
Motility Dynamics of T Cells in Tumor-Draining Lymph Nodes: A Rational Indicator of Antitumor Response and Immune Checkpoint Blockade


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
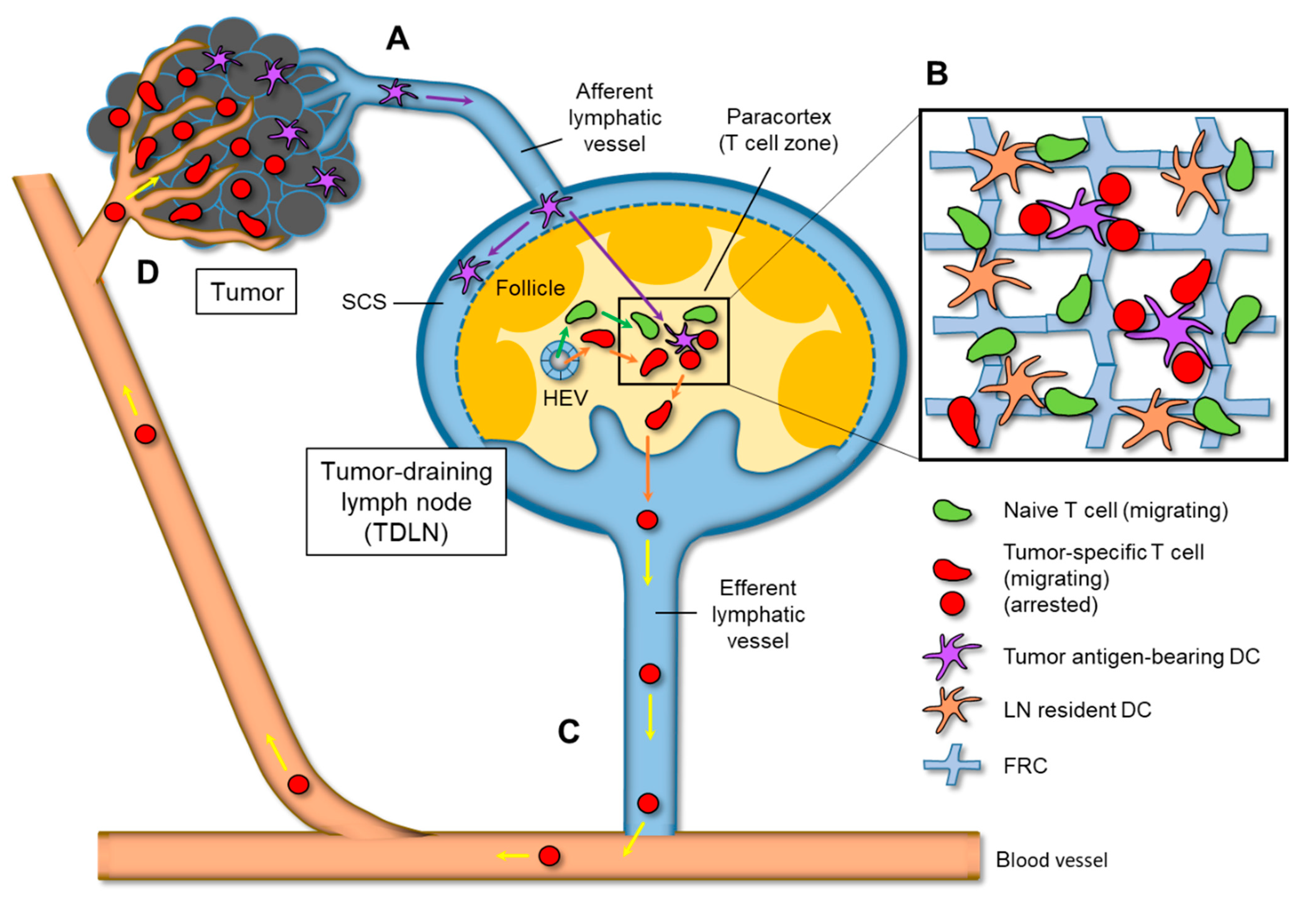
2. Antigen Transport from Primary Tumor to TDLNs via Lymphatic Vessels
3. Interstitial T Cell Migration in LNs
4. Antigen Recognition by T–DC Interaction and Changes in T Cell Motility
5. Immunological Synapse Formation and Stop Signal
6. Inhibition of T Cell Activation by CTLA-4 and PD-1
7. Immune Checkpoint Blockade and Activation of Antitumor Responses
8. Immune Checkpoint Inhibition and T Cell Motility
9. CTLA-4 Inhibition and T Cell Dynamics in Tumors
10. PD-1/PD-L1 Inhibition and T Cell Dynamics in Tumors
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APC | Antigen presenting cell |
| ATX | Autotaxin |
| CAR | Chimeric antigen receptor |
| CTLA-4 | Cytotoxic T-lymphocyte antigen 4 |
| DC | Dendritic cell |
| FRC | Fibroblastic reticular cell |
| HEV | High endothelial venule |
| HMG-box | High mobility group box |
| ICAM-1 | Intercellular adhesion molecule 1 |
| ICB | Immune checkpoint blockade |
| ICOS | Inducible T-cell costimulator |
| ICOSL | Inducible costimulator-ligand |
| IS | Immunological synapse |
| ITIM | Immunoreceptor tyrosine-based inhibitory motif |
| ITSM | Immunoreceptor-tyrosine-switch motif |
| JNK | c-Jun N-terminal kinase |
| LCMV | Lymphocytic choriomeningitis virus |
| LFA-1 | Lymphocyte function-associated antigen 1 |
| LN | Lymph node |
| LPA | Lysophosphatidic acid |
| MHC | Major histocompatibility complex |
| NFAT | Nuclear factor of activated T cells |
| NF-κB | Nuclear factor-kappa B |
| NKG2D | Natural-killer group 2, member D |
| NR4A | Nuclear receptor 4A |
| PD-1 | Programmed cell death 1 |
| PD-L1 | Programmed cell death 1 ligand 1 |
| PI3K | Phosphatidyl inositol 3-kinase |
| PKCθ | Protein kinase C θ |
| PP2A | Protein phosphatase 2A |
| SCS | Subcapsular sinus |
| SHP-1 | Src homology 2 domain-containing phosphatase 1 |
| SLO | Secondary lymphoid organ |
| SMAC | Supramolecular activation complex |
| TAM | Tumor-associated macrophage |
| TCF-1 | T cell factor-1 |
| TCR | T cell receptor |
| TDLN | Tumor-draining lymph node |
| ZAP-70 | Zeta-chain-associated protein kinase 70 |
References
- Lian, J.; Luster, A.D. Chemokine-guided cell positioning in the lymph node orchestrates the generation of adaptive immune responses. Curr. Opin. Cell Biol. 2015, 36, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Von Andrian, U.H.; Mempel, T.R. Homing and cellular traffic in lymph nodes. Nat. Rev. Immunol. 2003, 3, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Willard-Mack, C.L. Normal structure, function, and histology of lymph nodes. Toxicol. Pathol. 2006, 34, 409–424. [Google Scholar] [CrossRef] [Green Version]
- Katakai, T.; Hara, T.; Lee, J.H.; Gonda, H.; Sugai, M.; Shimizu, A. A novel reticular stromal structure in lymph node cortex: An immuno-platform for interactions among dendritic cells, T cells and B cells. Int. Immunol. 2004, 16, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Katakai, T.; Hara, T.; Sugai, M.; Gonda, H.; Shimizu, A. Lymph node fibroblastic reticular cells construct the stromal reticulum via contact with lymphocytes. J. Exp. Med. 2004, 200, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Cyster, J.G. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu. Rev. Immunol. 2005, 23, 127–159. [Google Scholar] [CrossRef] [PubMed]
- Bajenoff, M.; Egen, J.G.; Koo, L.Y.; Laugier, J.P.; Brau, F.; Glaichenhaus, N.; Germain, R.N. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity 2006, 25, 989–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gretz, J.E.; Anderson, A.O.; Shaw, S. Cords, channels, corridors and conduits: Critical architectural elements facilitating cell interactions in the lymph node cortex. Immunol. Rev. 1997, 156, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Gretz, J.E.; Kaldjian, E.P.; Anderson, A.O.; Shaw, S. Commentary—Sophisticated strategies for information encounter in the lymph node—The reticular network as a conduit of soluble information and a highway for cell traffic. J. Immunol. 1996, 157, 495–499. [Google Scholar]
- Kaldjian, E.P.; Gretz, J.E.; Anderson, A.O.; Shi, Y.; Shaw, S. Spatial and molecular organization of lymph node T cell cortex: A labyrinthine cavity bounded by an epithelium-like monolayer of fibroblastic reticular cells anchored to basement membrane-like extracellular matrix. Int. Immunol. 2001, 13, 1243–1253. [Google Scholar] [CrossRef]
- Miller, M.J.; Wei, S.H.; Parker, I.; Cahalan, M.D. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science 2002, 296, 1869–1873. [Google Scholar] [CrossRef] [Green Version]
- Gerner, M.Y.; Torabi-Parizi, P.; Germain, R.N. Strategically localized dendritic cells promote rapid T cell responses to lymph-borne particulate antigens. Immunity 2015, 42, 172–185. [Google Scholar] [CrossRef] [Green Version]
- Woodruff, M.C.; Turley, S.J. Chemokine ‘grooming’ by cLECs directs DC migration. Nat. Immunol. 2014, 15, 595–596. [Google Scholar] [CrossRef]
- Miller, M.J.; Safrina, O.; Parker, I.; Cahalan, M.D. Imaging the single cell dynamics of CD4+ T cell activation by dendritic cells in lymph nodes. J. Exp. Med. 2004, 200, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Kitano, M.; Yamazaki, C.; Takumi, A.; Ikeno, T.; Hemmi, H.; Takahashi, N.; Shimizu, K.; Fraser, S.E.; Hoshino, K.; Kaisho, T.; et al. Imaging of the cross-presenting dendritic cell subsets in the skin-draining lymph node. Proc. Natl. Acad. Sci. USA 2016, 113, 1044–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, Y.; Takeuchi, A.; Ozawa, M.; Kurosawa, Y.; Kawamura, T.; Bogdanova, D.; Iioka, H.; Kondo, E.; Kitazawa, Y.; Ueta, H.; et al. Visualizing the Rapid and Dynamic Elimination of Allogeneic T Cells in Secondary Lymphoid Organs. J. Immunol. 2018, 201, 1062–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katakai, T.; Kinashi, T. Microenvironmental Control of High-Speed Interstitial T Cell Migration in the Lymph Node. Front. Immunol. 2016, 7, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asano, K.; Nabeyama, A.; Miyake, Y.; Qiu, C.H.; Kurita, A.; Tomura, M.; Kanagawa, O.; Fujii, S.; Tanaka, M. CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity 2011, 34, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louie, D.A.P.; Liao, S. Lymph Node Subcapsular Sinus Macrophages as the Frontline of Lymphatic Immune Defense. Front. Immunol. 2019, 10, 347. [Google Scholar] [CrossRef] [Green Version]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [Green Version]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Paulete, A.R.; Cueto, F.J.; Martinez-Lopez, M.; Labiano, S.; Morales-Kastresana, A.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, M.A.; Quetglas, J.I.; et al. Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov. 2016, 6, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Paulete, A.R.; Teijeira, A.; Cueto, F.J.; Garasa, S.; Perez-Gracia, J.L.; Sanchez-Arraez, A.; Sancho, D.; Melero, I. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann. Oncol. 2017, 28, xii44–xii55. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mocellin, S.; Nitti, D. CTLA-4 blockade and the renaissance of cancer immunotherapy. Biochim. Biophys. Acta 2013, 1836, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Renkawitz, J.; Sixt, M. Mechanisms of force generation and force transmission during interstitial leukocyte migration. EMBO Rep. 2010, 11, 744–750. [Google Scholar] [CrossRef] [Green Version]
- Rudd, C.E. The reverse stop-signal model for CTLA4 function. Nat. Rev. Immunol. 2008, 8, 153–160. [Google Scholar] [CrossRef]
- Fife, B.T.; Pauken, K.E.; Eagar, T.N.; Obu, T.; Wu, J.; Tang, Q.; Azuma, M.; Krummel, M.F.; Bluestone, J.A. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol. 2009, 10, 1185–1192. [Google Scholar] [CrossRef]
- Ferris, S.T.; Durai, V.; Wu, R.; Theisen, D.J.; Ward, J.P.; Bern, M.D.; Davidson, J.T.; Bagadia, P.; Liu, T.; Briseno, C.G.; et al. cDC1 prime and are licensed by CD4(+) T cells to induce anti-tumour immunity. Nature 2020, 584, 624–629. [Google Scholar] [CrossRef]
- Wolkers, M.C.; Stoetter, G.; Vyth-Dreese, F.A.; Schumacher, T.N. Redundancy of direct priming and cross-priming in tumor-specific CD8+ T cell responses. J. Immunol. 2001, 167, 3577–3584. [Google Scholar] [CrossRef] [Green Version]
- Diao, J.; Zhao, J.; Winter, E.; Cattral, M.S. Recruitment and differentiation of conventional dendritic cell precursors in tumors. J. Immunol. 2010, 184, 1261–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.A.; Jackson, D.G. The chemokine CX3CL1 promotes trafficking of dendritic cells through inflamed lymphatics. J. Cell Sci. 2013, 126 Pt 22, 5259–5270. [Google Scholar] [CrossRef] [Green Version]
- Ruhland, M.K.; Roberts, E.W.; Cai, E.; Mujal, A.M.; Marchuk, K.; Beppler, C.; Nam, D.; Serwas, N.K.; Binnewies, M.; Krummel, M.F. Visualizing Synaptic Transfer of Tumor Antigens among Dendritic Cells. Cancer Cell 2020, 37, 786–799.e5. [Google Scholar] [CrossRef] [PubMed]
- Allan, R.S.; Waithman, J.; Bedoui, S.; Jones, C.M.; Villadangos, J.A.; Zhan, Y.; Lew, A.M.; Shortman, K.; Heath, W.R.; Carbone, F.R. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity 2006, 25, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4(+) T Cell Immunity. Cell 2019, 177, 556–571.e16. [Google Scholar] [CrossRef] [PubMed]
- Brightman, S.E.; Naradikian, M.S.; Miller, A.M.; Schoenberger, S.P. Harnessing neoantigen specific CD4 T cells for cancer immunotherapy. J. Leukoc. Biol. 2020, 107, 625–633. [Google Scholar] [CrossRef]
- Stoll, S.; Delon, J.; Brotz, T.M.; Germain, R.N. Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science 2002, 296, 1873–1876. [Google Scholar] [CrossRef]
- Katakai, T.; Habiro, K.; Kinashi, T. Dendritic cells regulate high-speed interstitial T cell migration in the lymph node via LFA-1/ICAM-1. J. Immunol. 2013, 191, 1188–1199. [Google Scholar] [CrossRef]
- Katakai, T.; Kondo, N.; Ueda, Y.; Kinashi, T. Autotaxin produced by stromal cells promotes LFA-1-independent and Rho-dependent interstitial T cell motility in the lymph node paracortex. J. Immunol. 2014, 193, 617–626. [Google Scholar] [CrossRef] [Green Version]
- Woolf, E.; Grigorova, I.; Sagiv, A.; Grabovsky, V.; Feigelson, S.W.; Shulman, Z.; Hartmann, T.; Sixt, M.; Cyster, J.G.; Alon, R. Lymph node chemokines promote sustained T lymphocyte motility without triggering stable integrin adhesiveness in the absence of shear forces. Nat. Immunol. 2007, 8, 1076–1085. [Google Scholar] [CrossRef]
- Stachowiak, A.N.; Wang, Y.; Huang, Y.C.; Irvine, D.J. Homeostatic lymphoid chemokines synergize with adhesion ligands to trigger T and B lymphocyte chemokinesis. J. Immunol. 2006, 177, 2340–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luther, S.A.; Tang, H.L.; Hyman, P.L.; Farr, A.G.; Cyster, J.G. Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc. Natl. Acad. Sci. USA 2000, 97, 12694–12699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunn, M.D.; Kyuwa, S.; Tam, C.; Kakiuchi, T.; Matsuzawa, A.; Williams, L.T.; Nakano, H. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J. Exp. Med. 1999, 189, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, T.; Cyster, J.G. CC chemokine receptor 7 contributes to Gi-dependent T cell motility in the lymph node. J. Immunol. 2007, 178, 2973–2978. [Google Scholar] [CrossRef] [Green Version]
- Worbs, T.; Mempel, T.R.; Bolter, J.; von Andrian, U.H.; Forster, R. CCR7 ligands stimulate the intranodal motility of T lymphocytes in vivo. J. Exp. Med. 2007, 204, 489–495. [Google Scholar] [CrossRef] [Green Version]
- An, S.; Dickens, M.A.; Bleu, T.; Hallmark, O.G.; Goetzl, E.J. Molecular cloning of the human Edg2 protein and its identification as a functional cellular receptor for lysophosphatidic acid. Biochem. Biophys. Res. Commun. 1997, 231, 619–622. [Google Scholar] [CrossRef]
- An, S.; Bleu, T.; Hallmark, O.G.; Goetzl, E.J. Characterization of a novel subtype of human G protein-coupled receptor for lysophosphatidic acid. J. Biol. Chem. 1998, 273, 7906–7910. [Google Scholar] [CrossRef] [Green Version]
- Bandoh, K.; Aoki, J.; Hosono, H.; Kobayashi, S.; Kobayashi, T.; Murakami-Murofushi, K.; Tsujimoto, M.; Arai, H.; Inoue, K. Molecular cloning and characterization of a novel human G-protein-coupled receptor, EDG7, for lysophosphatidic acid. J. Biol. Chem. 1999, 274, 27776–27785. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, K.; Ishii, S.; Shimizu, T. Identification of p2y9/GPR23 as a novel G protein-coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. J. Biol. Chem. 2003, 278, 25600–25606. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.W.; Rivera, R.; Gardell, S.; Dubin, A.E.; Chun, J. GPR92 as a new G12/13- and Gq-coupled lysophosphatidic acid receptor that increases cAMP, LPA5. J. Biol. Chem. 2006, 281, 23589–23597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagida, K.; Masago, K.; Nakanishi, H.; Kihara, Y.; Hamano, F.; Tajima, Y.; Taguchi, R.; Shimizu, T.; Ishii, S. Identification and characterization of a novel lysophosphatidic acid receptor, p2y5/LPA6. J. Biol. Chem. 2009, 284, 17731–17741. [Google Scholar] [CrossRef] [Green Version]
- Takeda, A.; Kobayashi, D.; Aoi, K.; Sasaki, N.; Sugiura, Y.; Igarashi, H.; Tohya, K.; Inoue, A.; Hata, E.; Akahoshi, N.; et al. Fibroblastic reticular cell-derived lysophosphatidic acid regulates confined intranodal T-cell motility. Elife 2016, 5, e10561. [Google Scholar] [CrossRef] [PubMed]
- Knowlden, S.A.; Capece, T.; Popovic, M.; Chapman, T.J.; Rezaee, F.; Kim, M.; Georas, S.N. Regulation of T cell motility in vitro and in vivo by LPA and LPA2. PLoS ONE 2014, 9, e101655. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.J.; Hejazi, A.S.; Wei, S.H.; Cahalan, M.D.; Parker, I. T cell repertoire scanning is promoted by dynamic dendritic cell behavior and random T cell motility in the lymph node. Proc. Natl. Acad. Sci. USA 2004, 101, 998–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dustin, M.L. Stop and go traffic to tune T cell responses. Immunity 2004, 21, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Mempel, T.R.; Henrickson, S.E.; Von Andrian, U.H. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 2004, 427, 154–159. [Google Scholar] [CrossRef] [Green Version]
- Clark, C.E.; Hasan, M.; Bousso, P. A role for the immediate early gene product c-fos in imprinting T cells with short-term memory for signal summation. PLoS ONE 2011, 6, e18916. [Google Scholar] [CrossRef]
- Marangoni, F.; Murooka, T.T.; Manzo, T.; Kim, E.Y.; Carrizosa, E.; Elpek, N.M.; Mempel, T.R. The transcription factor NFAT exhibits signal memory during serial T cell interactions with antigen-presenting cells. Immunity 2013, 38, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Fife, B.T.; Pauken, K.E. The role of the PD-1 pathway in autoimmunity and peripheral tolerance. Ann. N. Y. Acad. Sci. 2011, 1217, 45–59. [Google Scholar] [CrossRef]
- Celli, S.; Lemaitre, F.; Bousso, P. Real-time manipulation of T cell-dendritic cell interactions in vivo reveals the importance of prolonged contacts for CD4+ T cell activation. Immunity 2007, 27, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Duckworth, B.C.; Lafouresse, F.; Wimmer, V.C.; Broomfield, B.J.; Dalit, L.; Alexandre, Y.O.; Sheikh, A.A.; Qin, R.Z.; Alvarado, C.; Mielke, L.A.; et al. Effector and stem-like memory cell fates are imprinted in distinct lymph node niches directed by CXCR3 ligands. Nat. Immunol. 2021, 22, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, T.; Sakata-Sogawa, K.; Kobayashi, W.; Hiroshima, M.; Hashimoto-Tane, A.; Tokunaga, M.; Dustin, M.L.; Saito, T. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat. Immunol. 2005, 6, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Yokosuka, T. Immunological synapse and microclusters: The site for recognition and activation of T cells. Curr. Opin. Immunol. 2006, 18, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Bousso, P.; Robey, E. Dynamics of CD8+ T cell priming by dendritic cells in intact lymph nodes. Nat. Immunol. 2003, 4, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Hugues, S.; Fetler, L.; Bonifaz, L.; Helft, J.; Amblard, F.; Amigorena, S. Distinct T cell dynamics in lymph nodes during the induction of tolerance and immunity. Nat. Immunol. 2004, 5, 1235–1242. [Google Scholar] [CrossRef]
- Slavik, J.M.; Hutchcroft, J.E.; Bierer, B.E. CD28/CTLA-4 and CD80/CD86 families: Signaling and function. Immunol. Res. 1999, 19, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.F.; Bartumeus, F.; Gerard, A. T cell migration, search strategies and mechanisms. Nat. Rev. Immunol. 2016, 16, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Schneider, H.; Rudd, C.E. Murine regulatory T cells differ from conventional T cells in resisting the CTLA-4 reversal of TCR stop-signal. Blood 2012, 120, 4560–4570. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Li, X.; Liu, D.; Li, J.; Zhang, X.; Chen, X.; Hou, S.; Peng, L.; Xu, C.; Liu, W.; et al. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature 2013, 496, 523–527. [Google Scholar] [CrossRef]
- Occhipinti, S.; Dianzani, C.; Chiocchetti, A.; Boggio, E.; Clemente, N.; Gigliotti, C.L.; Soluri, M.F.; Minelli, R.; Fantozzi, R.; Yagi, J.; et al. Triggering of B7h by the ICOS modulates maturation and migration of monocyte-derived dendritic cells. J. Immunol. 2013, 190, 1125–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dianzani, C.; Minelli, R.; Gigliotti, C.L.; Occhipinti, S.; Giovarelli, M.; Conti, L.; Boggio, E.; Shivakumar, Y.; Baldanzi, G.; Malacarne, V.; et al. B7h triggering inhibits the migration of tumor cell lines. J. Immunol. 2014, 192, 4921–4931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raineri, D.; Dianzani, C.; Cappellano, G.; Maione, F.; Baldanzi, G.; Iacobucci, I.; Clemente, N.; Baldone, G.; Boggio, E.; Gigliotti, C.L.; et al. Osteopontin binds ICOSL promoting tumor metastasis. Commun. Biol. 2020, 3, 615. [Google Scholar] [CrossRef]
- Schneider, H.; Downey, J.; Smith, A.; Zinselmeyer, B.H.; Rush, C.; Brewer, J.M.; Wei, B.; Hogg, N.; Garside, P.; Rudd, C.E. Reversal of the TCR stop signal by CTLA-4. Science 2006, 313, 1972–1975. [Google Scholar] [CrossRef] [Green Version]
- Schneider, H.; Smith, X.; Liu, H.; Bismuth, G.; Rudd, C.E. CTLA-4 disrupts ZAP70 microcluster formation with reduced T cell/APC dwell times and calcium mobilization. Eur. J. Immunol. 2008, 38, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, R.; Sugiura, D.; Shimizu, K.; Maruhashi, T.; Watada, M.; Okazaki, I.M.; Okazaki, T. PD-1 Primarily Targets TCR Signal in the Inhibition of Functional T Cell Activation. Front. Immunol. 2019, 10, 630. [Google Scholar] [CrossRef] [Green Version]
- Linsley, P.S.; Brady, W.; Urnes, M.; Grosmaire, L.S.; Damle, N.K.; Ledbetter, J.A. CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 1991, 174, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Linsley, P.S.; Greene, J.L.; Tan, P.; Bradshaw, J.; Ledbetter, J.A.; Anasetti, C.; Damle, N.K. Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J. Exp. Med. 1992, 176, 1595–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Collins, A.V.; Brodie, D.W.; Gilbert, R.J.C.; Iaboni, A.; Manso-Sancho, R.; Walse, B.; Stuart, D.I.; van der Merwe, P.A.; Davis, S.J. The Interaction Properties of Costimulatory Molecules Revisited. Immunity 2002, 17, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.M.; Chuang, E.; Griffin, M.; Khattri, R.; Hong, D.K.; Zhang, W.; Straus, D.; Samelson, L.E.; Thompson, C.B.; Bluestone, J.A. Molecular basis of T cell inactivation by CTLA-4. Science 1998, 282, 2263–2266. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Rudd, C.E.; Schneider, H. Src kinases Fyn and Lck facilitate the accumulation of phosphorylated CTLA-4 and its association with PI-3 kinase in intracellular compartments of T-cells. Biochem. Biophys. Res. Commun. 2001, 288, 573–578. [Google Scholar] [CrossRef]
- Schneider, H.; Dias, S.d.R.; Hu, H.; Rudd, C.E. A regulatory role for cytoplasmic YVKM motif in CTLA-4 inhibition of TCR signaling. Eur. J. Immunol. 2001, 31, 2042–2050. [Google Scholar] [CrossRef]
- Marengere, L.E.; Waterhouse, P.; Duncan, G.S.; Mittrucker, H.W.; Feng, G.S.; Mak, T.W. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science 1996, 272, 1170–1173. [Google Scholar] [CrossRef] [PubMed]
- Chuang, E.; Fisher, T.S.; Morgan, R.W.; Robbins, M.D.; Duerr, J.M.; Vander Heiden, M.G.; Gardner, J.P.; Hambor, J.E.; Neveu, M.J.; Thompson, C.B. The CD28 and CTLA-4 Receptors Associate with the Serine/Threonine Phosphatase PP2A. Immunity 2000, 13, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Schneider, H.; Rudd, C.E. Tyrosine phosphatase SHP-2 binding to CTLA-4: Absence of direct YVKM/YFIP motif recognition. Biochem. Biophys. Res. Commun. 2000, 269, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, T.; Kobayashi, W.; Takamatsu, M.; Sakata-Sogawa, K.; Zeng, H.; Hashimoto-Tane, A.; Yagita, H.; Tokunaga, M.; Saito, T. Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity 2010, 33, 326–339. [Google Scholar] [CrossRef] [Green Version]
- Baroja, M.L.; Vijayakrishnan, L.; Bettelli, E.; Darlington, P.J.; Chau, T.A.; Ling, V.; Collins, M.; Carreno, B.M.; Madrenas, J.; Kuchroo, V.K. Inhibition of CTLA-4 function by the regulatory subunit of serine/threonine phosphatase 2A. J. Immunol. 2002, 168, 5070–5078. [Google Scholar] [CrossRef]
- Calvo, C.R.; Amsen, D.; Kruisbeek, A.M. Cytotoxic T lymphocyte antigen 4 (CTLA-4) interferes with extracellular signal-regulated kinase (ERK) and Jun NH2-terminal kinase (JNK) activation, but does not affect phosphorylation of T cell receptor zeta and ZAP70. J. Exp. Med. 1997, 186, 1645–1653. [Google Scholar] [CrossRef] [Green Version]
- Stein, P.H.; Fraser, J.D.; Weiss, A. The cytoplasmic domain of CD28 is both necessary and sufficient for costimulation of interleukin-2 secretion and association with phosphatidylinositol 3′-kinase. Mol. Cell Biol. 1994, 14, 3392–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovcinnikovs, V.; Ross, E.M.; Petersone, L.; Edner, N.M.; Heuts, F.; Ntavli, E.; Kogimtzis, A.; Kennedy, A.; Wang, C.J.; Bennett, C.L.; et al. CTLA-4–mediated transendocytosis of costimulatory molecules primarily targets migratory dendritic cells. Sci. Immunol. 2019, 4, eaaw0902. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, H.; Agata, Y.; Kawasaki, A.; Sato, M.; Imamura, S.; Minato, N.; Yagita, H.; Nakano, T.; Honjo, T. Developmentally regulated expression of the PD-1 protein on the surface of double-negative (CD4-CD8-) thymocytes. Int. Immunol. 1996, 8, 773–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of Lupus-like Autoimmune Diseases by Disruption of the PD-1 Gene Encoding an ITIM Motif-Carrying Immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, T.; Iwai, Y.; Honjo, T. New regulatory co-receptors: Inducible co-stimulator and PD-1. Curr. Opin. Immunol. 2002, 14, 779–782. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Okazaki, T.; Honjo, T. Rejuvenating exhausted T cells during chronic viral infection. Cell 2006, 124, 459–461. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.; Tanaka, S.; Chu, F.; Nurieva, R.I.; Martinez, G.J.; Rawal, S.; Wang, Y.H.; Lim, H.; Reynolds, J.M.; Zhou, X.H.; et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat. Med. 2011, 17, 983–988. [Google Scholar] [CrossRef]
- Jie, H.B.; Gildener-Leapman, N.; Li, J.; Srivastava, R.M.; Gibson, S.P.; Whiteside, T.L.; Ferris, R.L. Intratumoral regulatory T cells upregulate immunosuppressive molecules in head and neck cancer patients. Br. J. Cancer 2013, 109, 2629–2635. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Akiba, H.; Iwai, H.; Matsuda, H.; Aoki, M.; Tanno, Y.; Shin, T.; Tsuchiya, H.; Pardoll, D.M.; Okumura, K.; et al. Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol. 2002, 169, 5538–5545. [Google Scholar] [CrossRef] [Green Version]
- Ishida, M.; Iwai, Y.; Tanaka, Y.; Okazaki, T.; Freeman, G.J.; Minato, N.; Honjo, T. Differential expression of PD-L1 and PD-L2, ligands for an inhibitory receptor PD-1, in the cells of lymphohematopoietic tissues. Immunol. Lett. 2002, 84, 57–62. [Google Scholar] [CrossRef]
- Blank, C.; Brown, I.; Peterson, A.C.; Spiotto, M.; Iwai, Y.; Honjo, T.; Gajewski, T.F. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004, 64, 1140–1145. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef] [Green Version]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Duke-Cohan, J.S.; Chaudhri, A.; Aksoylar, H.I.; Wang, Q.; Council, A.; Berg, A.; Freeman, G.J.; Boussiotis, V.A. Interaction of SHP-2 SH2 domains with PD-1 ITSM induces PD-1 dimerization and SHP-2 activation. Commun. Biol. 2020, 3, 128. [Google Scholar] [CrossRef]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef] [Green Version]
- Sheppard, K.A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef] [Green Version]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, D.; Maruhashi, T.; Okazaki, I.-M.; Shimizu, K.; Maeda, T.K.; Takemoto, T.; Okazaki, T. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science 2019, 364, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lee, C.K.; Lin, C.-H.; Gassen, R.B.; Xu, X.; Huang, Z.; Xiao, C.; Bonorino, C.; Lu, L.-F.; Bui, J.D.; et al. PD-L1:CD80 cis-heterodimer triggers the co-stimulatory receptor CD28 while repressing the inhibitory PD-1 and CTLA-4 pathways. Immunity 2019, 51, 1059–1073.e1059. [Google Scholar] [CrossRef] [PubMed]
- Chikuma, S.; Terawaki, S.; Hayashi, T.; Nabeshima, R.; Yoshida, T.; Shibayama, S.; Okazaki, T.; Honjo, T. PD-1-mediated suppression of IL-2 production induces CD8+ T cell anergy in vivo. J. Immunol. 2009, 182, 6682–6689. [Google Scholar] [CrossRef] [Green Version]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Pedicord, V.A.; Montalvo, W.; Leiner, I.M.; Allison, J.P. Single dose of anti-CTLA-4 enhances CD8+ T-cell memory formation, function, and maintenance. Proc. Natl. Acad. Sci. USA 2011, 108, 266–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, F.; Du, X.; Liu, M.; Zheng, P.; Liu, Y. Anti-CTLA-4 antibodies in cancer immunotherapy: Selective depletion of intratumoral regulatory T cells or checkpoint blockade? Cell Biosci. 2018, 8, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selby, M.J.; Engelhardt, J.J.; Quigley, M.; Henning, K.A.; Chen, T.; Srinivasan, M.; Korman, A.J. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 2013, 1, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulliard, Y.; Jolicoeur, R.; Windman, M.; Rue, S.M.; Ettenberg, S.; Knee, D.A.; Wilson, N.S.; Dranoff, G.; Brogdon, J.L. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J. Exp. Med. 2013, 210, 1685–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Tang, F.; Liu, M.; Su, J.; Zhang, Y.; Wu, W.; Devenport, M.; Lazarski, C.A.; Zhang, P.; Wang, X.; et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. 2018, 28, 416–432. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Peng, Q.; Qiu, X.; Zhang, Z.; Zhang, S.; Zhang, Y.; Liang, Y.; Guo, J.; Peng, H.; Chen, M.; Fu, Y.X.; et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat. Commun. 2020, 11, 4835. [Google Scholar] [CrossRef]
- Perez, V.L.; Van Parijs, L.; Biuckians, A.; Zheng, X.X.; Strom, T.B.; Abbas, A.K. Induction of Peripheral T Cell Tolerance In Vivo Requires CTLA-4 Engagement. Immunity 1997, 6, 411–417. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, T.; Honjo, T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006, 27, 195–201. [Google Scholar] [CrossRef]
- Fife, B.T.; Guleria, I.; Gubbels Bupp, M.; Eagar, T.N.; Tang, Q.; Bour-Jordan, H.; Yagita, H.; Azuma, M.; Sayegh, M.H.; Bluestone, J.A. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway. J. Exp. Med. 2006, 203, 2737–2747. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef]
- Alfei, F.; Kanev, K.; Hofmann, M.; Wu, M.; Ghoneim, H.E.; Roelli, P.; Utzschneider, D.T.; von Hoesslin, M.; Cullen, J.G.; Fan, Y.; et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 2019, 571, 265–269. [Google Scholar] [CrossRef]
- Scott, A.C.; Dundar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef]
- Yao, C.; Sun, H.W.; Lacey, N.E.; Ji, Y.; Moseman, E.A.; Shih, H.Y.; Heuston, E.F.; Kirby, M.; Anderson, S.; Cheng, J.; et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8(+) T cell persistence in chronic infection. Nat. Immunol. 2019, 20, 890–901. [Google Scholar] [CrossRef]
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef] [Green Version]
- Kim, P.S.; Ahmed, R. Features of responding T cells in cancer and chronic infection. Curr. Opin. Immunol. 2010, 22, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinselmeyer, B.H.; Heydari, S.; Sacristan, C.; Nayak, D.; Cammer, M.; Herz, J.; Cheng, X.; Davis, S.J.; Dustin, M.L.; McGavern, D.B. PD-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. J. Exp. Med. 2013, 210, 757–774. [Google Scholar] [CrossRef] [Green Version]
- Odagiu, L.; May, J.; Boulet, S.; Baldwin, T.A.; Labrecque, N. Role of the Orphan Nuclear Receptor NR4A Family in T-Cell Biology. Front. Endocrinol. 2020, 11, 624122. [Google Scholar] [CrossRef] [PubMed]
- Pentcheva-Hoang, T.; Simpson, T.R.; Montalvo-Ortiz, W.; Allison, J.P. Cytotoxic T lymphocyte antigen-4 blockade enhances antitumor immunity by stimulating melanoma-specific T-cell motility. Cancer Immunol Res. 2014, 2, 970–980. [Google Scholar] [CrossRef] [Green Version]
- Ruocco, M.G.; Pilones, K.A.; Kawashima, N.; Cammer, M.; Huang, J.; Babb, J.S.; Liu, M.; Formenti, S.C.; Dustin, M.L.; Demaria, S. Suppressing T cell motility induced by anti-CTLA-4 monotherapy improves antitumor effects. J. Clin. Investig. 2012, 122, 3718–3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, H.; Valk, E.; da Rocha Dias, S.; Wei, B.; Rudd, C.E. CTLA-4 up-regulation of lymphocyte function-associated antigen 1 adhesion and clustering as an alternate basis for coreceptor function. Proc. Natl. Acad. Sci. USA 2005, 102, 12861–12866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, T.J.; Carey, K.D.; Wetzel, S.A.; Parker, D.C.; Stork, P.J. Regulation of the small GTPase Rap1 and extracellular signal-regulated kinases by the costimulatory molecule CTLA-4. Mol. Cell Biol. 2005, 25, 4117–4128. [Google Scholar] [CrossRef] [Green Version]
- Deguine, J.; Breart, B.; Lemaitre, F.; Di Santo, J.P.; Bousso, P. Intravital imaging reveals distinct dynamics for natural killer and CD8(+) T cells during tumor regression. Immunity 2010, 33, 632–644. [Google Scholar] [CrossRef] [Green Version]
- Boissonnas, A.; Fetler, L.; Zeelenberg, I.S.; Hugues, S.; Amigorena, S. In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J. Exp. Med. 2007, 204, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunner-Weinzierl, M.C.; Rudd, C.E. CTLA-4 and PD-1 Control of T-Cell Motility and Migration: Implications for Tumor Immunotherapy. Front. Immunol. 2018, 9, 2737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigelin, B.; Bolanos, E.; Teijeira, A.; Martinez-Forero, I.; Labiano, S.; Azpilikueta, A.; Morales-Kastresana, A.; Quetglas, J.I.; Wagena, E.; Sanchez-Paulete, A.R.; et al. Focusing and sustaining the antitumor CTL effector killer response by agonist anti-CD137 mAb. Proc. Natl. Acad. Sci. USA 2015, 112, 7551–7556. [Google Scholar] [CrossRef] [Green Version]
- Knieke, K.; Hoff, H.; Maszyna, F.; Kolar, P.; Schrage, A.; Hamann, A.; Debes, G.F.; Brunner-Weinzierl, M.C. CD152 (CTLA-4) determines CD4 T cell migration in vitro and in vivo. PLoS ONE 2009, 4, e5702. [Google Scholar] [CrossRef] [Green Version]
- Fehlings, M.; Simoni, Y.; Penny, H.L.; Becht, E.; Loh, C.Y.; Gubin, M.M.; Ward, J.P.; Wong, S.C.; Schreiber, R.D.; Newell, E.W. Checkpoint blockade immunotherapy reshapes the high-dimensional phenotypic heterogeneity of murine intratumoural neoantigen-specific CD8(+) T cells. Nat. Commun. 2017, 8, 562. [Google Scholar] [CrossRef] [PubMed]
- Lau, D.; Garcon, F.; Chandra, A.; Lechermann, L.M.; Aloj, L.; Chilvers, E.R.; Corrie, P.G.; Okkenhaug, K.; Gallagher, F.A. Intravital Imaging of Adoptive T-Cell Morphology, Mobility and Trafficking Following Immune Checkpoint Inhibition in a Mouse Melanoma Model. Front. Immunol. 2020, 11, 1514. [Google Scholar] [CrossRef]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guerin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Wang, Y.; Han, W. Chimeric antigen receptor-modified T cells for solid tumors: Challenges and prospects. J. Immunol. Res. 2016, 2016, 3850839. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Liu, B. Critical factors in chimeric antigen receptor-modified T-cell (CAR-T) therapy for solid tumors. Onco. Targets Ther. 2019, 12, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Rui, W.; Zheng, H.; Huang, D.; Yu, F.; Zhang, Y.; Dong, J.; Zhao, X.; Lin, X. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur. J. Immunol. 2020, 50, 712–724. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, S.; Togashi, Y.; Kamada, T.; Sugiyama, E.; Nishinakamura, H.; Takeuchi, Y.; Vitaly, K.; Itahashi, K.; Maeda, Y.; Matsui, S.; et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol. 2020, 21, 1346–1358. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e10. [Google Scholar] [CrossRef] [Green Version]
- Kurtulus, S.; Madi, A.; Escobar, G.; Klapholz, M.; Nyman, J.; Christian, E.; Pawlak, M.; Dionne, D.; Xia, J.; Rozenblatt-Rosen, O.; et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1(-)CD8(+) Tumor-Infiltrating T Cells. Immunity 2019, 50, 181–194.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardenas, M.A.; Prokhnevska, N.; Kissick, H.T. Organized immune cell interactions within tumors sustain a productive T-cell response. Int. Immunol. 2021, 33, 27–37. [Google Scholar] [CrossRef]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Jansen, C.S.; Prokhnevska, N.; Master, V.A.; Sanda, M.G.; Carlisle, J.W.; Bilen, M.A.; Cardenas, M.; Wilkinson, S.; Lake, R.; Sowalsky, A.G.; et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 2019, 576, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Dammeijer, F.; van Gulijk, M.; Mulder, E.E.; Lukkes, M.; Klaase, L.; van den Bosch, T.; van Nimwegen, M.; Lau, S.P.; Latupeirissa, K.; Schetters, S.; et al. The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell 2020, 38, 685–700.e8. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.A.; Wu, D.-C.; Cheung, J.; Navarro, A.; Xiong, H.; Cubas, R.; Totpal, K.; Chiu, H.; Wu, Y.; Comps-Agrar, L.; et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat. Cancer 2020, 1, 681–691. [Google Scholar] [CrossRef]
- Rotman, J.; Koster, B.D.; Jordanova, E.S.; Heeren, A.M.; de Gruijl, T.D. Unlocking the therapeutic potential of primary tumor-draining lymph nodes. Cancer Immunol. Immunother. 2019, 68, 1681–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamoto, K.; Chowdhury, P.S.; Kumar, A.; Sonomura, K.; Matsuda, F.; Fagarasan, S.; Honjo, T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E761–E770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fransen, M.F.; Schoonderwoerd, M.; Knopf, P.; Camps, M.G.; Hawinkels, L.J.; Kneilling, M.; van Hall, T.; Ossendorp, F. Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI Insight 2018, 3, e124507. [Google Scholar] [CrossRef] [Green Version]
- Francis, D.M.; Manspeaker, M.P.; Schudel, A.; Sestito, L.F.; O’Melia, M.J.; Kissick, H.T.; Pollack, B.P.; Waller, E.K.; Thomas, S.N. Blockade of immune checkpoints in lymph nodes through locoregional delivery augments cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaay3575. [Google Scholar] [CrossRef]
- Fransen, M.F.; van der Sluis, T.C.; Ossendorp, F.; Arens, R.; Melief, C.J. Controlled local delivery of CTLA-4 blocking antibody induces CD8+ T-cell-dependent tumor eradication and decreases risk of toxic side effects. Clin. Cancer Res. 2013, 19, 5381–5389. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanda, Y.; Okazaki, T.; Katakai, T. Motility Dynamics of T Cells in Tumor-Draining Lymph Nodes: A Rational Indicator of Antitumor Response and Immune Checkpoint Blockade. Cancers 2021, 13, 4616. https://doi.org/10.3390/cancers13184616
Kanda Y, Okazaki T, Katakai T. Motility Dynamics of T Cells in Tumor-Draining Lymph Nodes: A Rational Indicator of Antitumor Response and Immune Checkpoint Blockade. Cancers. 2021; 13(18):4616. https://doi.org/10.3390/cancers13184616
Chicago/Turabian StyleKanda, Yasuhiro, Taku Okazaki, and Tomoya Katakai. 2021. "Motility Dynamics of T Cells in Tumor-Draining Lymph Nodes: A Rational Indicator of Antitumor Response and Immune Checkpoint Blockade" Cancers 13, no. 18: 4616. https://doi.org/10.3390/cancers13184616
APA StyleKanda, Y., Okazaki, T., & Katakai, T. (2021). Motility Dynamics of T Cells in Tumor-Draining Lymph Nodes: A Rational Indicator of Antitumor Response and Immune Checkpoint Blockade. Cancers, 13(18), 4616. https://doi.org/10.3390/cancers13184616