HSP90α Mediates Sorafenib Resistance in Human Hepatocellular Carcinoma by Necroptosis Inhibition under Hypoxia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
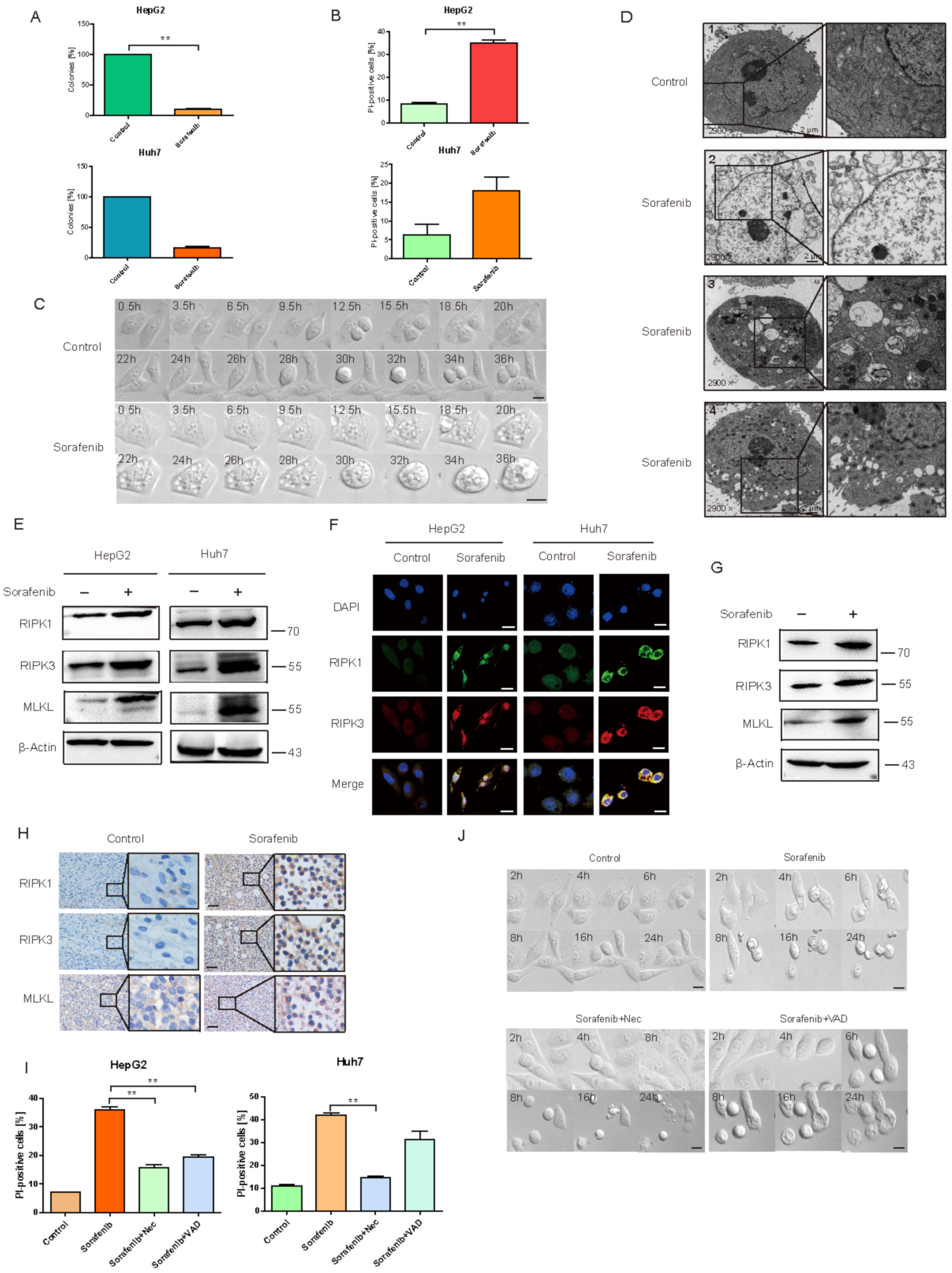
2.1. Sorafenib Induced HCC Necroptosis
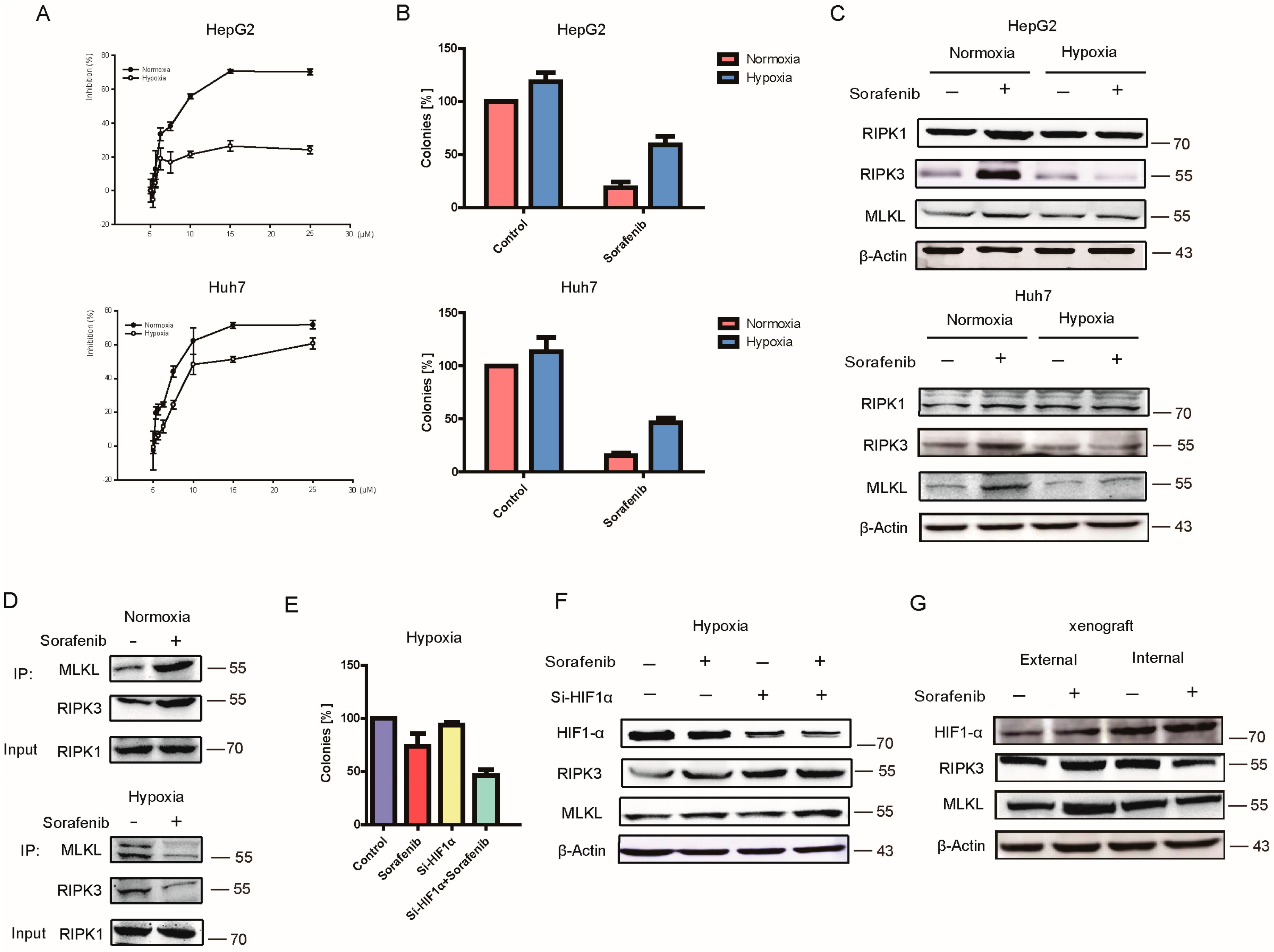
2.2. Hypoxia Contributed to HCC Resistance to Sorafenib
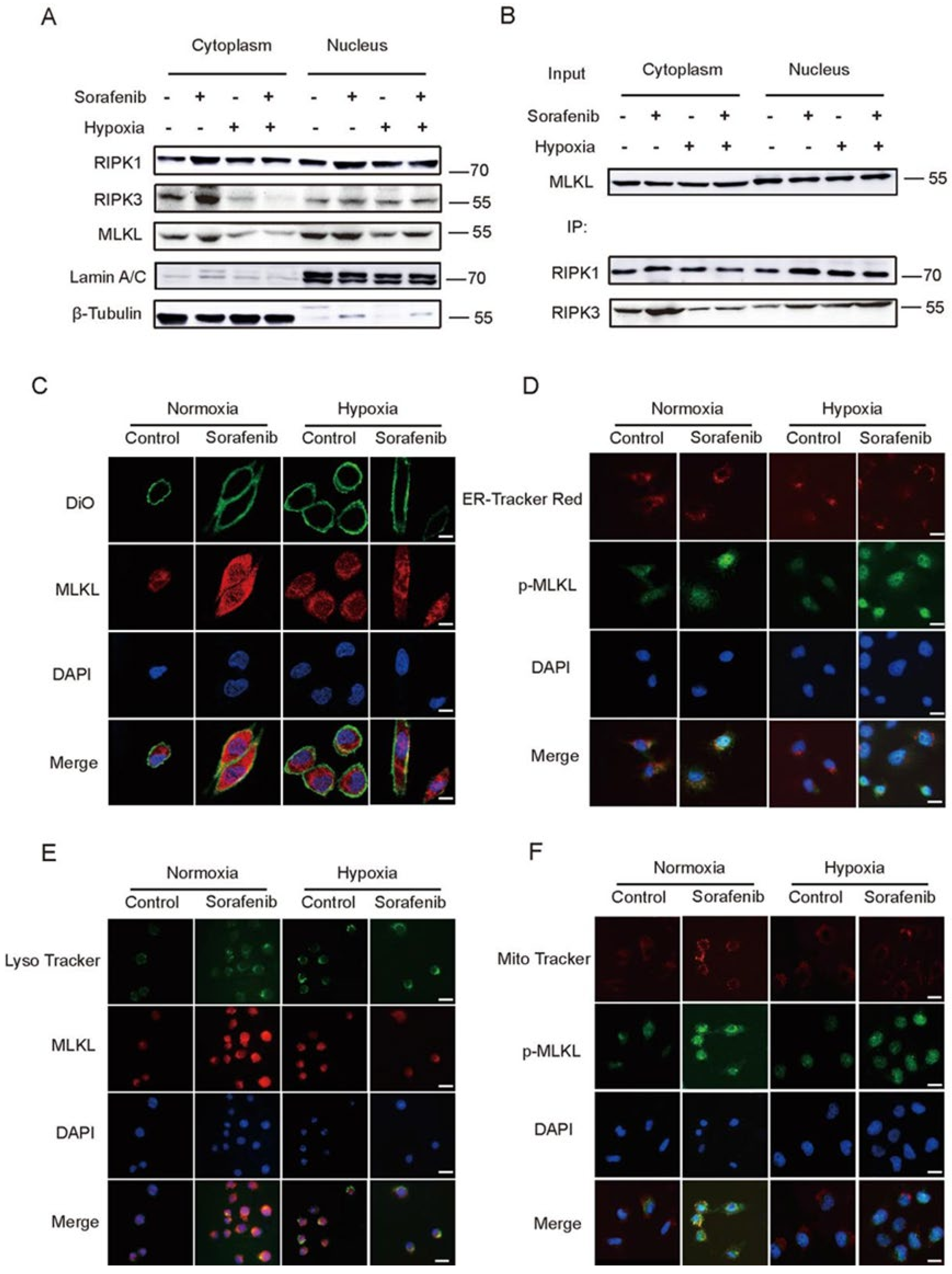
2.3. Hypoxia Impeded the Distribution of RIPK1/RIPK3/MLKL Complex in Cytoplasm
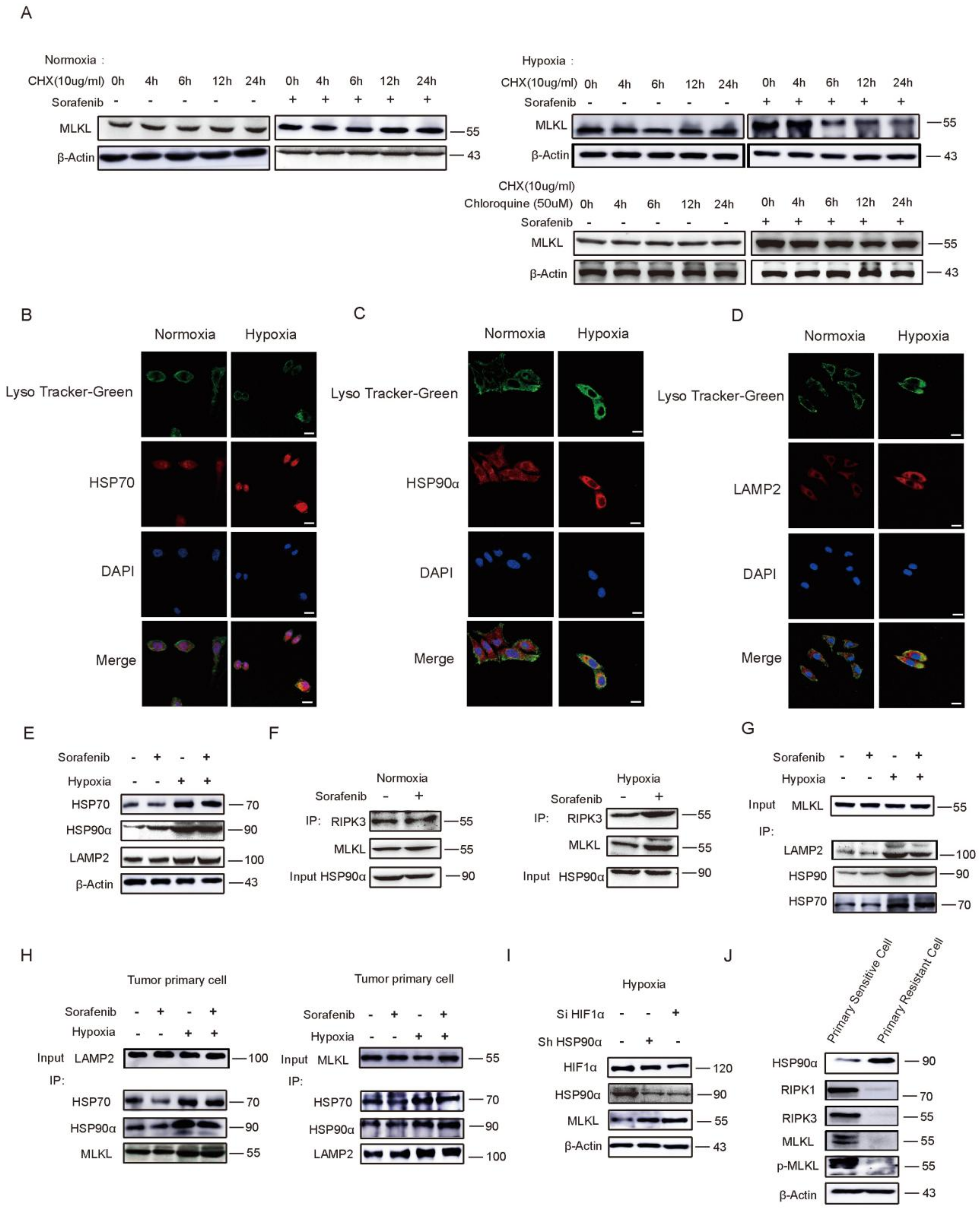
2.4. HSP90α Promotes Chaperone-Mediated Autophagy (CMA) Degradation by Directly Binding to MLKL in Hypoxia
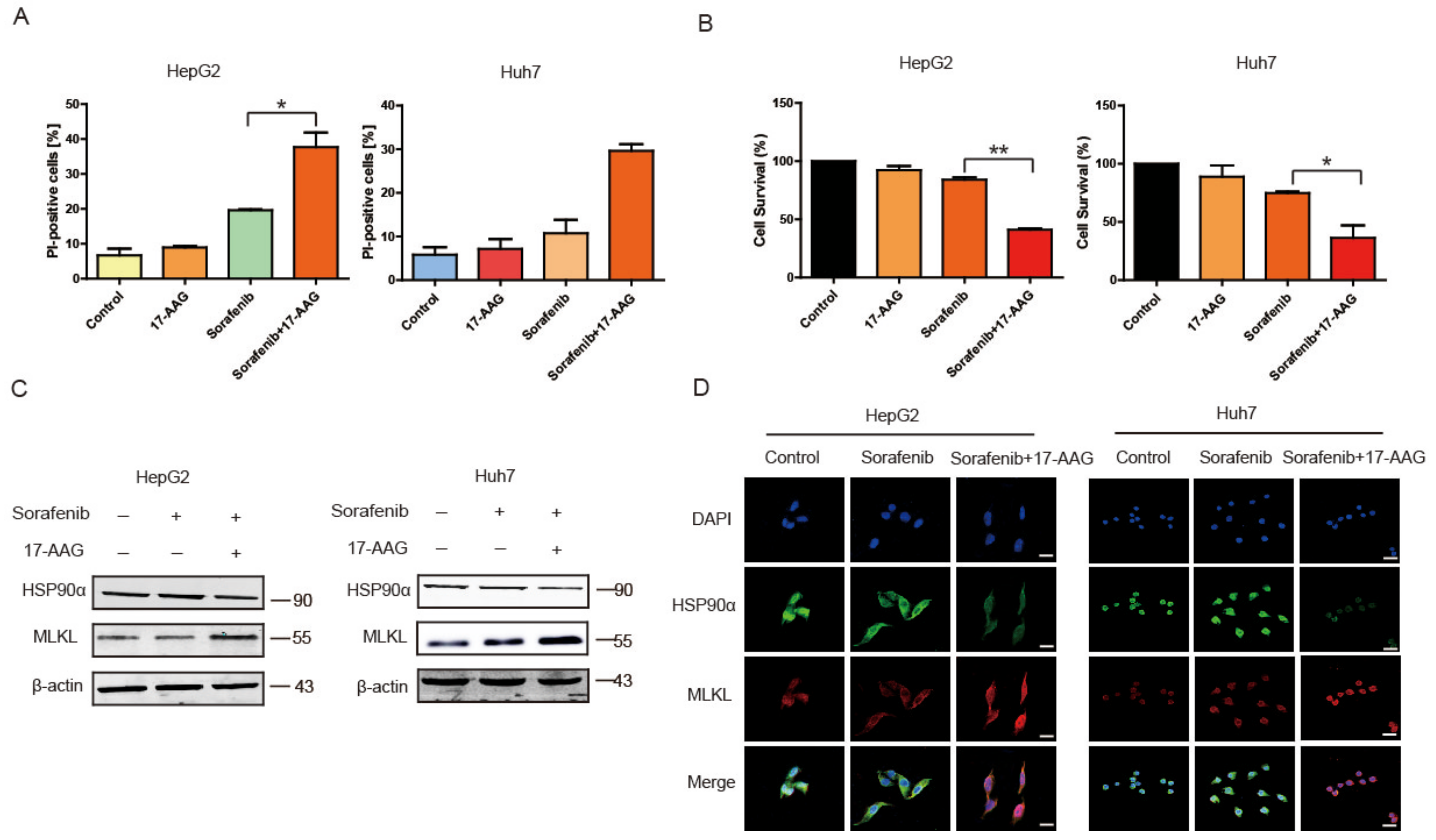
2.5. 17-AAG Combining with Sorafenib Enhanced Necroptosis Pathway In Vitro
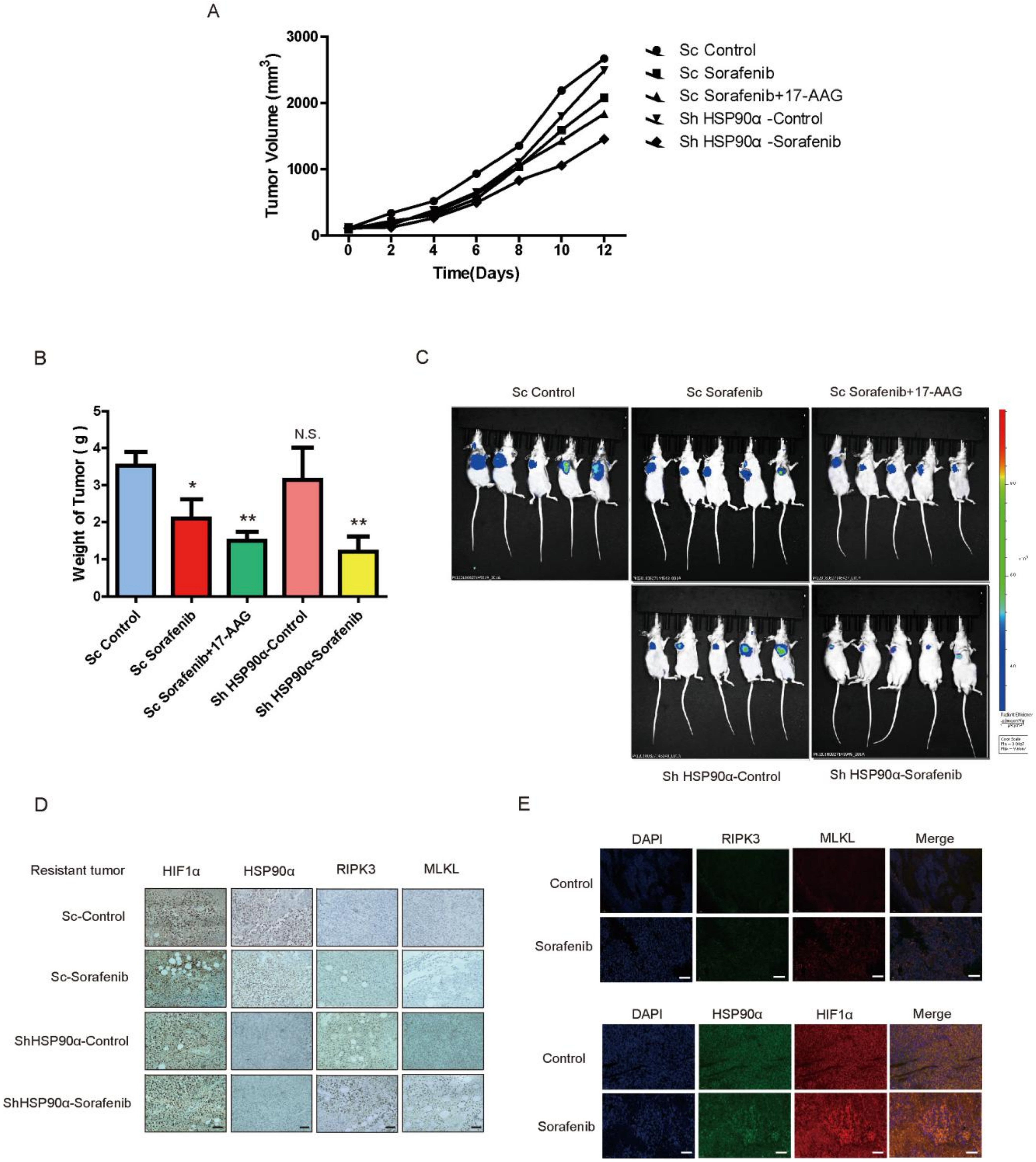
2.6. HSP90α Could Be an Important Target in Sorafenib Resistance In Vivo
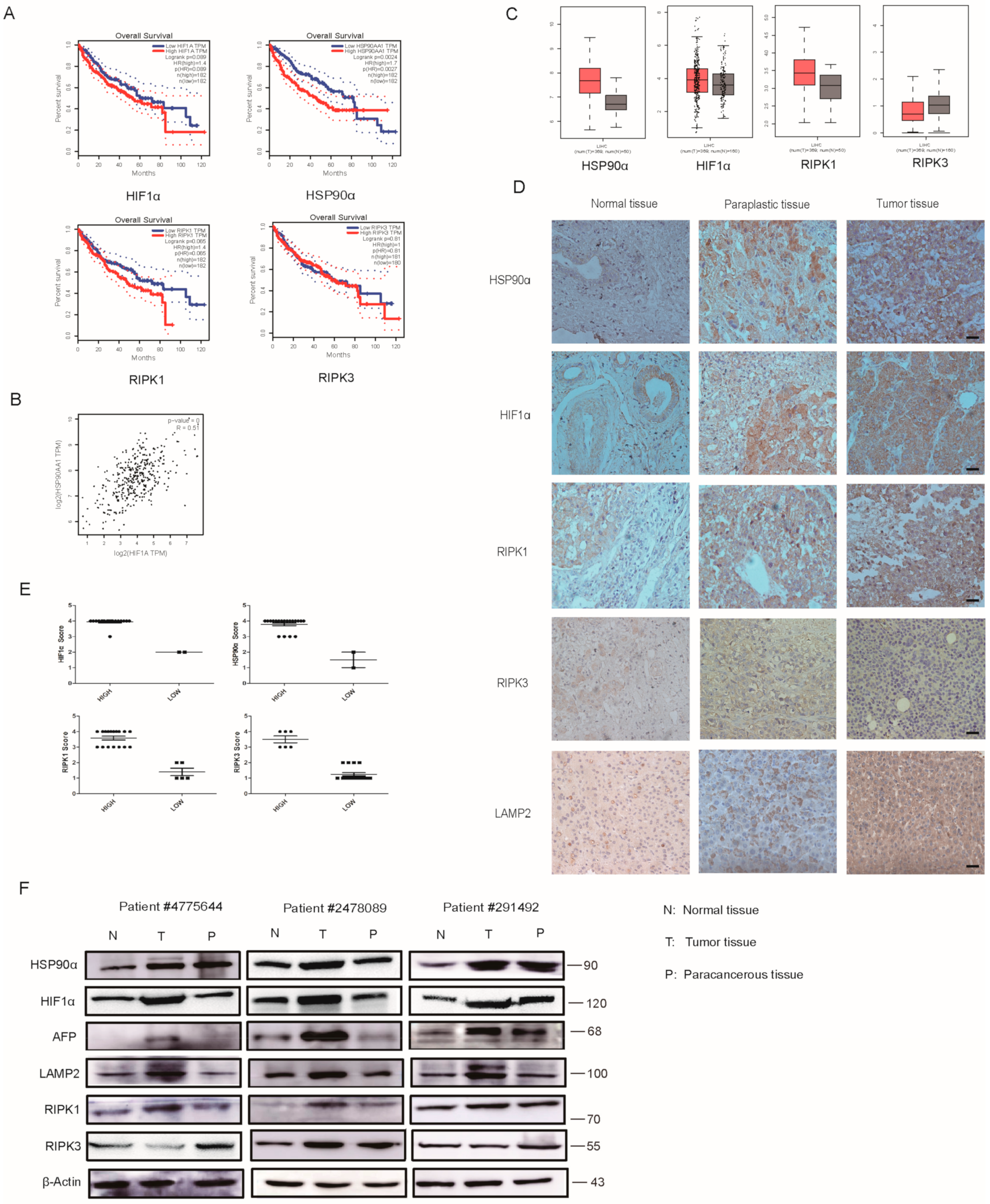
2.7. Clinical Analysis of HIF1α/HSP90α as a Therapeutic Target
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Clinical Samples
4.4. PDX (Patient-Derived Tumor Xenograft) Model and Sorafenib—Resistance Model In Vivo
4.5. Animal Studies
4.6. Western Blot Analysis
4.7. Immunohistochemistry (IHC)
4.8. Immunofluorescence Assay
4.9. Cell Transfection
4.10. Cytoplasmic and Nuclear Protein Extraction
4.11. Co-Immunoprecipitation (Co-IP)
4.12. Flowcytometry Analysis
4.13. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Ethics Approval and Consent to Participate
Abbreviations
| HCC | Hepatocellular carcinoma |
| 17-AAG | Demethoxygeldanamycin |
| IF | Immunofluorescence |
| WB | Western Blot |
| IHC | Immunohistochemistry |
| i.g. | intragastrically |
| Co-IP | Coimmunoprecipitation |
| CMA | chaperone-mediated autophagy |
| DAB | 3, 3′-diaminobenzidine |
| DAPI | 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride |
| PI | Propidium Iodide |
| PBS | Phosphate-Buffered Saline |
| MTT | 3- (4, 5-Dimethylthiazol-2-yl) -2, 5-diphenyltetrazolium bromide |
| DMSO | Dimethyl sulfoxide |
| Nec | necrostatin-1 |
| VAD | Z-VAD-FMK |
| RIPK1 | Receptor-interacting protein kinase 1 |
| MLKL | Mixed lineage kinase domain-like |
| RIPK3 | Receptor-interacting protein kinase-3 |
| HSP90α | Heat shock proteins 90α |
| HIF1α | Hypoxia inducible factor-1α |
| AFP | Alpha fetoprotein |
| LAMP2 | Lysosomal-associated membrane protein 2 |
| VEGF-R | Vascular endothelial growth factor receptors |
| PDGF-R | Platelet-derived growth factor receptor |
| PDX | Patient-Derived tumor Xenograft |
| GEPIA | Gene Expression Profiling Interactive Analysis |
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012: Global Cancer Statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, L.J.; Tong, M.J.; Busuttil, R.W.; Hiatt, J.R. Acetaminophen Hepatotoxicity and Acute Liver Failure. J. Clin. Gastroenterol. 2009, 43, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Macek-Jilkova, Z.; Kuyucu, A.Z.; Kurma, K.; Pour, S.T.A.; Roth, G.S.; Abbadessa, G.; Yu, Y.; Schwartz, B.; Sturm, N.; Marche, P.N.; et al. Combination of AKT inhibitor ARQ 092 and sorafenib potentiates inhibition of tumor progression in cirrhotic rat model of hepatocellular carcinoma. Oncotarget 2018, 9, 11145–11158. [Google Scholar] [CrossRef] [PubMed]
- Zhai, B.; Hu, F.; Jiang, X.; Xu, J.; Zhao, D.; Liu, B.; Pan, S.; Dong, X.; Tan, G.; Wei, Z.; et al. Inhibition of Akt Reverses the Acquired Resistance to Sorafenib by Switching Protective Autophagy to Autophagic Cell Death in Hepatocellular Carcinoma. Mol. Cancer Ther. 2014, 13, 1589–1598. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the RAF/MEK/ERK Pathway and Receptor Tyrosine Kinases Involved in Tumor Progression and Angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [Green Version]
- Ciamporcero, E.; Miles, K.M.; Adelaiye, R.; Ramakrishnan, S.; Shengyu, K.; Ku, S.Y.; Pizzimenti, S.; Sennino, B.; Barrera, G.; Pili, R. Combination Strategy Targeting VEGF and HGF/c-met in Human Renal Cell Carcinoma Models. Mol. Cancer Ther. 2015, 14, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Van Malenstein, H.; Dekervel, J.; Verslype, C.; Van Cutsem, E.; Windmolders, P.; Nevens, F.; Van Pelt, J. Long-term exposure to sorafenib of liver cancer cells induces resistance with epithelial-to-mesenchymal transition, increased invasion and risk of rebound growth. Cancer Lett. 2013, 329, 74–83. [Google Scholar] [CrossRef]
- Villanueva, A.; Llovet, J.M. Second-Line Therapies in Hepatocellular Carcinoma: Emergence of Resistance to Sorafenib: Figure 1. Clin. Cancer Res. 2012, 18, 1824–1826. [Google Scholar] [CrossRef] [Green Version]
- Farazi, P.A.; Depinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef]
- Kharaziha, P.; Chioureas, D.; Baltatzis, G.; Fonseca, P.; Rodriguez, P.; Gogvadze, V.; Lennartsson, L.; Björklund, A.-C.; Zhivotovsky, B.; Grandér, D.; et al. Sorafenib-induced defective autophagy promotes cell death by necroptosis. Oncotarget 2015, 6, 37066–37082. [Google Scholar] [CrossRef] [Green Version]
- Ramírez-Labrada, A.; López-Royuela, N.; Jarauta, V.; Galán-Malo, P.; Azaceta, G.; Palomera, L.; Pardo, J.; Anel, A.; Marzo, I.; Naval, J. Two death pathways induced by sorafenib in myeloma cells: Puma-mediated apoptosis and necroptosis. Clin. Transl. Oncol. 2014, 17, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, S.L.; Cleris, L.; Stirparo, G.G.; Tartari, S.; Saba, E.; Pierdominici, M.; Malorni, W.; Carbone, A.; Anichini, A.; Carlo-Stella, C. BIM upregulation and ROS-dependent necroptosis mediate the antitumor effects of the HDACi Givinostat and Sorafenib in Hodgkin lymphoma cell line xenografts. Leukemia 2014, 28, 1861–1871. [Google Scholar] [CrossRef] [PubMed]
- Martens, S.; Jeong, M.; Tonnus, W.; Feldmann, F.; Hofmans, S.; Goossens, V.; Takahashi, N.; Braesen, J.H.; Lee, E.-W.; Van Der Veken, P.; et al. Sorafenib tosylate inhibits directly necrosome complex formation and protects in mouse models of inflammation and tissue injury. Cell Death Dis. 2017, 8, e2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Zheng, T.; Song, R.; Wang, J.; Yin, D.; Wang, L.; Liu, H.; Tian, L.; Fang, X.; Meng, X.; et al. Hypoxia-mediated sorafenib resistance can be overcome by EF24 through Von Hippel-Lindau tumor suppressor-dependent HIF-1α inhibition in hepatocellular carcinoma. Hepatology 2013, 57, 1847–1857. [Google Scholar] [CrossRef]
- Unruh, A.; Ressel, A.; Mohamed, H.G.; Johnson, R.S.; Nadrowitz, R.; Richter, E.; Katschinski, D.M.; Wenger, R.H. The hypoxia-inducible factor-1α is a negative factor for tumor therapy. Oncogene 2003, 22, 3213–3220. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Liu, Y.; Yao, Q.; Zheng, R.; Zhang, L.; Lin, J.; Guo, Z.; Du, S.; Ren, C.; Yuan, Q.; et al. Hypoxia-induced angiotensin II by the lactate-chymase-dependent mechanism mediates radioresistance of hypoxic tumor cells. Sci. Rep. 2017, 7, 42396. [Google Scholar] [CrossRef] [Green Version]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nat. Cell Biol. 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Verba, K.A.; Wang, R.Y.-R.; Arakawa, A.; Liu, Y.; Shirouzu, M.; Yokoyama, S.; Agard, D. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Li, Y.; Guan, S.; Fan, J.; Cheng, C.; Bright, A.; Chin, C.; Chen, M.; Woodley, D. Extracellular heat shock protein-90alpha: Linking hypoxia to skin cell motility and wound healing. J. Invest. Dermatol. 2007, 127, S36. [Google Scholar]
- Fike, C.D.; Pfister, S.L.; Slaughter, J.C.; Kaplowitz, M.R.; Zhang, Y.; Zeng, H.; Frye, N.R.; Aschner, J.L. Protein complex formation with heat shock protein 90 in chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am. J. Physiol. Circ. Physiol. 2010, 299, H1190–H1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohwer, N.; Cramer, T. Hypoxia-mediated drug resistance: Novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist. Updates 2011, 14, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Xiao, L.; Wang, L.; Ruden, D.M. Hsp90 inhibitors and drug resistance in cancer: The potential benefits of combination therapies of Hsp90 inhibitors and other anti-cancer drugs. Biochem. Pharmacol. 2012, 83, 995–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Li, H.; Huang, Z.; He, Y.; Zhou, X.; Huang, T.; Dai, P.; Duan, D.; Ma, X.; Yin, Q.; et al. Hypoxia attenuates Hsp90 inhibitor 17-DMAG-induced cyclin B1 accumulation in hepatocellular carcinoma cells. Cell Stress Chaperones 2016, 21, 339–348. [Google Scholar] [CrossRef]
- Pelicano, H.; Carew, J.S.; McQueen, T.J.; Andreeff, M.; Plunkett, W.; Keating, M.J.; Huang, P. Targeting Hsp90 by 17-AAG in leukemia cells: Mechanisms for synergistic and antagonistic drug combinations with arsenic trioxide and Ara-C. Leukemia 2006, 20, 610–619. [Google Scholar] [CrossRef] [Green Version]
- Neckers, L. Heat shock protein 90 inhibition by 17-allylamino-17-demethoxygeldanamycin: A novel therapeutic approach for treating hormone-refractory prostate cancer. Clin. Cancer Res. 2002, 8, 962–966. [Google Scholar]
- Kim, S.H.; Kang, J.G.; Kim, C.S.; Ihm, S.-H.; Choi, M.G.; Yoo, H.J.; Lee, S.J. The effect of 17-allylamino-17-demethoxygeldanamycin alone or in combination with paclitaxel on anaplastic thyroid carcinoma cells. Endocrine 2014, 48, 886–893. [Google Scholar] [CrossRef]
- Mohammadian, M.; Zeynali-Moghaddam, S.; Ansari, M.H.K.; Rami, Y.; Azarbayjani, A.F.; Kheradmand, F. Dihydropyrimidine Dehydrogenase Levels in Colorectal Cancer Cells Treated with a Combination of Heat Shock Protein 90 Inhibitor and Oxaliplatin or Capecitabine. Adv. Pharm. Bull. 2019, 9, 439–444. [Google Scholar] [CrossRef] [Green Version]
- Kheradmand, F.; Mohammadian, M.; Zeynali, S.; Azarbaijani, A.F.; Ansari, M.H.K. Cytotoxic effects of the newly-developed chemotherapeutic agents 17-AAG in combination with oxaliplatin and capecitabine in colorectal cancer cell lines. Res. Pharm. Sci. 2017, 12, 517–525. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, R.L.; Zhu, L. Chaperone-Mediated Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 435–452. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.W.-L.; Leung, C.-T.; Liu, H.; Pang, S.Y.-Y.; Lam, C.S.-C.; Xian, J.; Li, L.; Kung, M.H.-W.; Ramsden, D.B.; Ho, S.-L. Age-dependent accumulation of oligomeric SNCA/α-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: Role for therapeutic activation of chaperone-mediated autophagy (CMA). Autophagy 2020, 16, 347–370. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Kacal, M.; Ouchida, A.T.; Zhang, B.; Norberg, E.; Vakifahmetoglu-Norberg, H. Targetome analysis of chaperone-mediated autophagy in cancer cells. Autophagy 2019, 15, 1558–1571. [Google Scholar] [CrossRef]
- Dash, S.; Aydin, Y.; Moroz, K. Chaperone-Mediated Autophagy in the Liver: Good or Bad? Cells 2019, 8, 1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Zefu, L.; Liu, Z.; Li, M.; Sui, D.; Liu, Y.; Shao, W.; Wang, B.; Liu, P.; Li, G. 17AAG improves histological and functional outcomes in a rat CCI model through autophagy activation and apoptosis attenuation. Neurosci. Lett. 2015, 599, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-H.; Chang, J.T.; Li, L.-A.; Tsai, H.-T.; Shen, M.-Y.; Lin, P. Aryl Hydrocarbon Receptor is a Target of 17-Allylamino-17-demethoxygeldanamycin and Enhances its Anticancer Activity in Lung Adenocarcinoma Cells. Mol. Pharmacol. 2012, 83, 605–612. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, Y.; Yang, Y.; Pan, D.; Ding, Y.; Zhang, H.; Ye, Y.; Li, J.; Zhao, L. HSP90α Mediates Sorafenib Resistance in Human Hepatocellular Carcinoma by Necroptosis Inhibition under Hypoxia. Cancers 2021, 13, 243. https://doi.org/10.3390/cancers13020243
Liao Y, Yang Y, Pan D, Ding Y, Zhang H, Ye Y, Li J, Zhao L. HSP90α Mediates Sorafenib Resistance in Human Hepatocellular Carcinoma by Necroptosis Inhibition under Hypoxia. Cancers. 2021; 13(2):243. https://doi.org/10.3390/cancers13020243
Chicago/Turabian StyleLiao, Yan, Yue Yang, Di Pan, Youxiang Ding, Heng Zhang, Yuting Ye, Jia Li, and Li Zhao. 2021. "HSP90α Mediates Sorafenib Resistance in Human Hepatocellular Carcinoma by Necroptosis Inhibition under Hypoxia" Cancers 13, no. 2: 243. https://doi.org/10.3390/cancers13020243
APA StyleLiao, Y., Yang, Y., Pan, D., Ding, Y., Zhang, H., Ye, Y., Li, J., & Zhao, L. (2021). HSP90α Mediates Sorafenib Resistance in Human Hepatocellular Carcinoma by Necroptosis Inhibition under Hypoxia. Cancers, 13(2), 243. https://doi.org/10.3390/cancers13020243