
PD-L1 Expression Is Significantly Associated with Tumor Mutation Burden and Microsatellite Instability Score

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Tumor Samples
2.2. DNA Extraction
2.3. Library Preparation, Sequencing, and Data Analysis
2.4. Immunohistochemistry
2.5. MSI Analysis
2.6. Statistical Analysis
3. Results
3.1. Overall Sequencing Quality
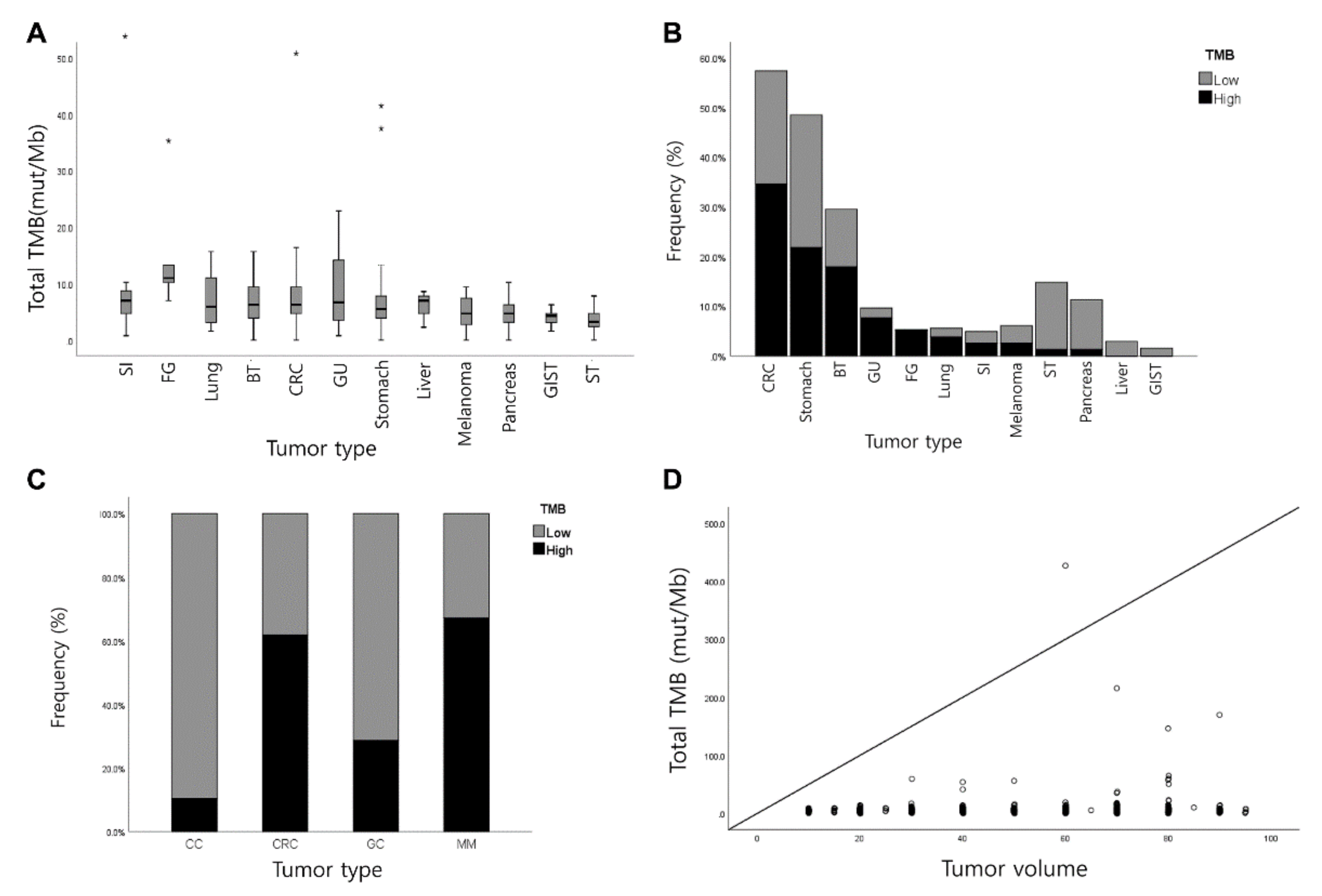
3.2. Measurement of TMB
3.3. Validation and Correlation of TMB between TSO 500 and WES
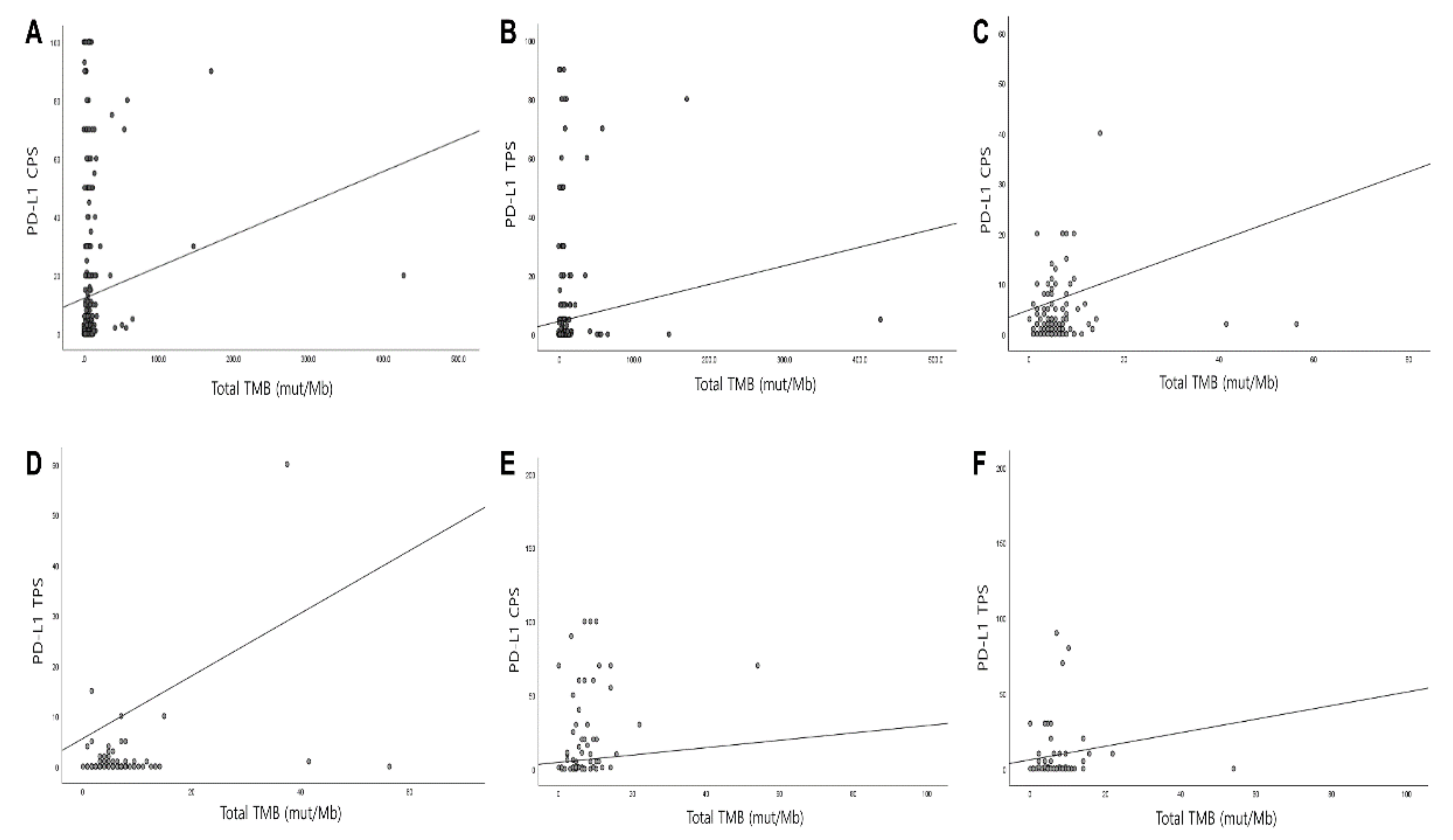
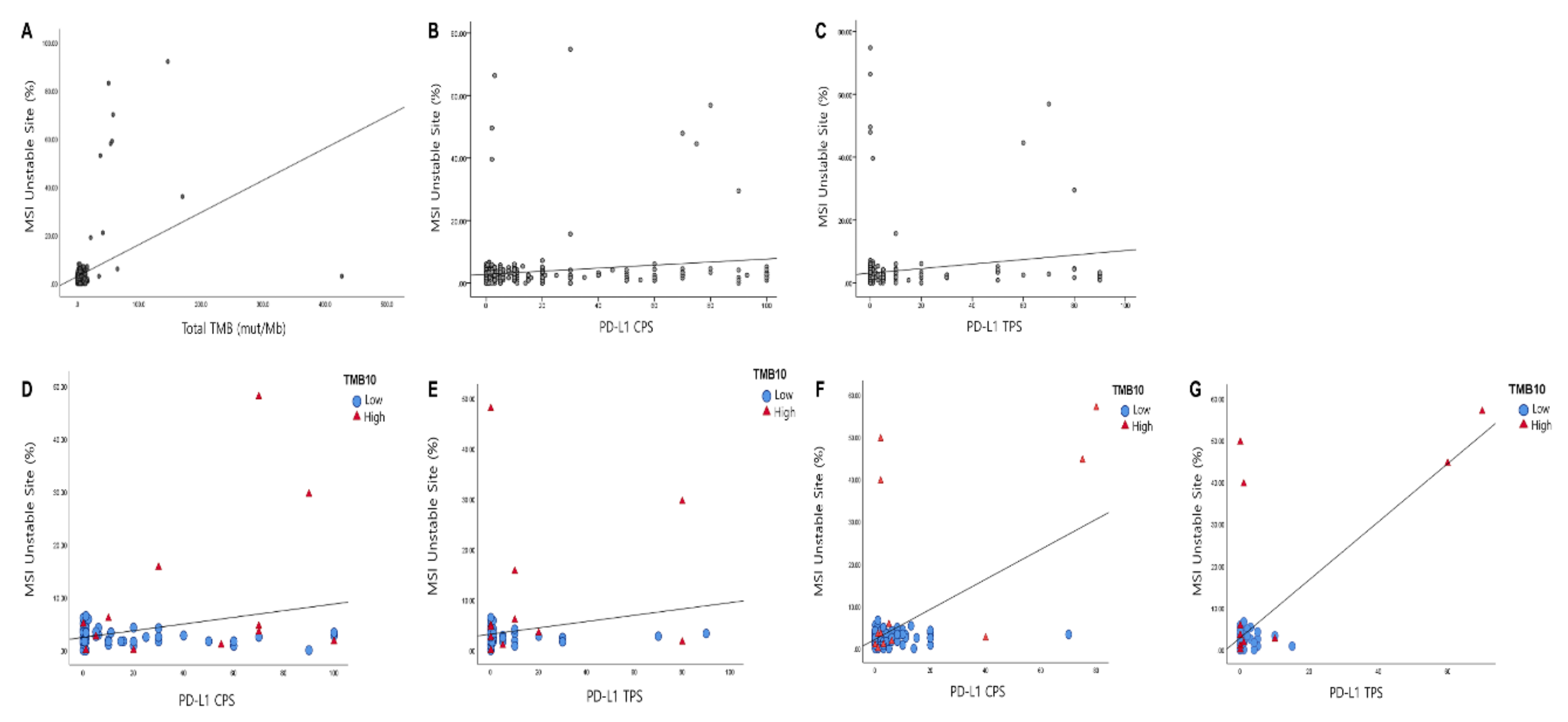
3.4. Correlation of PD-L1 with TMB and MSI
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kroeze, L.I.; de Voer, R.M.; Kamping, E.J.; von Rhein, D.; Jansen, E.A.M.; Hermsen, M.J.W.; Barberis, M.C.P.; Botling, J.; Garrido-Martin, E.M.; Haller, F.; et al. Evaluation of a Hybrid Capture-Based Pan-Cancer Panel for Analysis of Treatment Stratifying Oncogenic Aberrations and Processes. J. Mol. Diagn. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Vokes, E.E.; Ready, N.; Felip, E.; Horn, L.; Burgio, M.A.; Antonia, S.J.; Aren Frontera, O.; Gettinger, S.; Holgado, E.; Spigel, D.; et al. Nivolumab versus docetaxel in previously treated advanced non-small-cell lung cancer (CheckMate 017 and CheckMate 057): 3-year update and outcomes in patients with liver metastases. Ann. Oncol. 2018, 29, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers (Basel) 2020, 12, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, D.E.; Banerji, S. Biomarkers of immune checkpoint inhibitor efficacy in cancer. Curr. Oncol. 2020, 27, S106–S114. [Google Scholar] [CrossRef]
- Hsu, J.C.; Lin, J.-Y.; Hsu, M.-Y.; Lin, P.-C. Effectiveness and safety of immune checkpoint inhibitors: A retrospective study in Taiwan. PLoS ONE 2018, 13, e0202725. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer 2015, 14, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenbacher, L.; Otto, G.; Malboeuf, C.; et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24, 1441–1448. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Callahan, M.K.; Awad, M.M.; Calvo, E.; Ascierto, P.A.; Atmaca, A.; Rizvi, N.A.; Hirsch, F.R.; Selvaggi, G.; Szustakowski, J.D.; et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell 2018, 33, 853–861.e854. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, p.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Hegde, P.S.; Clynes, R.; Foukas, P.G.; Harari, A.; Kleen, T.O.; Kvistborg, P.; Maccalli, C.; Maecker, H.T.; Page, D.B.; et al. Novel technologies and emerging biomarkers for personalized cancer immunotherapy. J. Immunother. Cancer 2016, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fancello, L.; Gandini, S.; Pelicci, P.G.; Mazzarella, L. Tumor mutational burden quantification from targeted gene panels: Major advancements and challenges. J. Immunother. Cancer 2019, 7, 183. [Google Scholar] [CrossRef] [Green Version]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Karasaki, T.; Nakajima, J.; Kakimi, K. Neoantigens and Whole-Exome Sequencing. Gan Kagaku Ryoho. Cancer Chemother. 2016, 43, 791–797. [Google Scholar]
- Allgauer, M.; Budczies, J.; Christopoulos, P.; Endris, V.; Lier, A.; Rempel, E.; Volckmar, A.L.; Kirchner, M.; von Winterfeld, M.; Leichsenring, J.; et al. Implementing tumor mutational burden (TMB) analysis in routine diagnostics-a primer for molecular pathologists and clinicians. Transl. Lung Cancer Res. 2018, 7, 703–715. [Google Scholar] [CrossRef]
- Chilamakuri, C.S.; Lorenz, S.; Madoui, M.A.; Vodak, D.; Sun, J.; Hovig, E.; Myklebost, O.; Meza-Zepeda, L.A. Performance comparison of four exome capture systems for deep sequencing. BMC Genom. 2014, 15, 449. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Boussios, S.; Ozturk, M.A.; Moschetta, M.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Katsanos, K.H.; Christodoulou, D.K.; Pavlidis, N. The Developing Story of Predictive Biomarkers in Colorectal Cancer. J. Pers. Med. 2019, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, R.; Meagher, A.; Tomlinson, I.; O’Connor, T.; Norrie, M.; Wu, R.; Hawkins, N. Microsatellite instability and the clinicopathological features of sporadic colorectal cancer. Gut 2001, 48, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.M.; Zhang, S.; Geiger, T.; Hafez, M.J.; Bacher, J.; Berg, K.D.; Eshleman, J.R. Comparison of the microsatellite instability analysis system and the Bethesda panel for the determination of microsatellite instability in colorectal cancers. J. Mol. Diagn. 2006, 8, 305–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beamer, L.C.; Grant, M.L.; Espenschied, C.R.; Blazer, K.R.; Hampel, H.L.; Weitzel, J.N.; MacDonald, D.J. Reflex immunohistochemistry and microsatellite instability testing of colorectal tumors for Lynch syndrome among US cancer programs and follow-up of abnormal results. J. Clin. Oncol. 2012, 30, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Ribic, C.M.; Sargent, D.J.; Moore, M.J.; Thibodeau, S.N.; French, A.J.; Goldberg, R.M.; Hamilton, S.R.; Laurent-Puig, P.; Gryfe, R.; Shepherd, L.E.; et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N. Engl. J. Med. 2003, 349, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Sankila, R.; Aaltonen, L.A.; Järvinen, H.J.; Mecklin, J.P. Better survival rates in patients with MLH1-associated hereditary colorectal cancer. Gastroenterology 1996, 110, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar]
- Laghi, L.; Bianchi, P.; Malesci, A. Differences and evolution of the methods for the assessment of microsatellite instability. Oncogene 2008, 27, 6313–6321. [Google Scholar] [CrossRef] [Green Version]
- Baudrin, L.G.; Deleuze, J.F.; How-Kit, A. Molecular and Computational Methods for the Detection of Microsatellite Instability in Cancer. Front. Oncol. 2018, 8, 621. [Google Scholar] [CrossRef] [PubMed]
- Waalkes, A.; Smith, N.; Penewit, K.; Hempelmann, J.; Konnick, E.Q.; Hause, R.J.; Pritchard, C.C.; Salipante, S.J. Accurate Pan-Cancer Molecular Diagnosis of Microsatellite Instability by Single-Molecule Molecular Inversion Probe Capture and High-Throughput Sequencing. Clin. Chem. 2018, 64, 950–958. [Google Scholar] [CrossRef]
- Salipante, S.J.; Scroggins, S.M.; Hampel, H.L.; Turner, E.H.; Pritchard, C.C. Microsatellite instability detection by next generation sequencing. Clin. Chem. 2014, 60, 1192–1199. [Google Scholar] [CrossRef]
- Pestinger, V.; Smith, M.; Sillo, T.; Findlay, J.M.; Laes, J.F.; Martin, G.; Middleton, G.; Taniere, P.; Beggs, A.D. Use of an Integrated Pan-Cancer Oncology Enrichment Next-Generation Sequencing Assay to Measure Tumour Mutational Burden and Detect Clinically Actionable Variants. Mol. Diagn. 2020. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, F.R.; McElhinny, A.; Stanforth, D.; Ranger-Moore, J.; Jansson, M.; Kulangara, K.; Richardson, W.; Towne, P.; Hanks, D.; Vennapusa, B.; et al. PD-L1 Immunohistochemistry Assays for Lung Cancer: Results from Phase 1 of the Blueprint PD-L1 IHC Assay Comparison Project. J. Thorac. Oncol. 2017, 12, 208–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarchoan, M.; Albacker, L.A.; Hopkins, A.C.; Montesion, M.; Murugesan, K.; Vithayathil, T.T.; Zaidi, N.; Azad, N.S.; Laheru, D.A.; Frampton, G.M.; et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.S.P.; Haberberger, J.; Severson, E.; Duncan, D.L.; Hemmerich, A.; Edgerly, C.; Ferguson, N.L.; Williams, E.; Elvin, J.; Vergilio, J.-A.; et al. A pan-cancer analysis of PD-L1 immunohistochemistry and gene amplification, tumor mutation burden and microsatellite instability in 48,782 cases. Mod. Pathol. 2021, 34, 252–263. [Google Scholar] [CrossRef]
- Min, B.H.; Tae, C.H.; Ahn, S.M.; Kang, S.Y.; Woo, S.Y.; Kim, S.; Kim, K.M. Epstein-Barr virus infection serves as an independent predictor of survival in patients with lymphoepithelioma-like gastric carcinoma. Gastric Cancer 2016, 19, 852–859. [Google Scholar] [CrossRef] [Green Version]
- Skidmore, Z.L.; Wagner, A.H.; Lesurf, R.; Campbell, K.M.; Kunisaki, J.; Griffith, O.L.; Griffith, M. GenVisR: Genomic Visualizations in R. Bioinformatics 2016, 32, 3012–3014. [Google Scholar] [CrossRef] [Green Version]
- Lowery, M.A.; Ptashkin, R.; Jordan, E.; Berger, M.F.; Zehir, A.; Capanu, M.; Kemeny, N.E.; O’Reilly, E.M.; El-Dika, I.; Jarnagin, W.R.; et al. Comprehensive Molecular Profiling of Intrahepatic and Extrahepatic Cholangiocarcinomas: Potential Targets for Intervention. Clin. Cancer Res. 2018, 24, 4154–4161. [Google Scholar] [CrossRef] [Green Version]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2019, 29, 3766. [Google Scholar] [CrossRef]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; Yusko, E.; Xu, Y.; Guo, X.; Ennis, R.C.; Fabrizio, D.; Chalmers, Z.R.; Greenbowe, J.; et al. Targeted Next Generation Sequencing Identifies Markers of Response to PD-1 Blockade. Cancer Immunol. Res. 2016, 4, 959–967. [Google Scholar] [CrossRef] [Green Version]
- Peggs, K.S.; Segal, N.H.; Allison, J.P. Targeting immunosupportive cancer therapies: Accentuate the positive, eliminate the negative. Cancer Cell 2007, 12, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Segal, N.H.; Parsons, D.W.; Peggs, K.S.; Velculescu, V.; Kinzler, K.W.; Vogelstein, B.; Allison, J.P. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008, 68, 889–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchhalter, I.; Rempel, E.; Endris, V.; Allgauer, M.; Neumann, O.; Volckmar, A.L.; Kirchner, M.; Leichsenring, J.; Lier, A.; von Winterfeld, M.; et al. Size matters: Dissecting key parameters for panel-based tumor mutational burden analysis. Int. J. Cancer 2019, 144, 848–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heydt, C.; Rehker, J.; Pappesch, R.; Buhl, T.; Ball, M.; Siebolts, U.; Haak, A.; Lohneis, P.; Buttner, R.; Hillmer, A.M.; et al. Analysis of tumor mutational burden: Correlation of five large gene panels with whole exome sequencing. Sci. Rep. 2020, 10, 11387. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Carlson, J.A.; Elvin, J.A.; Vergilio, J.A.; Suh, J.; Ramkissoon, S.; Daniel, S.; Fabrizio, D.; Frampton, G.M.; Ali, S.M.; et al. Landscape of genomic alterations (GA) and tumor mutational burden (TMB) in different metastatic melanoma (MM) subtypes. J. Clin. Oncol. 2017, 35, 9536. [Google Scholar] [CrossRef]
- Forschner, A.; Battke, F.; Hadaschik, D.; Schulze, M.; Weissgraeber, S.; Han, C.T.; Kopp, M.; Frick, M.; Klumpp, B.; Tietze, N.; et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma—Results of a prospective biomarker study. J. Immunother. Cancer 2019, 7, 180. [Google Scholar] [CrossRef]
- Chun, Y.S.; Javle, M. Systemic and Adjuvant Therapies for Intrahepatic Cholangiocarcinoma. Cancer Control. 2017, 24, 1073274817729241. [Google Scholar] [CrossRef]
- Jain, A.; Kwong, L.N.; Javle, M. Genomic Profiling of Biliary Tract Cancers and Implications for Clinical Practice. Curr. Treat. Options Oncol. 2016, 17, 58. [Google Scholar] [CrossRef]
- Cao, J.; Hu, J.; Liu, S.; Meric-Bernstam, F.; Abdel-Wahab, R.; Xu, J.; Li, Q.; Yan, M.; Feng, Y.; Lin, J.; et al. Intrahepatic Cholangiocarcinoma: Genomic Heterogeneity Between Eastern and Western Patients. JCO Precis. Oncol. 2020, 4. [Google Scholar] [CrossRef]
- Labriola, M.K.; Zhu, J.; Gupta, R.; McCall, S.; Jackson, J.; Kong, E.F.; White, J.R.; Cerqueira, G.; Gerding, K.; Simmons, J.K.; et al. Characterization of tumor mutation burden, PD-L1 and DNA repair genes to assess relationship to immune checkpoint inhibitors response in metastatic renal cell carcinoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Liu, Q.; Chen, Z.; Wang, Y.; Yang, W.; Hu, Y.; Han, W.; Zeng, H.; Ma, H.; Dai, J.; et al. PD-L1 expression and tumor mutational burden status for prediction of response to chemotherapy and targeted therapy in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2019, 38, 193. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhang, T.; Li, J.; Lin, J.; Liang, W.; Huang, W.; Wan, N.; Jiang, J. Association Between Tumor Mutation Burden (TMB) and Outcomes of Cancer Patients Treated With PD-1/PD-L1 Inhibitions: A Meta-Analysis. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Lin, G.; Huang, C.; Zhu, W.; Miao, Q.; Fan, X.; Wu, B.; Zheng, X.; Lin, X.; Jiang, K.; et al. Assessment of Concordance between 22C3 and SP142 Immunohistochemistry Assays regarding PD-L1 Expression in Non-Small Cell Lung Cancer. Sci. Rep. 2017, 7, 16956. [Google Scholar] [CrossRef] [Green Version]
- Rimm, D.L.; Han, G.; Taube, J.M.; Yi, E.S.; Bridge, J.A.; Flieder, D.B.; Homer, R.; West, W.W.; Wu, H.; Roden, A.C.; et al. A Prospective, Multi-institutional, Pathologist-Based Assessment of 4 Immunohistochemistry Assays for PD-L1 Expression in Non-Small Cell Lung Cancer. JAMA Oncol. 2017, 3, 1051–1058. [Google Scholar] [CrossRef]
- Salem, M.E.; Puccini, A.; Grothey, A.; Raghavan, D.; Goldberg, R.M.; Xiu, J.; Korn, W.M.; Weinberg, B.A.; Hwang, J.J.; Shields, A.F.; et al. Landscape of Tumor Mutation Load, Mismatch Repair Deficiency, and PD-L1 Expression in a Large Patient Cohort of Gastrointestinal Cancers. Mol. Cancer Res. 2018, 16, 805–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Category | Variables | Number of Cases (n = 588, %) |
|---|---|---|
| Age (years) | Mean ± SD (Range, median) | 58.26 ± 11.84 (18–87, 59) |
| Sex | Female | 230 (39.1) |
| Male | 358 (60.9) | |
| Source of tissue | Our institute | 534 (90.8) |
| Other institutes | 54 (9.2) | |
| Paraffin block age (days) | Mean ± SD (Range, median) | 250.72 ± 439.11 (2–3536, 53) |
| Pathologic diagnosis | Gastric cancer | 153 (26.0) |
| Tubular adenocarcinoma | 48 | |
| Poorly cohesive carcinoma | 97 | |
| Papillary adenocarcinoma | 2 | |
| Mucinous adenocarcinoma | 2 | |
| Medullary carcinoma with lymphoid stroma | 1 | |
| Squamous cell carcinoma | 1 | |
| Neuroendocrine carcinoma | 1 | |
| Mixed neuroendocrine-nonneuroendocrine neoplasm | 1 | |
| Colorectal cancer | 143 (24.3) | |
| Adenocarcinoma | 140 | |
| Mucinous adenocarcinoma | 1 | |
| Undifferentiated carcinoma | 1 | |
| Neuroendocrine carcinoma | 1 | |
| Malignant melanoma | 20 (3.4) | |
| Cutaneous | 3 | |
| Acral and subungal | 8 | |
| Mucosal | 8 | |
| Unknown | 1 | |
| GIST * | 8 (1.4) | |
| Small intestine | 5 | |
| Stomach | 1 | |
| Extragastrointestinal | 2 | |
| Hepatobiliary carcinoma | 88 (15.0) | |
| Cholangiocarcinoma | 47 | |
| Hepatocellular carcinoma | 11 | |
| Combined hepatocellular carcinoma and cholangiocarcinoma | 2 | |
| Adenocarcinoma, NOS | 19 | |
| Adenosquamous carcinoma | 2 | |
| Intracystic papillary neoplasm with associated invasive carcinoma | 1 | |
| Carcinoma, undifferentiated, NOS | 4 | |
| Neuroendocrine carcinoma, NOS | 1 | |
| Epithelioid hemangioendothelioma | 1 | |
| Pancreas carcinoma | 52 (8.8) | |
| Ductal adenocarcinoma | 47 | |
| Undifferentiated carcinoma with osteoclast-like giant cells | 2 | |
| Acinar cell carcinoma | 1 | |
| Neuroendocrine tumor, NOS | 2 | |
| Small intestine carcinoma | 14 (2.4) | |
| Adenocarcinoma, NOS | 14 | |
| Kidney and Genitourinary tract cancer | 16 (2.7) | |
| Infiltrating urothelial cell carcinoma | 15 | |
| Renal cell carcinoma | 1 | |
| Mesenchymal tumor | 69 (11.7) | |
| Alveolar soft part sarcoma | 2 | |
| Angiosarcoma | 5 | |
| Chondrosarcoma | 1 | |
| Clear cell sarcoma | 1 | |
| Dedifferentiated liposarcoma | 14 | |
| Epithelioid sarcoma | 2 | |
| Ewing sarcoma | 5 | |
| Extraskeletal myxoid chondrosarcoma | 2 | |
| Intimal sarcoma | 1 | |
| Leiomyosarcoma | 12 | |
| Malignant peripheral nerve sheath tumor | 1 | |
| Malignant perivascular epithelioid cell neoplasm | 1 | |
| Mesenchymal chondrosarcoma | 1 | |
| Myxofibrosarcoma | 1 | |
| Myxoid liposarcoma | 3 | |
| Osteosarcoma | 1 | |
| Primary intimal sarcoma | 1 | |
| Rhabdomyosarcoma | 1 | |
| Solitary fibrous tumor | 4 | |
| Synovial sarcoma | 1 | |
| Undifferentiated pleomoprhic sarcoma | 2 | |
| Undifferentiated pleomorphic sarcoma | 4 | |
| Undifferentiated spindle cell sarcoma | 2 | |
| Well differentiated liposarcoma | 1 | |
| Female genital tract cancer | 6 (1.0) | |
| Ovarian serous carcinoma | 2 | |
| Squamous cell carcinoma, uterine cervix | 1 | |
| Endometrioid adenocarcinoma, uterine corpus | 1 | |
| Endometrial stromal sarcoma | 1 | |
| Adenosarcoma, uterine corpus | 1 | |
| Lung cancer | 12 (2.0) | |
| Adenocarcinoma | 6 | |
| Mucinous adenocarcinoma | 2 | |
| Squamous cell carcinoma | 2 | |
| Combined large cell neuroendocrine carcinoma | 1 | |
| Small cell neuroendocrine carcinoma | 1 | |
| Other carcinoma | 7 (1.2) | |
| Adrenocortical carcinoma | 2 | |
| Extramammary Paget disease | 1 | |
| Appendiceal goblet cell adenocarcinoma | 1 | |
| Appendiceal signet ring cell adenocarcinoma | 1 | |
| Mucinous adenocarcinoma of retroperitoneum | 1 | |
| Unknown primary | 1 | |
| Primary vs. metastasis | Primary | 478 (81.3) |
| Metastasis | 110 (18.7) | |
| Specimen type | Biopsy | 289 (49.1) |
| Resection | 299 (50.9) |
| PD-L1 CPS * | Total (n = 413) | Gastric Cancer (n = 126) | Biliary Tract Cancer (n = 73) | |||||||||
| Negative (n = 105) | Low (n = 265) | High (n = 43) | p-value | Negative (n = 41) | Low (n = 82) | High (n=2) | p-value | Negative (n = 15) | Low (n = 44) | High (n = 14) | p-value | |
| TMB < 10 | 100 (95.2%) | 230 (86.8%) | 33 (76.6%) | 0.005 | 40 (97.6%) | 73 (89.0%) | 1 (33.3%) | 0.001 | 14 (93.3%) | 37 (84.1%) | 8 (57.1%) | 0.032 |
| TMB ≥ 10 | 5 (4.8%) | 35 (13.2%) | 10 (23.3%) | 1 (2.4%) | 9 (11.0%) | 2 (66.7%) | 1 (6.7%) | 7 (15.9%) | 6 (42.9%) | |||
| PD-L1 CPS ** | Total (n = 413) | Gastric Cancer (n = 126) | Biliary Tract Cancer (n = 73) | |||||||||
| Negative (n = 107) | Positive (n = 306) | p-value | Negative (n = 41) | Positive (n = 85) | p-value | Negative (n = 16) | Positive (n = 57) | p-value | ||||
| TMB < 10 | 101 (94.4%) | 262 (85.6%) | 0.017 | 40 (97.6%) | 74 (87.1%) | 0.060 | 15 (93.8%) | 44 (77.2%) | 0.137 | |||
| TMB ≥ 10 | 6 (5.6%) | 44 (14.4%) | 1 (2.4%) | 11 (12.9%) | 1 (6.2%) | 13 (22.8%) | ||||||
| PD-L1 TPS * | Total (n = 413) | Gastric Cancer (n = 126) | Biliary Tract Cancer (n = 73) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Negative (n = 289) | Low (n = 106) | High (n = 18) | p-value | Negative (n = 98) | Low (n = 26) | High (n = 2) | p-value | Negative (n = 46) | Low (n = 23) | High (n = 4) | p-value | |
| TMB < 10 | 257 (88.9%) | 92 (86.8%) | 14 (77.8%) | 0.343 | 91 (92.9%) | 23 (88.5%) | 0 (0.0%) | <0.001 | 38 (82.6%) | 19 (82.6%) | 2 (50.0%) | 0.273 |
| TMB ≥ 10 | 32 (11.1%) | 14 (13.2%) | 4 (22.2%) | 7 (7.1%) | 3 (11.5%) | 2 (100.0%) | 8 (17.4%) | 4 (17.4%) | 2 (50.0%) | |||
| CPS * | TPS * | TMB | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Negative (<1) (n = 107) | Positive (≥1) (n = 306) | p-value | Negative (<1) (n = 290) | Low (1–49) (n = 105) | High (≥50) (n = 18) | p-value | TMB-L (<10) (n = 363) | TMB-H (≥10) (n = 51) | p-value | |
| MSI-L | 107 (100%) | 298 (97.4%) | 0.091 | 286 (98.6%) | 104 (99.0%) | 15 (83.3%) | <0.001 | 363 (100.0%) | 43 (84.3%) | <0.001 |
| MSI-H | 0 (0.0%) | 8 (2.6%) | 4 (1.4%) | 1 (1.0%) | 3 (16.7%) | 0 (0.0%) | 8 (15.7%) | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, Y.A.; Lee, H.; Kim, D.G.; Kim, H.; Ha, S.Y.; Choi, Y.-L.; Jang, K.-T.; Kim, K.-M. PD-L1 Expression Is Significantly Associated with Tumor Mutation Burden and Microsatellite Instability Score. Cancers 2021, 13, 4659. https://doi.org/10.3390/cancers13184659
Cho YA, Lee H, Kim DG, Kim H, Ha SY, Choi Y-L, Jang K-T, Kim K-M. PD-L1 Expression Is Significantly Associated with Tumor Mutation Burden and Microsatellite Instability Score. Cancers. 2021; 13(18):4659. https://doi.org/10.3390/cancers13184659
Chicago/Turabian StyleCho, Yoon Ah, Hyunwoo Lee, Deok Geun Kim, Hyunjin Kim, Sang Yun Ha, Yoon-La Choi, Kee-Taek Jang, and Kyoung-Mee Kim. 2021. "PD-L1 Expression Is Significantly Associated with Tumor Mutation Burden and Microsatellite Instability Score" Cancers 13, no. 18: 4659. https://doi.org/10.3390/cancers13184659
APA StyleCho, Y. A., Lee, H., Kim, D. G., Kim, H., Ha, S. Y., Choi, Y. -L., Jang, K. -T., & Kim, K. -M. (2021). PD-L1 Expression Is Significantly Associated with Tumor Mutation Burden and Microsatellite Instability Score. Cancers, 13(18), 4659. https://doi.org/10.3390/cancers13184659