]
- Thompson, L.D.R.; Gill, A.J.; Asa, S.L.; Clifton-Bligh, R.J.; de Krijger, R.R.; Kimura, N.; Komminoth, P.; Lack, E.E.; Lenders, J.W.M.; Lloyd, R.V.; et al. Data set for the reporting of pheochromocytoma and paraganglioma: Explanations and recommendations of the guidelines from the International Collaboration on Cancer Reporting. Hum. Pathol. 2021, 110, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L. Pheochromocytoma and Paraganglioma: Genetics, Diagnosis, and Treatment. Hematol. Oncol. Clin. N. Am. 2016, 30, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Del Rivero, J.; Else, T.; Howe, J.R.; Asa, S.L.; Cohen, D.L.; Dahia, P.L.M.; Fraker, D.L.; Goodman, K.A.; Hope, T.A.; et al. The North American Neuroendocrine Tumor Society Consensus Guidelines for Surveillance and Management of Metastatic and/or Unresectable Pheochromocytoma and Paraganglioma. Pancreas 2021, 50, 469–493. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Ramirez, M.; Feng, L.; Johnson, M.M.; Ejaz, S.; Habra, M.A.; Rich, T.; Busaidy, N.; Cote, G.J.; Perrier, N.; Phan, A.; et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: Primary tumor size and primary tumor location as prognostic indicators. J. Clin. Endocrinol. Metab. 2011, 96, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Dahia, P.L. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Rev. Cancer 2014, 14, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Remacha, L.; Currás-Freixes, M.; Torres-Ruiz, R.; Schiavi, F.; Torres-Pérez, R.; Calsina, B.; Letón, R.; Comino-Méndez, I.; Roldán-Romero, J.M.; Montero-Conde, C.; et al. Gain-of-function mutations in DNMT3A in patients with paraganglioma. Genet. Med. 2018, 20, 1644–1651. [Google Scholar] [CrossRef]
- Remacha, L.; Pirman, D.; Mahoney, C.E.; Coloma, J.; Calsina, B.; Currás-Freixes, M.; Letón, R.; Torres-Pérez, R.; Richter, S.; Pita, G.; et al. Recurrent Germline DLST Mutations in Individuals with Multiple Pheochromocytomas and Paragangliomas. Am. J. Hum. Genet. 2019, 104, 651–664. [Google Scholar] [CrossRef] [Green Version]
- Curras-Freixes, M.; Pineiro-Yanez, E.; Montero-Conde, C.; Apellaniz-Ruiz, M.; Calsina, B.; Mancikova, V.; Remacha, L.; Richter, S.; Ercolino, T.; Rogowski-Lehmann, N.; et al. PheoSeq: A Targeted Next-Generation Sequencing Assay for Pheochromocytoma and Paraganglioma Diagnostics. J. Mol. Diagn. 2017, 19, 575–588. [Google Scholar] [CrossRef] [Green Version]
- Buffet, A.; Burnichon, N.; Favier, J.; Gimenez-Roqueplo, A.P. An overview of 20 years of genetic studies in pheochromocytoma and paraganglioma. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101416. [Google Scholar] [CrossRef]
- Castro-Vega, L.J.; Lepoutre-Lussey, C.; Gimenez-Roqueplo, A.P.; Favier, J. Rethinking pheochromocytomas and paragangliomas from a genomic perspective. Oncogene 2016, 35, 1080–1089. [Google Scholar] [CrossRef]
- Favier, J.; Amar, L.; Gimenez-Roqueplo, A.P. Paraganglioma and phaeochromocytoma: From genetics to personalized medicine. Nat. Rev. Endocrinol. 2015, 11, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahia, P.L. The genetic landscape of pheochromocytomas and paragangliomas: Somatic mutations take center stage. J. Clin. Endocrinol. Metab. 2013, 98, 2679–2681. [Google Scholar] [CrossRef] [PubMed]
- Ben Aim, L.; Pigny, P.; Castro-Vega, L.J.; Buffet, A.; Amar, L.; Bertherat, J.; Drui, D.; Guilhem, I.; Baudin, E.; Lussey-Lepoutre, C.; et al. Targeted next-generation sequencing detects rare genetic events in pheochromocytoma and paraganglioma. J. Med. Genet. 2019, 56, 513–520. [Google Scholar] [CrossRef]
- Welander, J.; Larsson, C.; Backdahl, M.; Hareni, N.; Sivler, T.; Brauckhoff, M.; Soderkvist, P.; Gimm, O. Integrative genomics reveals frequent somatic NF1 mutations in sporadic pheochromocytomas. Hum. Mol. Genet. 2012, 21, 5406–5416. [Google Scholar] [CrossRef] [Green Version]
- Toledo, R.A.; Qin, Y.; Cheng, Z.M.; Gao, Q.; Iwata, S.; Silva, G.M.; Prasad, M.L.; Ocal, I.T.; Rao, S.; Aronin, N.; et al. Recurrent Mutations of Chromatin-Remodeling Genes and Kinase Receptors in Pheochromocytomas and Paragangliomas. Clin. Cancer Res. 2016, 22, 2301–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascón, A.; Remacha, L.; Calsina, B.; Robledo, M. Pheochromocytomas and Paragangliomas: Bypassing Cellular Respiration. Cancers 2019, 11, 683. [Google Scholar] [CrossRef] [Green Version]
- Castro-Vega, L.J.; Buffet, A.; de Cubas, A.A.; Cascon, A.; Menara, M.; Khalifa, E.; Amar, L.; Azriel, S.; Bourdeau, I.; Chabre, O.; et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum. Mol. Genet. 2014, 23, 2440–2446. [Google Scholar] [CrossRef] [Green Version]
- Dahia, P.L.; Ross, K.N.; Wright, M.E.; Hayashida, C.Y.; Santagata, S.; Barontini, M.; Kung, A.L.; Sanso, G.; Powers, J.F.; Tischler, A.S.; et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005, 1, e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koopman, K.; Gaal, J.; de Krijger, R.R. Pheochromocytomas and Paragangliomas: New Developments with Regard to Classification, Genetics, and Cell of Origin. Cancers 2019, 11, 1070. [Google Scholar] [CrossRef] [Green Version]
- Job, S.; Draskovic, I.; Burnichon, N.; Buffet, A.; Cros, J.; Lepine, C.; Venisse, A.; Robidel, E.; Verkarre, V.; Meatchi, T.; et al. Telomerase Activation and ATRX Mutations Are Independent Risk Factors for Metastatic Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2019, 25, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Fishbein, L.; Khare, S.; Wubbenhorst, B.; De Sloover, D.; D’Andrea, K.; Merrill, S.; Cho, N.W.; Greenberg, R.A.; Else, T.; Montone, K.; et al. Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat. Commun. 2015, 6, 6140. [Google Scholar] [CrossRef] [Green Version]
- Lenders, J.W.M.; Kerstens, M.N.; Amar, L.; Prejbisz, A.; Robledo, M.; Taieb, D.; Pacak, K.; Crona, J.; Zelinka, T.; Mannelli, M.; et al. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: A position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J. Hypertens. 2020, 38, 1443–1456. [Google Scholar] [CrossRef]
- Crona, J.; Taieb, D.; Pacak, K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr. Rev. 2017, 38, 489–515. [Google Scholar] [CrossRef]
- Nolting, S.; Grossman, A.; Pacak, K. Metastatic Phaeochromocytoma: Spinning Towards More Promising Treatment Options. Exp. Clin. Endocrinol. Diabetes 2019, 127, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Eisenhofer, G.; Huynh, T.-T.; Hiroi, M.; Pacak, K. Understanding Catecholamine Metabolism as a Guide to the Biochemical Diagnosis of Pheochromocytoma. Rev. Endocr. Metab. Disord. 2001, 2, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Nolting, S.; Bechmann, N.; Taieb, D.; Beuschlein, F.; Fassnacht, M.; Kroiss, M.; Eisenhofer, G.; Grossman, A.; Pacak, K. Personalized management of pheochromocytoma and paraganglioma. Endocr. Rev. 2021, bnab019. [Google Scholar] [CrossRef] [PubMed]
- Wachtel, H.; Fishbein, L. Genetics of pheochromocytoma and paraganglioma. Curr. Opin. Endocrinol. Diabetes Obes. 2021, 28, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Taieb, D.; Pacak, K. New Insights into the Nuclear Imaging Phenotypes of Cluster 1 Pheochromocytoma and Paraganglioma. Trends Endocrinol. Metab. 2017, 28, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Timmers, H.J.; Chen, C.C.; Carrasquillo, J.A.; Whatley, M.; Ling, A.; Havekes, B.; Eisenhofer, G.; Martiniova, L.; Adams, K.T.; Pacak, K. Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J. Clin. Endocrinol. Metab. 2009, 94, 4757–4767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Suh, C.H.; Woo, S.; Kim, Y.J.; Lee, J.J. Performance of (68)Ga-DOTA-Conjugated Somatostatin Receptor-Targeting Peptide PET in Detection of Pheochromocytoma and Paraganglioma: A Systematic Review and Metaanalysis. J. Nucl. Med. 2019, 60, 369–376. [Google Scholar] [CrossRef] [Green Version]
- Janssen, I.; Blanchet, E.M.; Adams, K.; Chen, C.C.; Millo, C.M.; Herscovitch, P.; Taieb, D.; Kebebew, E.; Lehnert, H.; Fojo, A.T.; et al. Superiority of [68Ga]-DOTATATE PET/CT to Other Functional Imaging Modalities in the Localization of SDHB-Associated Metastatic Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2015, 21, 3888–3895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassnacht, M.; Assie, G.; Baudin, E.; Eisenhofer, G.; de la Fouchardiere, C.; Haak, H.R.; de Krijger, R.; Porpiglia, F.; Terzolo, M.; Berruti, A.; et al. Adrenocortical carcinomas and malignant phaeochromocytomas: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 1476–1490. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.P.H.; Tsoy, U.; Bancos, I.; Amodru, V.; Walz, M.K.; Tirosh, A.; Kaur, R.J.; McKenzie, T.; Qi, X.; Bandgar, T.; et al. Comparison of Pheochromocytoma-Specific Morbidity and Mortality Among Adults With Bilateral Pheochromocytomas Undergoing Total Adrenalectomy vs Cortical-Sparing Adrenalectomy. JAMA Netw. Open 2019, 2, e198898. [Google Scholar] [CrossRef]
- Pryma, D.A.; Chin, B.B.; Noto, R.B.; Dillon, J.S.; Perkins, S.; Solnes, L.; Kostakoglu, L.; Serafini, A.N.; Pampaloni, M.H.; Jensen, J.; et al. Efficacy and Safety of High-Specific-Activity (131)I-MIBG Therapy in Patients with Advanced Pheochromocytoma or Paraganglioma. J. Nucl. Med. 2019, 60, 623–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, G.; Grozinsky-Glasberg, S.; Hofman, M.S.; Callahan, J.; Meirovitz, A.; Maimon, O.; Pattison, D.A.; Gross, D.J.; Hicks, R.J. Efficacy of Peptide Receptor Radionuclide Therapy for Functional Metastatic Paraganglioma and Pheochromocytoma. J. Clin. Endocrinol. Metab. 2017, 102, 3278–3287. [Google Scholar] [CrossRef] [Green Version]
- Ayala-Ramirez, M.; Feng, L.; Habra, M.A.; Rich, T.; Dickson, P.V.; Perrier, N.; Phan, A.; Waguespack, S.; Patel, S.; Jimenez, C. Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic extra-adrenal paragangliomas: Insights from the largest single-institutional experience. Cancer 2012, 118, 2804–2812. [Google Scholar] [CrossRef] [Green Version]
- Druce, M.R.; Kaltsas, G.A.; Fraenkel, M.; Gross, D.J.; Grossman, A.B. Novel and evolving therapies in the treatment of malignant phaeochromocytoma: Experience with the mTOR inhibitor everolimus (RAD001). Horm. Metab. Res. 2009, 41, 697–702. [Google Scholar] [CrossRef]
- Jimenez, C.; Fazeli, S.; Roman-Gonzalez, A. Antiangiogenic therapies for pheochromocytoma and paraganglioma. Endocr. Relat. Cancer 2020, 27, R239–R254. [Google Scholar] [CrossRef]
- Dahia, P.L.M.; Toledo, R.A. Recognizing hypoxia in phaeochromocytomas and paragangliomas. Nat. Rev. Endocrinol. 2020, 16, 191–192. [Google Scholar] [CrossRef]
- Toledo, R.A. New HIF2alpha inhibitors: Potential implications as therapeutics for advanced pheochromocytomas and paragangliomas. Endocr. Relat. Cancer 2017, 24, C9–C19. [Google Scholar] [CrossRef] [Green Version]
- Choueiri, T.K.; Bauer, T.M.; Papadopoulos, K.P.; Plimack, E.R.; Merchan, J.R.; McDermott, D.F.; Michaelson, M.D.; Appleman, L.J.; Thamake, S.; Perini, R.F.; et al. Inhibition of hypoxia-inducible factor-2alpha in renal cell carcinoma with belzutifan: A phase 1 trial and biomarker analysis. Nat. Med. 2021, 27, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, C.; Subbiah, V.; Stephen, B.; Ma, J.; Milton, D.; Xu, M.; Zarifa, A.; Akhmedzhanov, F.O.; Tsimberidou, A.; Habra, M.A.; et al. Phase II Clinical Trial of Pembrolizumab in Patients with Progressive Metastatic Pheochromocytomas and Paragangliomas. Cancers 2020, 12, 2307. [Google Scholar] [CrossRef] [PubMed]
- Buffet, A.; Ben Aim, L.; Leboulleux, S.; Drui, D.; Vezzosi, D.; Libe, R.; Ajzenberg, C.; Bernardeschi, D.; Cariou, B.; Chabolle, F.; et al. Positive Impact of Genetic Test on the Management and Outcome of Patients With Paraganglioma and/or Pheochromocytoma. J. Clin. Endocrinol. Metab. 2019, 104, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Group, N.G.S.i.P.S.; Toledo, R.A.; Burnichon, N.; Cascon, A.; Benn, D.E.; Bayley, J.P.; Welander, J.; Tops, C.M.; Firth, H.; Dwight, T.; et al. Consensus Statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat. Rev. Endocrinol. 2017, 13, 233–247. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Holcomb, D.; Hamasaki-Katagiri, N.; Laurie, K.; Katneni, U.; Kames, J.; Alexaki, A.; Bar, H.; Kimchi-Sarfaty, C. New approaches to predict the effect of co-occurring variants on protein characteristics. Am. J. Hum. Genet. 2021, 108, 1502–1511. [Google Scholar] [CrossRef]
- Gieldon, L.; William, D.; Hackmann, K.; Jahn, W.; Jahn, A.; Wagner, J.; Rump, A.; Bechmann, N.; Nölting, S.; Knösel, T.; et al. Optimizing Genetic Workup in Pheochromocytoma and Paraganglioma by Integrating Diagnostic and Research Approaches. Cancers 2019, 11, 809. [Google Scholar] [CrossRef] [Green Version]
- Flynn, A.; Dwight, T.; Harris, J.; Benn, D.; Zhou, L.; Hogg, A.; Catchpoole, D.; James, P.; Duncan, E.L.; Trainer, A.; et al. Pheo-Type: A Diagnostic Gene-expression Assay for the Classification of Pheochromocytoma and Paraganglioma. J. Clin. Endocrinol. Metab. 2016, 101, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Castro-Vega, L.J.; Letouze, E.; Burnichon, N.; Buffet, A.; Disderot, P.H.; Khalifa, E.; Loriot, C.; Elarouci, N.; Morin, A.; Menara, M.; et al. Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat. Commun. 2015, 6, 6044. [Google Scholar] [CrossRef] [Green Version]
- Creason, A.; Haan, D.; Dang, K.; Chiotti, K.E.; Inkman, M.; Lamb, A.; Yu, T.; Hu, Y.; Norman, T.C.; Buchanan, A.; et al. A community challenge to evaluate RNA-seq, fusion detection, and isoform quantification methods for cancer discovery. Cell Syst. 2021, 12, 827–838.e5. [Google Scholar] [CrossRef]
- Maher, C.A.; Kumar-Sinha, C.; Cao, X.; Kalyana-Sundaram, S.; Han, B.; Jing, X.; Sam, L.; Barrette, T.; Palanisamy, N.; Chinnaiyan, A.M. Transcriptome sequencing to detect gene fusions in cancer. Nature 2009, 458, 97–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Hüser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Rätsch, G. Comprehensive Analysis of Alternative Splicing Across Tumors from 8705 Patients. Cancer Cell 2018, 34, 211–224.e6. [Google Scholar] [CrossRef] [Green Version]
- Beaubier, N.; Bontrager, M.; Huether, R.; Igartua, C.; Lau, D.; Tell, R.; Bobe, A.M.; Bush, S.; Chang, A.L.; Hoskinson, D.C.; et al. Integrated genomic profiling expands clinical options for patients with cancer. Nat. Biotechnol. 2019, 37, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.Z.; Berger, M.F.; Adiconis, X.; Rogov, P.; Melnikov, A.; Fennell, T.; Nusbaum, C.; Garraway, L.A.; Gnirke, A. Targeted next-generation sequencing of a cancer transcriptome enhances detection of sequence variants and novel fusion transcripts. Genome Biol. 2009, 10, R115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papathomas, T.G.; Oudijk, L.; Persu, A.; Gill, A.J.; van Nederveen, F.; Tischler, A.S.; Tissier, F.; Volante, M.; Matias-Guiu, X.; Smid, M.; et al. SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: A multicenter interobserver variation analysis using virtual microscopy: A Multinational Study of the European Network for the Study of Adrenal Tumors (ENS@T). Mod. Pathol. 2015, 28, 807–821. [Google Scholar] [CrossRef] [Green Version]
- Richter, S.; Gieldon, L.; Pang, Y.; Peitzsch, M.; Huynh, T.; Leton, R.; Viana, B.; Ercolino, T.; Mangelis, A.; Rapizzi, E.; et al. Metabolome-guided genomics to identify pathogenic variants in isocitrate dehydrogenase, fumarate hydratase, and succinate dehydrogenase genes in pheochromocytoma and paraganglioma. Genet. Med. 2019, 21, 705–717. [Google Scholar] [CrossRef]
- Qiu, Z.; Lin, A.P.; Jiang, S.; Elkashef, S.M.; Myers, J.; Srikantan, S.; Sasi, B.; Cao, J.Z.; Godley, L.A.; Rakheja, D.; et al. MYC Regulation of D2HGDH and L2HGDH Influences the Epigenome and Epitranscriptome. Cell Chem. Biol. 2020, 27, 538–550.e7. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Yao, L.; King, E.E.; Buddavarapu, K.; Lenci, R.E.; Chocron, E.S.; Lechleiter, J.D.; Sass, M.; Aronin, N.; Schiavi, F.; et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat. Genet. 2010, 42, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Qin, Y.; Srikantan, S.; Luo, A.; Cheng, Z.M.; Flores, S.K.; Vogel, K.S.; Wang, E.; Dahia, P.L.M. The TMEM127 human tumor suppressor is a component of the mTORC1 lysosomal nutrient-sensing complex. Hum. Mol. Genet. 2018, 27, 1794–1808. [Google Scholar] [CrossRef] [PubMed]
- Flores, S.K.; Deng, Y.; Cheng, Z.; Zhang, X.; Tao, S.; Saliba, A.; Chu, I.; Burnichon, N.; Gimenez-Roqueplo, A.P.; Wang, E.; et al. Functional Characterization of TMEM127 Variants Reveals Novel Insights into Its Membrane Topology and Trafficking. J. Clin. Endocrinol. Metab. 2020, 105, e3142–e3156. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Deng, Y.; Ricketts, C.J.; Srikantan, S.; Wang, E.; Maher, E.R.; Dahia, P.L. The tumor susceptibility gene TMEM127 is mutated in renal cell carcinomas and modulates endolysosomal function. Hum. Mol. Genet. 2014, 23, 2428–2439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.; Schiavi, F.; Cascon, A.; Qin, Y.; Inglada-Perez, L.; King, E.E.; Toledo, R.A.; Ercolino, T.; Rapizzi, E.; Ricketts, C.J.; et al. Spectrum and prevalence of FP/TMEM127 gene mutations in pheochromocytomas and paragangliomas. JAMA 2010, 304, 2611–2619. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Flores, S.K.; Cheng, Z.; Jasper, A.M.; Natori, K.; Okamoto, T.; Tanabe, A.; Gotoh, K.; Shibata, H.; Sakurai, A.; Nakai, T.; et al. A synonymous VHL variant in exon 2 confers susceptibility to familial pheochromocytoma and von Hippel-Lindau disease. J. Clin. Endocrinol. Metab. 2019, 104, 3826–3834. [Google Scholar] [CrossRef]
- Lenglet, M.; Robriquet, F.; Schwarz, K.; Camps, C.; Couturier, A.; Hoogewijs, D.; Buffet, A.; Knight, S.J.L.; Gad, S.; Couve, S.; et al. Identification of a new VHL exon and complex splicing alterations in familial erythrocytosis or von Hippel-Lindau disease. Blood 2018, 132, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Dahia, P.L. Pheochromocytomas and Paragangliomas, Genetically Diverse and Minimalist, All at Once! Cancer Cell 2017, 31, 159–161. [Google Scholar] [CrossRef] [Green Version]
- Mucci, L.A.; Wedren, S.; Tamimi, R.M.; Trichopoulos, D.; Adami, H.O. The role of gene-environment interaction in the aetiology of human cancer: Examples from cancers of the large bowel, lung and breast. J. Intern. Med. 2001, 249, 477–493. [Google Scholar] [CrossRef]
- Hoffman, J.I.; Kaplan, S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef] [Green Version]
- Opotowsky, A.R.; Moko, L.E.; Ginns, J.; Rosenbaum, M.; Greutmann, M.; Aboulhosn, J.; Hageman, A.; Kim, Y.; Deng, L.X.; Grewal, J.; et al. Pheochromocytoma and paraganglioma in cyanotic congenital heart disease. J. Clin. Endocrinol. Metab. 2015, 100, 1325–1334. [Google Scholar] [CrossRef]
- Saldana, M.J.; Salem, L.E.; Travezan, R. High altitude hypoxia and chemodectomas. Hum. Pathol. 1973, 4, 251–263. [Google Scholar] [CrossRef]
- Rodriguez-Cuevas, H.; Lau, I.; Rodriguez, H.P. High-altitude paragangliomas diagnostic and therapeutic considerations. Cancer 1986, 57, 672–676. [Google Scholar] [CrossRef]
- Vaidya, A.; Flores, S.K.; Cheng, Z.M.; Nicolas, M.; Deng, Y.; Opotowsky, A.R.; Lourenco, D.M., Jr.; Barletta, J.A.; Rana, H.Q.; Pereira, M.A.; et al. EPAS1 Mutations and Paragangliomas in Cyanotic Congenital Heart Disease. N. Engl. J. Med. 2018, 378, 1259–1261. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. The VHL Tumor Suppressor Gene: Insights into Oxygen Sensing and Cancer. Trans. Am. Clin. Climatol. Assoc. 2017, 128, 298–307. [Google Scholar] [PubMed]
- Toledo, R.A.; Qin, Y.; Srikantan, S.; Morales, N.P.; Li, Q.; Deng, Y.; Kim, S.W.; Pereira, M.A.; Toledo, S.P.; Su, X.; et al. In vivo and in vitro oncogenic effects of HIF2A mutations in pheochromocytomas and paragangliomas. Endocr. Relat. Cancer 2013, 20, 349–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Lotti, L.V.; Vespa, S.; Pantalone, M.R.; Perconti, S.; Esposito, D.L.; Visone, R.; Veronese, A.; Paties, C.T.; Sanna, M.; Verginelli, F.; et al. A Developmental Perspective on Paragangliar Tumorigenesis. Cancers 2019, 11, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerecer-Gil, N.Y.; Figuera, L.E.; Llamas, F.J.; Lara, M.; Escamilla, J.G.; Ramos, R.; Estrada, G.; Hussain, A.K.; Gaal, J.; Korpershoek, E.; et al. Mutation of SDHB is a Cause of Hypoxia-Related High-Altitude Paraganglioma. Clin. Cancer Res. 2010, 16, 4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astrom, K.; Cohen, J.E.; Willett-Brozick, J.E.; Aston, C.E.; Baysal, B.E. Altitude is a phenotypic modifier in hereditary paraganglioma type 1: Evidence for an oxygen-sensing defect. Hum. Genet. 2003, 113, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Jech, M.; Alvarado-Cabrero, I.; Albores-Saavedra, J.; Dahia, P.L.; Tischler, A.S. Genetic analysis of high altitude paragangliomas. Endocr. Pathol. 2006, 17, 201–202. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores, S.K.; Estrada-Zuniga, C.M.; Thallapureddy, K.; Armaiz-Peña, G.; Dahia, P.L.M. Insights into Mechanisms of Pheochromocytomas and Paragangliomas Driven by Known or New Genetic Drivers. Cancers 2021, 13, 4602. https://doi.org/10.3390/cancers13184602
Flores SK, Estrada-Zuniga CM, Thallapureddy K, Armaiz-Peña G, Dahia PLM. Insights into Mechanisms of Pheochromocytomas and Paragangliomas Driven by Known or New Genetic Drivers. Cancers. 2021; 13(18):4602. https://doi.org/10.3390/cancers13184602
Chicago/Turabian StyleFlores, Shahida K., Cynthia M. Estrada-Zuniga, Keerthi Thallapureddy, Gustavo Armaiz-Peña, and Patricia L. M. Dahia. 2021. "Insights into Mechanisms of Pheochromocytomas and Paragangliomas Driven by Known or New Genetic Drivers" Cancers 13, no. 18: 4602. https://doi.org/10.3390/cancers13184602
APA StyleFlores, S. K., Estrada-Zuniga, C. M., Thallapureddy, K., Armaiz-Peña, G., & Dahia, P. L. M. (2021). Insights into Mechanisms of Pheochromocytomas and Paragangliomas Driven by Known or New Genetic Drivers. Cancers, 13(18), 4602. https://doi.org/10.3390/cancers13184602