
Chimeric Antigen Receptor T cell Therapy and the Immunosuppressive Tumor Microenvironment in Pediatric Sarcoma

 ,
,  , , , ,
, , , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. CAR T Cells in Clinical Trials for Pediatric Sarcomas
3. CAR T Cells in Pre-Clinical Development for Pediatric Sarcoma
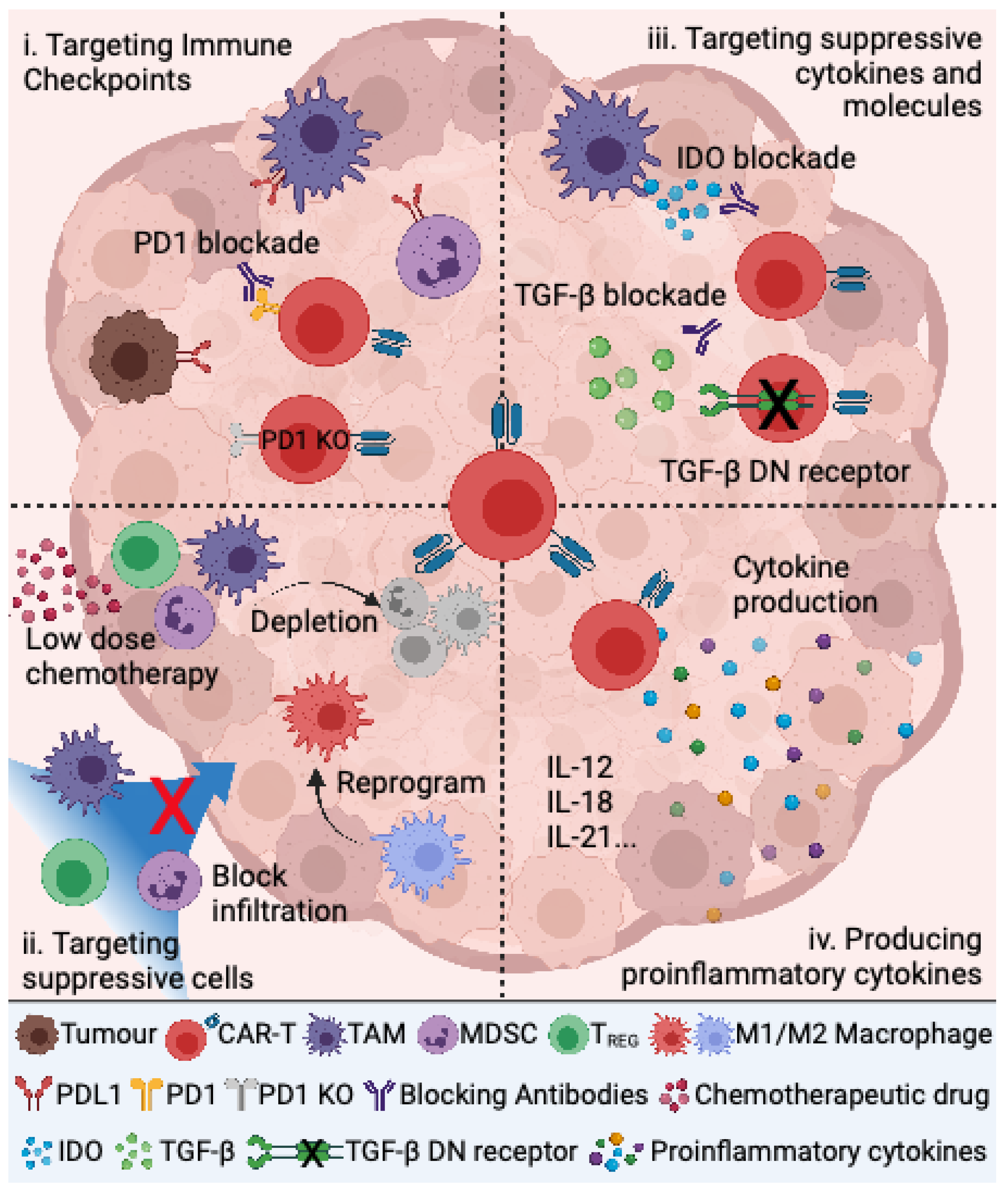
4. Targeting the Immunosuppressive TME to Improve CAR T Cell Therapy for Pediatric Sarcomas
4.1. Targeting Immune Checkpoints
4.2. Targeting Suppressive Immune Cells
4.3. Targeting Immunosuppressive Cytokines and Other Molecules
4.4. Production of Proinflammatory Cytokines
5. Understanding the Immunosuppressive TME Is Key to Improving CAR T Cell Therapy
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Steliarova-Foucher, E.; Colombet, M.; Ries, L.A.G.; Moreno, F.; Dolya, A.; Bray, F.; Hesseling, P.; Shin, H.Y.; Stiller, C.A.; Bouzbid, S.; et al. International incidence of childhood cancer, 2001–2010: A population-based registry study. Lancet Oncol. 2017, 18, 719–731. [Google Scholar] [CrossRef]
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2017; National Cancer Institute: Bethesda, MD, USA, 2020; Based on November 2019 SEER data submission, posted to the SEER web site, April 2020. [Google Scholar]
- Lewis, D.R.; Siembida, E.J.; Seibel, N.L.; Smith, A.W.; Mariotto, A.B. Survival outcomes for cancer types with the highest death rates for adolescents and young adults, 1975–2016. Cancer 2021. [Google Scholar] [CrossRef]
- Bhakta, N.; Liu, Q.; Ness, K.K.; Baassiri, M.; Eissa, H.; Yeo, F.; Chemaitilly, W.; Ehrhardt, M.J.; Bass, J.; Bishop, M.W.; et al. The cumulative burden of surviving childhood cancer: An initial report from the St Jude Lifetime Cohort Study (SJLIFE). Lancet 2017, 390, 2569–2582. [Google Scholar] [CrossRef]
- Keegan, T.H.; Ries, L.A.; Barr, R.D.; Geiger, A.M.; Dahlke, D.V.; Pollock, B.H.; Bleyer, W.A. National Cancer Institute Next Steps for Adolescent and Young Adult Oncology Epidemiology Working Group Comparison of cancer survival trends in the United States of adolescents and young adults with those in children and older adults. Cancer 2016, 122, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Dyson, K.A.; Stover, B.D.; Grippin, A.; Mendez-Gomez, H.R.; Lagmay, J.; Mitchell, D.A.; Sayour, E.J. Emerging trends in immunotherapy for pediatric sarcomas. J. Hematol. Oncol. 2019, 12, 78. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeller, M.; Haynes, N.M.; Trapani, J.A.; Teng, M.W.; Jackson, J.T.; Tanner, J.E.; Cerutti, L.; Jane, S.M.; Kershaw, M.H.; Smyth, M.J.; et al. A functional role for CD28 costimulation in tumor recognition by single-chain receptor-modified T cells. Cancer Gene Ther. 2004, 11, 371–379. [Google Scholar] [CrossRef]
- Petersen, C.T.; Krenciute, G. Next Generation CAR T Cells for the Immunotherapy of High-Grade Glioma. Front. Oncol. 2019, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Wang, G.; Cheng, H.; Wei, C.; Qi, K.; Sang, W.; Zhenyu, L.; Shi, M.; Li, H.; Qiao, J.; et al. Potent anti-leukemia activities of humanized CD19-targeted Chimeric antigen receptor T (CAR-T) cells in patients with relapsed/refractory acute lymphoblastic leukemia. Am. J. Hematol. 2018, 93, 851–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochenderfer, J.N.; Somerville, R.P.T.; Lu, T.; Yang, J.C.; Sherry, R.M.; Feldman, S.A.; McIntyre, L.; Bot, A.; Rossi, J.; Lam, N.; et al. Long-Duration Complete Remissions of Diffuse Large B Cell Lymphoma after Anti-CD19 Chimeric Antigen Receptor T Cell Therapy. Mol. Ther. 2017, 25, 2245–2253. [Google Scholar] [CrossRef] [Green Version]
- Chavez, J.C.; Bachmeier, C.; Kharfan-Dabaja, M.A. CAR T-cell therapy for B-cell lymphomas: Clinical trial results of available products. Ther. Adv. Hematol. 2019, 10, 2040620719841581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesfaye, M.; Savoldo, B. Adoptive Cell Therapy in Treating Pediatric Solid Tumors. Curr. Oncol. Rep. 2018, 20, 73. [Google Scholar] [CrossRef]
- Fleuren, E.D.; Versleijen-Jonkers, Y.M.; Boerman, O.C.; van der Graaf, W.T. Targeting receptor tyrosine kinases in osteosarcoma and Ewing sarcoma: Current hurdles and future perspectives. Biochim. Biophys. Acta 2014, 1845, 266–276. [Google Scholar] [CrossRef]
- Scotlandi, K.; Manara, M.C.; Hattinger, C.M.; Benini, S.; Perdichizzi, S.; Pasello, M.; Bacci, G.; Zanella, L.; Bertoni, F.; Picci, P.; et al. Prognostic and therapeutic relevance of HER2 expression in osteosarcoma and Ewing’s sarcoma. Eur. J. Cancer 2005, 41, 1349–1361. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef]
- Tabak, S.A.; Khalifa, S.E.; Fathy, Y. HER-2 Immunohistochemical Expression in Bone Sarcomas: A New Hope for Osteosarcoma Patients. Open Access Maced. J. Med. Sci. 2018, 6, 1555–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Navai, S.; Derenzo, C.; Joseph, S.; Sanber, K.; Byrd, T.; Zhang, H.; Mata, M.; Gerken, C.; Shree, A.; Mathew, P.; et al. Abstract LB-147: Administration of HER2-CAR T Cells after Lymphodepletion Safely Improves T Cell Expansion and Induces Clinical Responses in Patients with Advanced Sarcomas; 2019; p. LB-147. [Google Scholar]
- Hegde, M.; Joseph, S.K.; Pashankar, F.; DeRenzo, C.; Sanber, K.; Navai, S.; Byrd, T.T.; Hicks, J.; Xu, M.L.; Gerken, C.; et al. Tumor response and endogenous immune reactivity after administration of HER2 CAR T cells in a child with metastatic rhabdomyosarcoma. Nat. Commun. 2020, 11, 3549. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.H.; Koeppen, H.; Garcia, R.; Chiriboga, L.; Tarlow, B.D.; Peters, B.A.; Eigenbrot, C.; Yee, H.; Steiner, G.; Greco, M.A. Epidermal growth factor receptor in osteosarcoma: Expression and mutational analysis. Hum. Pathol. 2007, 38, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.L.; Hannan, M.T.; Russell, P.J.; Crowe, P.J. Expression of HER1/EGFR protein in human soft tissue sarcomas. Eur. J. Surg. Oncol. 2006, 32, 466–468. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Iwenofu, O.H. NY-ESO-1: A promising cancer testis antigen for sarcoma immunotherapy and diagnosis. Chin. Clin. Oncol. 2018, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Desar, I.M.E.; Fleuren, E.D.G.; van der Graaf, W.T.A. Systemic Treatment for Adults with Synovial Sarcoma. Curr. Treat. Options Oncol. 2018, 19, 13. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 (c259)T Cells in Synovial Sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, I.; Lowther, D.E.; Dryer-Minnerly, R.; Wang, R.; Fayngerts, S.; Nunez, D.; Betts, G.; Bath, N.; Tipping, A.J.; Melchiori, L.; et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J. Immunother. Cancer 2019, 7, 276. [Google Scholar] [CrossRef] [PubMed]
- Keung, E.Z.; Tawbi, H.A. Engineered T Cells in Synovial Sarcoma: Persistence Pays Off! Cancer Discov. 2018, 8, 914–917. [Google Scholar] [CrossRef] [Green Version]
- Van Tine, B.A.; Butler, M.O.; Araujo, D.; Johnson, M.L.; Clarke, J.; Liebner, D.; Odunsi, K.; Olszanski, A.J.; Basu, S.; Brophy, F.; et al. ADP-A2M4 (MAGE-A4) in patients with synovial sarcoma. Ann. Oncol. 2019, 30, v684–v685. [Google Scholar] [CrossRef]
- Dobrenkov, K.; Ostrovnaya, I.; Gu, J.; Cheung, I.Y.; Cheung, N.K. Oncotargets GD2 and GD3 are highly expressed in sarcomas of children, adolescents, and young adults. Pediatr. Blood Cancer 2016, 63, 1780–1785. [Google Scholar] [CrossRef] [Green Version]
- Nazha, B.; Inal, C.; Owonikoko, T.K. Disialoganglioside GD2 Expression in Solid Tumors and Role as a Target for Cancer Therapy. Front. Oncol. 2020, 10, 1000. [Google Scholar] [CrossRef]
- Poon, V.I.; Roth, M.; Piperdi, S.; Geller, D.; Gill, J.; Rudzinski, E.R.; Hawkins, D.S.; Gorlick, R. Ganglioside GD2 expression is maintained upon recurrence in patients with osteosarcoma. Clin. Sarcoma Res. 2015, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Charan, M.; Dravid, P.; Cam, M.; Audino, A.; Gross, A.C.; Arnold, M.A.; Roberts, R.D.; Cripe, T.P.; Pertsemlidis, A.; Houghton, P.J.; et al. GD2-directed CAR-T cells in combination with HGF-targeted neutralizing antibody (AMG102) prevent primary tumor growth and metastasis in Ewing sarcoma. Int. J. Cancer 2020, 146, 3184–3195. [Google Scholar] [CrossRef] [PubMed]
- Chulanetra, M.; Morchang, A.; Sayour, E.; Eldjerou, L.; Milner, R.; Lagmay, J.; Cascio, M.; Stover, B.; Slayton, W.; Chaicumpa, W.; et al. GD2 chimeric antigen receptor modified T cells in synergy with sub-toxic level of doxorubicin targeting osteosarcomas. Am. J. Cancer Res. 2020, 10, 674–687. [Google Scholar] [PubMed]
- Hsu, K.; Middlemiss, S.; Saletta, F.; Gottschalk, S.; McCowage, G.B.; Kramer, B. Chimeric Antigen Receptor-modified T cells targeting EphA2 for the immunotherapy of paediatric bone tumours. Cancer Gene Ther. 2021, 28, 321–334. [Google Scholar] [CrossRef]
- Lin, Q.; Ba, T.; Ho, J.; Chen, D.; Cheng, Y.; Wang, L.; Xu, G.; Xu, L.; Zhou, Y.; Wei, Y.; et al. First-in-Human Trial of EphA2-Redirected CAR T-Cells in Patients With Recurrent Glioblastoma: A Preliminary Report of Three Cases at the Starting Dose. Front. Oncol. 2021, 11, 694941. [Google Scholar] [CrossRef] [PubMed]
- Modak, S.; Kramer, K.; Gultekin, S.H.; Guo, H.F.; Cheung, N.K. Monoclonal antibody 8H9 targets a novel cell surface antigen expressed by a wide spectrum of human solid tumors. Cancer Res. 2001, 61, 4048–4054. [Google Scholar] [PubMed]
- Wang, L.; Zhang, Q.; Chen, W.; Shan, B.; Ding, Y.; Zhang, G.; Cao, N.; Liu, L.; Zhang, Y. B7-H3 is overexpressed in patients suffering osteosarcoma and associated with tumor aggressiveness and metastasis. PLoS ONE 2013, 8, e70689. [Google Scholar] [CrossRef]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Alijaj, N.; Moutel, S.; Gouveia, Z.L.; Gray, M.; Roveri, M.; Dzhumashev, D.; Weber, F.; Meier, G.; Luciani, P.; Rossler, J.K.; et al. Novel FGFR4-Targeting Single-Domain Antibodies for Multiple Targeted Therapies against Rhabdomyosarcoma. Cancers 2020, 12, 3313. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.; Wei, J.S.; Ringner, M.; Saal, L.H.; Ladanyi, M.; Westermann, F.; Berthold, F.; Schwab, M.; Antonescu, C.R.; Peterson, C.; et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat. Med. 2001, 7, 673–679. [Google Scholar] [CrossRef]
- Evans, C.H.; Liu, F.; Porter, R.M.; O’Sullivan, R.P.; Merghoub, T.; Lunsford, E.P.; Robichaud, K.; Van Valen, F.; Lessnick, S.L.; Gebhardt, M.C.; et al. EWS-FLI-1-targeted cytotoxic T-cell killing of multiple tumor types belonging to the Ewing sarcoma family of tumors. Clin. Cancer Res. 2012, 18, 5341–5351. [Google Scholar] [CrossRef] [Green Version]
- Gattenlohner, S.; Marx, A.; Markfort, B.; Pscherer, S.; Landmeier, S.; Juergens, H.; Muller-Hermelink, H.K.; Matthews, I.; Beeson, D.; Vincent, A.; et al. Rhabdomyosarcoma lysis by T cells expressing a human autoantibody-based chimeric receptor targeting the fetal acetylcholine receptor. Cancer Res. 2006, 66, 24–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.; Yu, L.; Cooper, L.J.; Hollomon, M.; Huls, H.; Kleinerman, E.S. Genetically modified T cells targeting interleukin-11 receptor alpha-chain kill human osteosarcoma cells and induce the regression of established osteosarcoma lung metastases. Cancer Res. 2012, 72, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Park, H.; Greene, J.; Pao, J.; Mulvey, E.; Zhou, S.X.; Albert, C.M.; Moy, F.; Sachdev, D.; Yee, D.; et al. IGF1R- and ROR1-Specific CAR T Cells as a Potential Therapy for High Risk Sarcomas. PLoS ONE 2015, 10, e0133152. [Google Scholar] [CrossRef]
- Simon-Keller, K.; Paschen, A.; Hombach, A.A.; Strobel, P.; Coindre, J.M.; Eichmuller, S.B.; Vincent, A.; Gattenlohner, S.; Hoppe, F.; Leuschner, I.; et al. Survivin blockade sensitizes rhabdomyosarcoma cells for lysis by fetal acetylcholine receptor-redirected T cells. Am. J. Pathol. 2013, 182, 2121–2131. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, L.; Metais, J.Y.; Escudero, A.; Vela, M.; Valentin, J.; Vallcorba, I.; Leivas, A.; Torres, J.; Valeri, A.; Patino-Garcia, A.; et al. Memory T Cells Expressing an NKG2D-CAR Efficiently Target Osteosarcoma Cells. Clin. Cancer Res. 2017, 23, 5824–5835. [Google Scholar] [CrossRef]
- Lehner, M.; Gotz, G.; Proff, J.; Schaft, N.; Dorrie, J.; Full, F.; Ensser, A.; Muller, Y.A.; Cerwenka, A.; Abken, H.; et al. Redirecting T cells to Ewing’s sarcoma family of tumors by a chimeric NKG2D receptor expressed by lentiviral transduction or mRNA transfection. PLoS ONE 2012, 7, e31210. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, A.; Thiede, M.; Grunewald, T.G.; Alba Rubio, R.; Richter, G.H.; Kirchner, T.; Busch, D.H.; Burdach, S.; Thiel, U. Pappalysin-1 T cell receptor transgenic allo-restricted T cells kill Ewing sarcoma in vitro and in vivo. Oncoimmunology 2017, 6, e1273301. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Wang, J.; Wen, X.; Xu, B.; Que, Y.; Yu, K.; Xu, L.; Zhao, J.; Pan, Q.; Zhou, P.; et al. Chimeric antigen receptor-modified T-cell therapy for platelet-derived growth factor receptor alpha-positive rhabdomyosarcoma. Cancer 2020, 126 (Suppl. 9), 2093–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Englisch, A.; Altvater, B.; Kailayangiri, S.; Hartmann, W.; Rossig, C. VEGFR2 as a target for CAR T cell therapy of Ewing sarcoma. Pediatr. Blood Cancer 2020, 67, e28313. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Devaud, C.; Duong, C.P.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhang, Q.; Tian, K.; Wang, H.; Yin, H.; Zheng, J. Current status and future prospects of the strategy of combining CART with PD1 blockade for antitumor therapy (Review). Mol. Med. Rep. 2018, 17, 2083–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGowan, E.; Lin, Q.; Ma, G.; Yin, H.; Chen, S.; Lin, Y. PD-1 disrupted CAR-T cells in the treatment of solid tumors: Promises and challenges. Biomed. Pharmacother. 2020, 121, 109625. [Google Scholar] [CrossRef]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Zou, Y.; Zhang, L.; Tang, J.; Niedermann, G.; Firat, E.; Huang, X.; Zhu, X. Nucleofection with Plasmid DNA for CRISPR/Cas9-Mediated Inactivation of Programmed Cell Death Protein 1 in CD133-Specific CAR T Cells. Hum. Gene Ther. 2019, 30, 446–458. [Google Scholar] [CrossRef]
- Zou, F.; Lu, L.; Liu, J.; Xia, B.; Zhang, W.; Hu, Q.; Liu, W.; Zhang, Y.; Lin, Y.; Jing, S.; et al. Engineered triple inhibitory receptor resistance improves anti-tumor CAR-T cell performance via CD56. Nat. Commun. 2019, 10, 4109. [Google Scholar] [CrossRef]
- Majzner, R.G.; Simon, J.S.; Grosso, J.F.; Martinez, D.; Pawel, B.R.; Santi, M.; Merchant, M.S.; Geoerger, B.; Hezam, I.; Marty, V.; et al. Assessment of programmed death-ligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer 2017, 123, 3807–3815. [Google Scholar] [CrossRef] [Green Version]
- Marin-Acevedo, J.A.; Kimbrough, E.O.; Lou, Y. Next generation of immune checkpoint inhibitors and beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef]
- Hingorani, P.; Maas, M.L.; Gustafson, M.P.; Dickman, P.; Adams, R.H.; Watanabe, M.; Eshun, F.; Williams, J.; Seidel, M.J.; Dietz, A.B. Increased CTLA-4(+) T cells and an increased ratio of monocytes with loss of class II (CD14(+) HLA-DR(lo/neg)) found in aggressive pediatric sarcoma patients. J. Immunother. Cancer 2015, 3, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocco, C.; Morandi, F.; Airoldi, I. Immune Checkpoints in Pediatric Solid Tumors: Targetable Pathways for Advanced Therapeutic Purposes. Cells 2021, 10, 927. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yang, D.; Yang, Q.; Lv, X.; Huang, W.; Zhou, Z.; Wang, Y.; Zhang, Z.; Yuan, T.; Ding, X.; et al. Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. Nat. Commun. 2020, 11, 6322. [Google Scholar] [CrossRef] [PubMed]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef] [Green Version]
- Ghiringhelli, F.; Larmonier, N.; Schmitt, E.; Parcellier, A.; Cathelin, D.; Garrido, C.; Chauffert, B.; Solary, E.; Bonnotte, B.; Martin, F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur. J. Immunol. 2004, 34, 336–344. [Google Scholar] [CrossRef]
- Srivastava, S.; Furlan, S.N.; Jaeger-Ruckstuhl, C.A.; Sarvothama, M.; Berger, C.; Smythe, K.S.; Garrison, S.M.; Specht, J.M.; Lee, S.M.; Amezquita, R.A.; et al. Immunogenic Chemotherapy Enhances Recruitment of CAR-T Cells to Lung Tumors and Improves Antitumor Efficacy when Combined with Checkpoint Blockade. Cancer Cell 2021, 39, 193–208.e110. [Google Scholar] [CrossRef]
- Argyle, D.; Kitamura, T. Targeting Macrophage-Recruiting Chemokines as a Novel Therapeutic Strategy to Prevent the Progression of Solid Tumors. Front. Immunol. 2018, 9, 2629. [Google Scholar] [CrossRef]
- Anfray, C.; Ummarino, A.; Andon, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2019, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Clappaert, E.J.; Murgaski, A.; Van Damme, H.; Kiss, M.; Laoui, D. Diamonds in the Rough: Harnessing Tumor-Associated Myeloid Cells for Cancer Therapy. Front. Immunol. 2018, 9, 2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, D.; Nishikawa, H.; Maeda, Y.; Nishioka, M.; Tanemura, A.; Katayama, I.; Ezoe, S.; Kanakura, Y.; Sato, E.; Fukumori, Y.; et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. USA 2013, 110, 17945–17950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guislain, A.; Gadiot, J.; Kaiser, A.; Jordanova, E.S.; Broeks, A.; Sanders, J.; van Boven, H.; de Gruijl, T.D.; Haanen, J.B.; Bex, A.; et al. Sunitinib pretreatment improves tumor-infiltrating lymphocyte expansion by reduction in intratumoral content of myeloid-derived suppressor cells in human renal cell carcinoma. Cancer Immunol. Immunother. 2015, 64, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Stiff, A.; Trikha, P.; Wesolowski, R.; Kendra, K.; Hsu, V.; Uppati, S.; McMichael, E.; Duggan, M.; Campbell, A.; Keller, K.; et al. Myeloid-Derived Suppressor Cells Express Bruton’s Tyrosine Kinase and Can Be Depleted in Tumor-Bearing Hosts by Ibrutinib Treatment. Cancer Res. 2016, 76, 2125–2136. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Liu, C.; Peng, W.; Lizee, G.; Overwijk, W.W.; Liu, Y.; Woodman, S.E.; Hwu, P. Antitumor T-cell responses contribute to the effects of dasatinib on c-KIT mutant murine mastocytoma and are potentiated by anti-OX40. Blood 2012, 120, 4533–4543. [Google Scholar] [CrossRef] [Green Version]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Nadella, V.; Singh, S.; Jain, A.; Jain, M.; Vasquez, K.M.; Sharma, A.; Tanwar, P.; Rath, G.K.; Prakash, H. Low dose radiation primed iNOS + M1macrophages modulate angiogenic programming of tumor derived endothelium. Mol. Carcinog. 2018, 57, 1664–1671. [Google Scholar] [CrossRef]
- Liu, R.; Xiong, S.; Zhang, L.; Chu, Y. Enhancement of antitumor immunity by low-dose total body irradiationis associated with selectively decreasing the proportion and number of T regulatory cells. Cell. Mol. Immunol. 2010, 7, 157–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Yin, Y.; Li, N.; Zhu, D.; Zhang, J.; Zhang, C.Y.; Zen, K. Re-polarization of tumor-associated macrophages to pro-inflammatory M1 macrophages by microRNA-155. J. Mol. Cell Biol. 2012, 4, 341–343. [Google Scholar] [CrossRef]
- Zhang, F.; Parayath, N.N.; Ene, C.I.; Stephan, S.B.; Koehne, A.L.; Coon, M.E.; Holland, E.C.; Stephan, M.T. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat. Commun. 2019, 10, 3974. [Google Scholar] [CrossRef]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truitt, M.; Olson, P.; et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 2016, 6, 270–285. [Google Scholar] [CrossRef] [Green Version]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Locatelli, S.L.; Careddu, G.; Serio, S.; Consonni, F.M.; Maeda, A.; Viswanadha, S.; Vakkalanka, S.; Castagna, L.; Santoro, A.; Allavena, P.; et al. Targeting Cancer Cells and Tumor Microenvironment in Preclinical and Clinical Models of Hodgkin Lymphoma Using the Dual PI3Kdelta/gamma Inhibitor RP6530. Clin. Cancer Res. 2019, 25, 1098–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flinsenberg, T.W.H.; Tromedjo, C.C.; Hu, N.; Liu, Y.; Guo, Y.; Thia, K.Y.T.; Noori, T.; Song, X.; Aw Yeang, H.X.; Tantalo, D.G.; et al. Differential effects of BTK inhibitors ibrutinib and zanubrutinib on NK-cell effector function in patients with mantle cell lymphoma. Haematologica 2020, 105, e76–e79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Wrzesinski, S.H.; Stern, E.; Look, M.; Criscione, J.; Ragheb, R.; Jay, S.M.; Demento, S.L.; Agawu, A.; Licona Limon, P.; et al. Combination delivery of TGF-beta inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat. Mater. 2012, 11, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-beta Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Xia, J.; Wang, L.; Wang, X.; Ma, X.; Deng, Q.; Lu, Y.; Kumar, M.; Zhou, Z.; Li, L.; et al. miR-153 suppresses IDO1 expression and enhances CAR T cell immunotherapy. J. Hematol. Oncol. 2018, 11, 58. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Trinchieri, G. Interleukin-12: A proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu. Rev. Immunol. 1995, 13, 251–276. [Google Scholar] [CrossRef] [PubMed]
- Pegram, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Curran, K.J.; Giralt, S.A.; Barker, J.N.; Brentjens, R.J. IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia 2015, 29, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Kansara, M.; Thomson, K.; Pang, P.; Dutour, A.; Mirabello, L.; Acher, F.; Pin, J.P.; Demicco, E.G.; Yan, J.; Teng, M.W.L.; et al. Infiltrating Myeloid Cells Drive Osteosarcoma Progression via GRM4 Regulation of IL23. Cancer Discov. 2019, 9, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 regulates both Th1 and Th2 responses. Annu. Rev. Immunol. 2001, 19, 423–474. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [Green Version]
- Spolski, R.; Leonard, W.J. Interleukin-21: A double-edged sword with therapeutic potential. Nat. Rev. Drug Discov. 2014, 13, 379–395. [Google Scholar] [CrossRef]
- Markley, J.C.; Sadelain, M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood 2010, 115, 3508–3519. [Google Scholar] [CrossRef] [Green Version]
- Terry, R.L.; Meyran, D.; Ziegler, D.S.; Haber, M.; Ekert, P.G.; Trapani, J.A.; Neeson, P.J. Immune profiling of pediatric solid tumors. J. Clin. Investig. 2020, 130, 3391–3402. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, S.; Barsan, V.; Mackall, C. Prospects and challenges for use of CAR T cell therapies in solid tumors. Expert Opin. Biol. Ther. 2020, 20, 503–516. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Trujillo, J.A.; Sweis, R.F.; Bao, R.; Luke, J.J. T Cell-Inflamed versus Non-T Cell-Inflamed Tumors: A Conceptual Framework for Cancer Immunotherapy Drug Development and Combination Therapy Selection. Cancer Immunol. Res. 2018, 6, 990–1000. [Google Scholar] [CrossRef] [Green Version]
- Bielack, S.S.; Smeland, S.; Whelan, J.S.; Marina, N.; Jovic, G.; Hook, J.M.; Krailo, M.D.; Gebhardt, M.; Papai, Z.; Meyer, J.; et al. Methotrexate, Doxorubicin, and Cisplatin (MAP) Plus Maintenance Pegylated Interferon Alfa-2b Versus MAP Alone in Patients With Resectable High-Grade Osteosarcoma and Good Histologic Response to Preoperative MAP: First Results of the EURAMOS-1 Good Response Randomized Controlled Trial. J. Clin. Oncol. 2015, 33, 2279–2287. [Google Scholar] [CrossRef]
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; Lewis, I.J.; Ferrari, S.; Le Deley, M.C.; Kovar, H.; Grimer, R.; Whelan, J.; Claude, L.; et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J. Clin. Oncol. 2015, 33, 3036–3046. [Google Scholar] [CrossRef] [PubMed]
- Meyran, D.; Terry, R.L.; Zhu, J.J.; Haber, M.; Ziegler, D.S.; Ekert, P.G.; Trapani, J.A.; Darcy, P.K.; Neeson, P.J. Early-phenotype CAR-T cells for the treatment of pediatric cancers. Ann. Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Nakatsugawa, M.; Yamashita, Y.; Ochi, T.; Guo, T.; Anczurowski, M.; Saso, K.; Butler, M.O.; Arrowsmith, C.H.; Hirano, N. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J. Clin. Investig. 2016, 126, 3479–3494. [Google Scholar] [CrossRef] [Green Version]
- Kong, W.; Dimitri, A.; Wang, W.; Jung, I.-Y.; Ott, C.J.; Fasolino, M.; Wang, Y.; Kulikovskaya, I.; Gupta, M.; Yoder, T.; et al. BET bromodomain protein inhibition reverses chimeric antigen receptor extinction and reinvigorates exhausted T cells in chronic lymphocytic leukemia. J. Clin. Investig. 2021, 131, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Nobles, C.L.; Sherrill-Mix, S.; Everett, J.K.; Reddy, S.; Fraietta, J.A.; Porter, D.L.; Frey, N.; Gill, S.I.; Grupp, S.A.; Maude, S.L.; et al. CD19-targeting CAR T cell immunotherapy outcomes correlate with genomic modification by vector integration. J. Clin. Investig. 2020, 130, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Stenger, D.; Stief, T.A.; Kaeuferle, T.; Willier, S.; Rataj, F.; Schober, K.; Vick, B.; Lotfi, R.; Wagner, B.; Grünewald, T.G.P.; et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood 2020, 136, 1407–1418. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Trial Number | Target Antigen | Sarcoma Subtype | Phase | Age (Years) | Dose | Lymphodepletion | Status |
|---|---|---|---|---|---|---|---|
| NCT00902044 | HER2 | All sarcoma | I/II | All | 1 × 104–1 × 108/m2 | Fludarabine, Cyclophosphamide | Completed, with results |
| NCT00889954 | HER2 | All cancers | I | 3+ | 1 × 104–1 × 108/m2 | No | Completed |
| NCT01343043 | NY-ESO-1 | Synovial | I | 4+ | >40 kg: 1 × 109–6 × 109 <40 kg: 0.025 × 109 cells/kg | Fludarabine, Cyclophosphamide | Completed, with results |
| NCT03638206 | NY-ESO-1 | All cancers | I/II | 4–70 | Not specified | Cyclophosphamide or Fludarabine | Recruiting |
| NCT02107963 | GD2 | All | I | <36 | 1 × 105–1 × 107 cells/kg | Cyclophosphamide | Completed |
| NCT1953900 | GD2 | Osteosarcoma | I | 1 × 106–1 × 109 | Fludarabine, Cyclophosphamide | Active | |
| NCT03635632 | GD2 | All | I | 1–74 | 1 × 107–1 × 108 | Fludarabine, Cyclophosphamide | Recruiting |
| NCT04539366 | GD2 | Osteosarcoma | I | <36 | Not specified | Fludarabine, Cyclophosphamide | Not yet recruiting |
| NCT03721068 | GD2 | Osteosarcoma | I | 1.5–18 | 0.5 × 106–1.5 × 106 | Fludarabine, Cyclophosphamide | Recruiting |
| NCT04483778 | B7-H3 | All | I | 0–27 | Not specified | Not specified | Recruiting |
| NCT04897321 | B7-H3 | All | I | 0–21 | Not specified | Fludarabine, Cyclophosphamide | Not yet recruiting |
| NCT04864821 | B7-H3 | All | I | 1–70 | Not specified | Not specified | Not yet recruiting |
| NCT03618381 | EGFR | All | I | 1–26 | Not specified | Not specified | Recruiting |
| NCT04377932 | GPC3 | All | I | 1–21 | 3 × 107–3 × 108/m2 | Fludarabine, Cyclophosphamide | Not yet recruiting |
| NCT04715191 | GPC3 | All | I | 1–21 | 1 × 108–1 × 109/m2 | Fludarabine, Cyclophosphamide | Not yet recruiting |
| Target Antigen | Sarcoma Subtype | In Vitro and In Vivo Responses | Reference |
|---|---|---|---|
| EWS-FLI-1 | Ewing’s sarcoma |
| [44] |
| EPHA2 | Osteosarcoma and Ewing’s sarcoma |
| [37] |
| fAchR | Rhabdomyosarcoma |
| [45] |
| fAchR | Rhabdomyosarcoma |
| [48] |
| FGFR1 |
| [42] | |
| IGF-1R | Rhabdomyosarcoma, Osteosarcoma, Ewing’s sarcoma, Fibrosarcoma |
| [47] |
| IL11Ra | Osteosarcoma |
| [46] |
| NKG2D | Ewing’s sarcoma |
| [50] |
| PAPPA | Ewing’s sarcoma |
| [51] |
| PDGFRA | Rhabdomyosarcoma |
| [52] |
| ROR1 | Osteosarcoma |
| [47] |
| VEGFR2 | Ewing sarcoma |
| [53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terry, R.L.; Meyran, D.; Fleuren, E.D.G.; Mayoh, C.; Zhu, J.; Omer, N.; Ziegler, D.S.; Haber, M.; Darcy, P.K.; Trapani, J.A.; et al. Chimeric Antigen Receptor T cell Therapy and the Immunosuppressive Tumor Microenvironment in Pediatric Sarcoma. Cancers 2021, 13, 4704. https://doi.org/10.3390/cancers13184704
Terry RL, Meyran D, Fleuren EDG, Mayoh C, Zhu J, Omer N, Ziegler DS, Haber M, Darcy PK, Trapani JA, et al. Chimeric Antigen Receptor T cell Therapy and the Immunosuppressive Tumor Microenvironment in Pediatric Sarcoma. Cancers. 2021; 13(18):4704. https://doi.org/10.3390/cancers13184704
Chicago/Turabian StyleTerry, Rachael L., Deborah Meyran, Emmy D. G. Fleuren, Chelsea Mayoh, Joe Zhu, Natacha Omer, David S. Ziegler, Michelle Haber, Phillip K. Darcy, Joseph A. Trapani, and et al. 2021. "Chimeric Antigen Receptor T cell Therapy and the Immunosuppressive Tumor Microenvironment in Pediatric Sarcoma" Cancers 13, no. 18: 4704. https://doi.org/10.3390/cancers13184704
APA StyleTerry, R. L., Meyran, D., Fleuren, E. D. G., Mayoh, C., Zhu, J., Omer, N., Ziegler, D. S., Haber, M., Darcy, P. K., Trapani, J. A., Neeson, P. J., & Ekert, P. G. (2021). Chimeric Antigen Receptor T cell Therapy and the Immunosuppressive Tumor Microenvironment in Pediatric Sarcoma. Cancers, 13(18), 4704. https://doi.org/10.3390/cancers13184704