Wilms’ Tumor 1 (WT1): A Novel Immunomarker of Dermatofibrosarcoma Protuberans—An Immunohistochemical Study on a Series of 114 Cases of Bland-Looking Mesenchymal Spindle Cell Lesions of the Dermis/Subcutaneous Tissues


 ,
, 
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Clinical Data of the Cohort of Cases Included in the Study
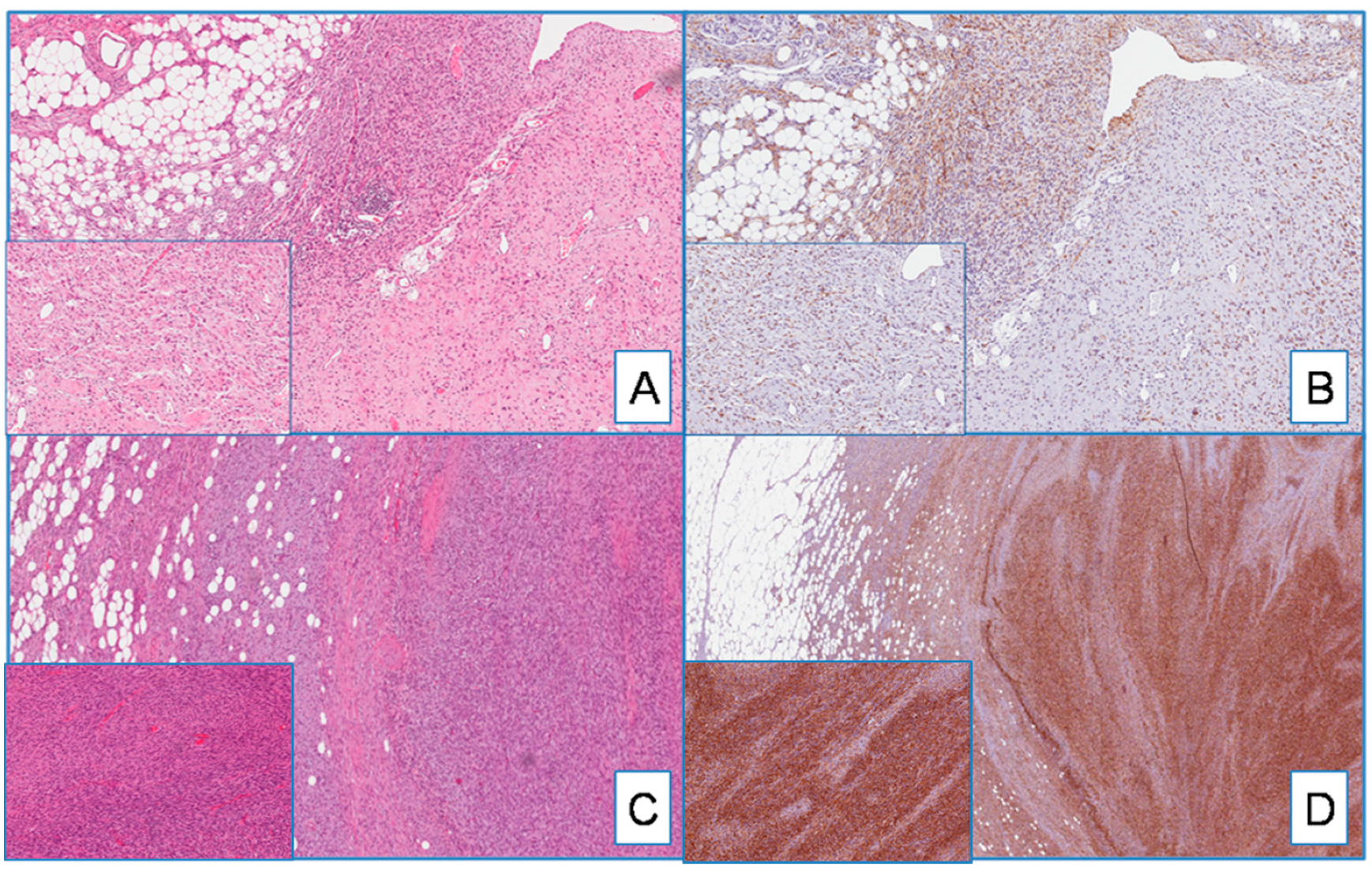
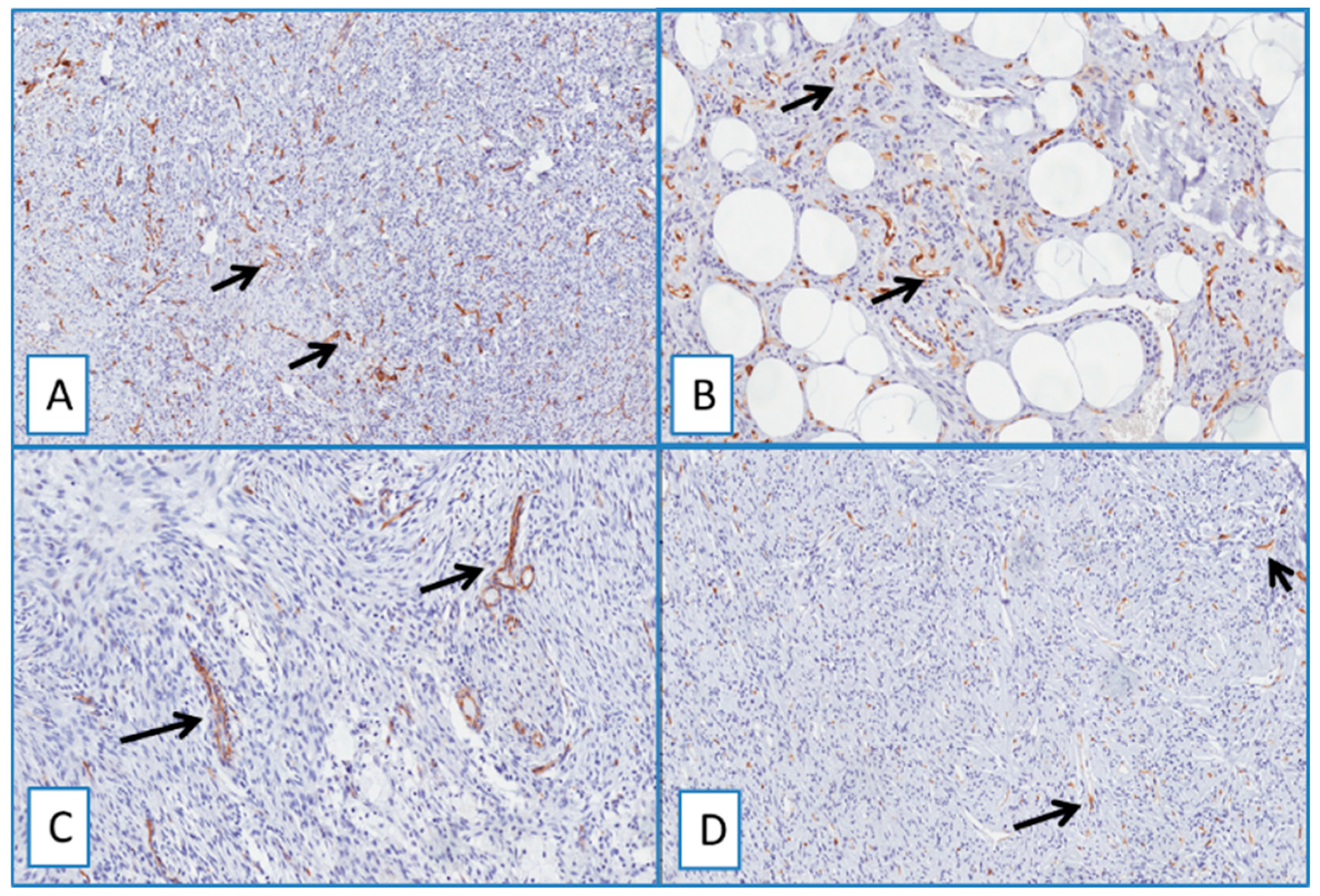
2.2. Evaluation of Immunohistochemical Expression of WT1
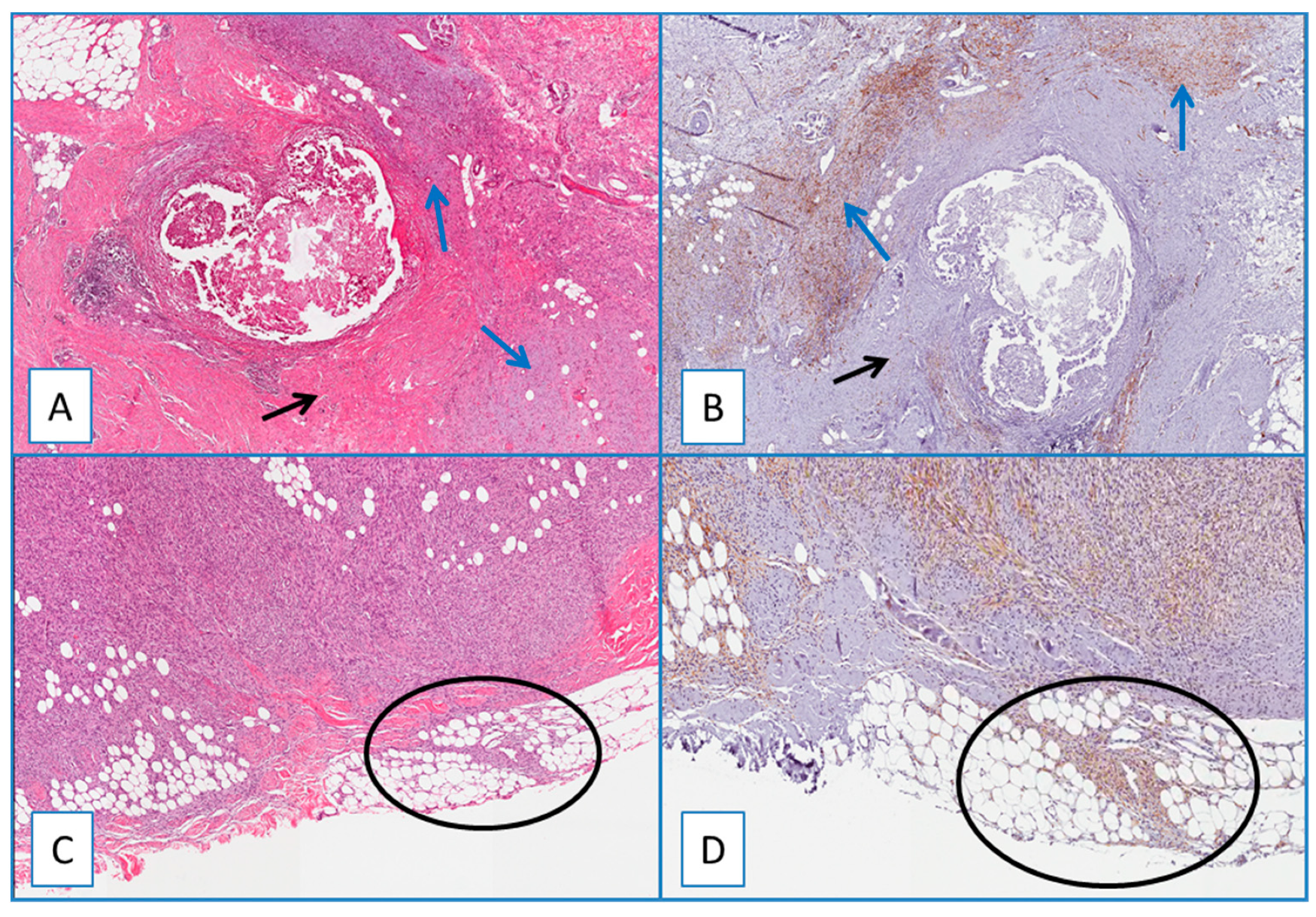
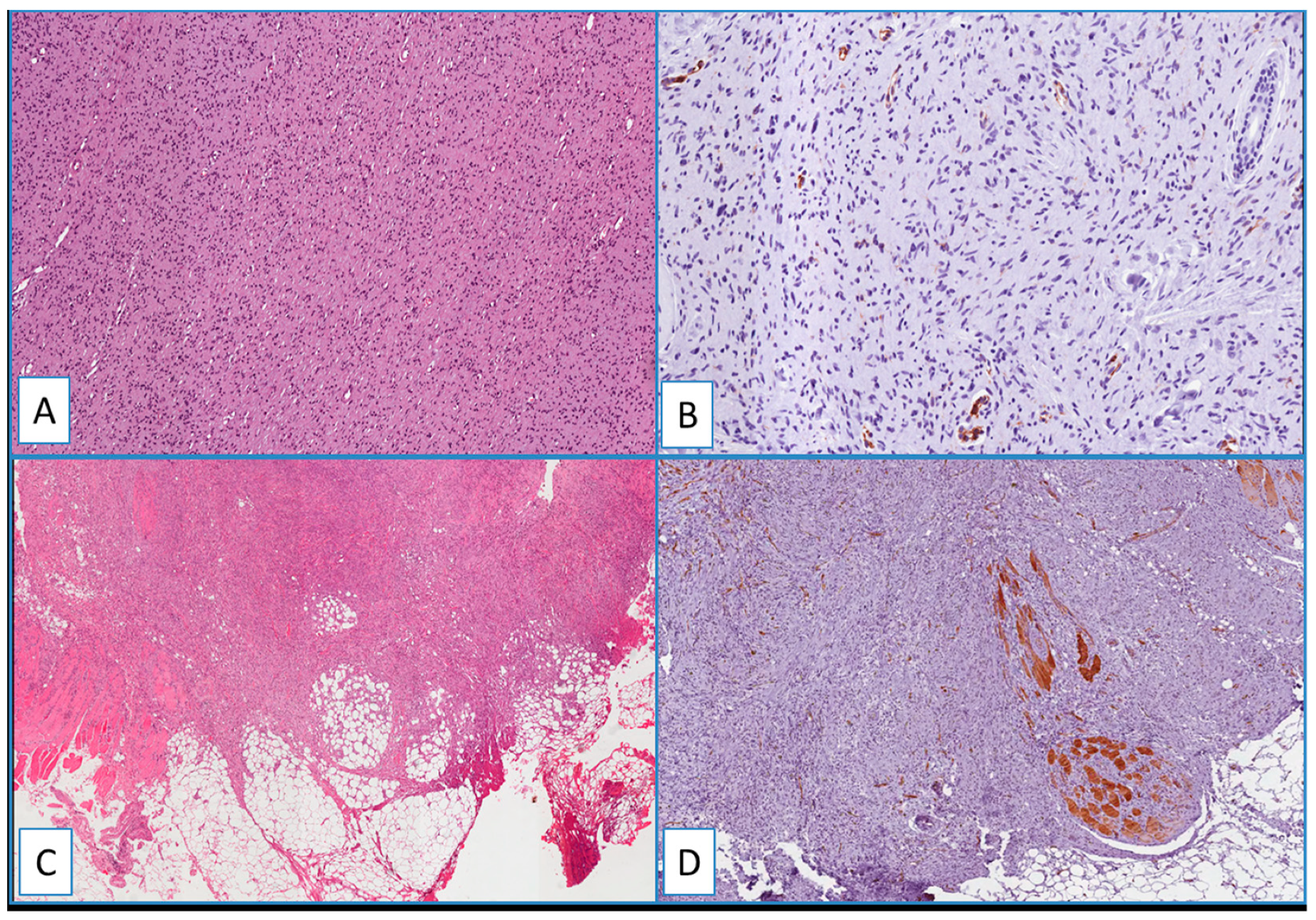
2.3. DFSP
2.4. Non-DFSP Lesions
3. Discussion
3.1. Diagnostic Utility of WT1 in Distinguishing DFSP from Its Morphological Mimickers
3.2. Diagnostic Utility of WT1 in Recurrent/Residual DFSP
4. Materials and Methods
- Fifty-seven cases of DFSP; 11 of these cases were recurrent lesions; 2 primary cases exhibited an additional fibrosarcomatous overgrowth, while 2 primary and one recurrent tumor contained a minority of giant cell fibroblastoma component);
- Fifteen cases of dermatofibroma (classic type and cellular variants);
- Five cases of deep fibrous histiocytoma
- Eight cases of dermal scars;
- Five cases of spindle cell lipoma;
- Six cases of nodular fasciitis;
- Five cases of cutaneous leiomyomas;
- Eight cases of neurofibroma;
- Five cases of solitary fibrous tumor.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Degos, R.; Mouly, R.; Civatte, J.; Chérif-Cheikh, J.L.; Lautmann, F. Dermato-fibro-sarcome de Darier-Ferrand, datant de 70 ans, opéré au stade ultime de tumeur monstrueuse [Darier-Ferrand dermato-fibrosarcoma of 70 years’ duration operated on in the last stage of massive tumor]. Bull. Soc. Fr. Dermatol. Syphiligr. 1967, 74, 190–191. [Google Scholar] [PubMed]
- McKee, P.H.; Fletcher, C.D. Dermatofibrosarcoma protuberans presenting in infancy and childhood. J. Cutan. Pathol. 1991, 18, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Petoin, D.S.; Verola, O.; Banzet, P.; Dufourmentel, C.; Servant, J.M. Dermatofibrosarcome de Darier et Ferrand. Etude de 96 cas sur 15 ans [Darier-Ferrand dermatofibrosarcoma. Study of 96 cases over 15 years]. Chirurgie 1985, 111, 132–138. [Google Scholar] [PubMed]
- Taylor, H.B.; Helwig, E.B. Dermatofibrosarcoma protuberans. A study of 115 cases. Cancer 1962, 15, 717–725. [Google Scholar] [CrossRef]
- Thway, K.; Noujaim, J.; Jones, R.L.; Fisher, C. Dermatofibrosarcoma protuberans: Pathology, genetics, and potential therapeutic strategies. Ann. Diagn. Pathol. 2016, 25, 64–71. [Google Scholar] [CrossRef]
- Allen, A.; Ahn, C.; Sangüeza, O.P. Dermatofibrosarcoma Protuberans. Dermatol. Clin. 2019, 37, 483–488. [Google Scholar] [CrossRef]
- Vecchio, G.M.; Broggi, G.; Mulè, A.; Piombino, E.; Magro, G. Dermatofibrosarcoma protuberans: A tumor in the wide spectrum of the bland-looking spindle cell lesions of the breast. Pathologica 2019, 111, 87–91. [Google Scholar] [CrossRef] [Green Version]
- Magro, G.; Salvatorelli, L.; Puzzo, L.; Piombino, E.; Bartoloni, G.; Broggi, G.; Vecchio, G.M. Practical approach to diagnosis of bland-looking spindle cell lesions of the breast. Pathologica 2019, 111, 344–360. [Google Scholar] [CrossRef]
- Sadullahoğlu, C.; Dere, Y.; Atasever, T.R.; Öztop, M.T.; Karaaslan, Ö. The Role of CD34 and D2-40 in the Differentiation of Dermatofibroma and Dermatofibrosarcoma Protuberans. Turk. Patoloji. Derg. 2017, 1, 223–227. [Google Scholar] [CrossRef] [Green Version]
- Morimitsu, Y.; Hisaoka, M.; Okamoto, S.; Hashimoto, H.; Ushijima, M. Dermatofibrosarcoma protuberans and its fibrosarcomatous variant with areas of myoid differentiation: A report of three cases. Histopathology 1998, 32, 547–551. [Google Scholar] [CrossRef]
- Sanz-Trelles, A.; Ayala-Carbonero, A.; Rodrigo-Fernández, I.; Weil-Lara, B. Leiomyomatous nodules and bundles of vascular origin in the fibrosarcomatous variant of dermatofibrosarcoma protuberans. J. Cutan. Pathol. 1998, 25, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Goldblum, J.R.; Reith, J.D.; Weiss, S.W. Sarcomas arising in dermatofibrosarcoma protuberans: A reappraisal of biologic behavior in eighteen cases treated by wide local excision with extended clinical follow up. Am. J. Surg. Pathol. 2000, 24, 1125–1130. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, A.; O’Brien, K.P.; Sjöblom, T.; Pietras, K.; Buchdunger, E.; Collins, V.P.; Heldin, C.H.; Dumanski, J.P.; Ostman, A. The dermatofibrosarcoma protuberans-associated collagen type Ialpha1/platelet-derived growth factor (PDGF) B-chain fusion gene generates a transforming protein that is processed to functional PDGF-BB. Cancer Res. 1999, 59, 3719–3723. [Google Scholar] [PubMed]
- Mentzel, T.; Beham, A.; Katenkamp, D.; Dei Tos, A.P.; Fletcher, C.D. Fibrosarcomatous (“high-grade”) dermatofibrosarcoma protuberans: Clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am. J. Surg. Pathol. 1998, 22, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Wrotnowski, U.; Cooper, P.H.; Shmookler, B.M. Fibrosarcomatous change in dermatofibrosarcoma protuberans. Am. J. Surg. Pathol. 1988, 12, 287–293. [Google Scholar] [CrossRef]
- Connelly, J.H.; Evans, H.L. Dermatofibrosarcoma protuberans. A clinicopathologic review with emphasis on fibrosarcomatous areas. Am. J. Surg. Pathol. 1992, 16, 921–925. [Google Scholar] [CrossRef]
- Lopez, L.V.; Yatsenko, S.A.; Burgess, M.; Schoedel, K.; Rao, U.N.M. Dermatofibrosarcoma protuberans with fibrosarcomatous transformation: Our experience, molecular evaluation of selected cases, and short literature review. Int. J. Dermatol. 2019, 58, 1246–1252. [Google Scholar] [CrossRef]
- Abbott, J.J.; Oliveira, A.M.; Nascimento, A.G. The prognostic significance of fibrosarcomatous transformation in dermatofibrosarcoma protuberans. Am. J. Surg. Pathol. 2006, 30, 436–443. [Google Scholar] [CrossRef]
- Broggi, G.; Salvatorelli, L.; Reibaldi, M.; Bonfiglio, V.; Longo, A.; Russo, A.; Caltabiano, R.; Magro, G. Solitary fibrous tumor of the orbital region: Report of a case with emphasis on the diagnostic utility of STAT-6. Pathologica 2020, 112, 195–199. [Google Scholar] [CrossRef]
- Magro, G.; Salvatorelli, L.; Piombino, E.; Broggi, G.; Castorina, S. Solitary fibrous tumor with atypical features of the paravesical space: Benign clinical course at the 10-years follow-up. Report of a case and review of the literature. Pathologica 2020, 112, 200–209. [Google Scholar] [CrossRef]
- Call, K.M.; Glaser, T.; Ito, C.Y.; Buckler, A.J.; Pelletier, J.; Haber, D.A.; Rose, E.A.; Kral, A.; Yeger, H.; Lewis, W.H.; et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell 1990, 60, 509–520. [Google Scholar] [CrossRef]
- Tsuta, K.; Kato, Y.; Tochigi, N.; Hoshino, T.; Takeda, Y.; Hosako, M.; Maeshima, A.M.; Asamura, H.; Kondo, T.; Matsuno, Y. Comparison of different clones (WT49 versus 6F-H2) of WT-1 antibodies for immunohistochemical diagnosis of malignant pleural mesothelioma. Appl. Immunohistochem. Mol. Morphol. 2009, 17, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Gessler, M.; Poustka, A.; Cavenee, W.; Neve, R.L.; Orkin, S.H.; Bruns, G.A. Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature 1990, 343, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Menke, A.L.; van der Eb, A.J.; Jochemsen, A.G. The Wilms’ tumor 1 gene: Oncogene or tumor suppressor gene? Int. Rev. Cytol. 1998, 181, 151–212. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Haber, D.A. Wilms tumor and the WT1 gene. Exp. Cell Res. 2001, 264, 74–99. [Google Scholar] [CrossRef] [PubMed]
- Huff, V. Wilms’ tumours: About tumour suppressor genes, an oncogene and a chameleon gene. Nat. Rev. Cancer 2011, 11, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Loke, S.L.; Neckers, L.M.; Schwab, G.; Jaffe, E.S. c-myc protein in normal tissue. Effects of fixation on its apparent subcellular distribution. Am. J. Pathol. 1988, 131, 29–37. [Google Scholar]
- Royds, J.A.; Sharrard, R.M.; Wagner, B.; Polacarz, S.V. Cellular localisation of c-myc product in human colorectal epithelial neoplasia. J. Pathol. 1992, 166, 225–233. [Google Scholar] [CrossRef]
- Ramani, P.; Cowell, J.K. The expression pattern of Wilms’ tumour gene (WT1) product in normal tissues and paediatric renal tumours. J. Pathol. 1996, 179, 162–168. [Google Scholar] [CrossRef]
- Lawley, L.P.; Cerimele, F.; Weiss, S.W.; North, P.; Cohen, C.; Kozakewich, H.P.; Mulliken, J.B.; Arbiser, J.L. Expression of Wilms tumor 1 gene distinguishes vascular malformations from proliferative endothelial lesions. Arch. Dermatol. 2005, 141, 1297–1300. [Google Scholar] [CrossRef] [Green Version]
- Timár, J.; Mészáros, L.; Orosz, Z.; Albini, A.; Rásó, E. WT1 expression in angiogenic tumours of the skin. Histopathology 2005, 47, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, S.; Oji, Y.; Horiuchi, T.; Kanda, T.; Kitagawa, M.; Takeuchi, T.; Kawano, K.; Kuwae, Y.; Yamauchi, A.; Okumura, M.; et al. Immunohistochemical detection of WT1 protein in a variety of cancer cells. Mod. Pathol. 2006, 19, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Salvatorelli, L.; Calabrese, G.; Parenti, R.; Vecchio, G.M.; Puzzo, L.; Caltabiano, R.; Musumeci, G.; Magro, G. Immunohistochemical Expression of Wilms’ Tumor 1 Protein in Human Tissues: From Ontogenesis to Neoplastic Tissues. Appl. Sci. 2020, 10, 40. [Google Scholar] [CrossRef] [Green Version]
- Parenti, R.; Puzzo, L.; Vecchio, G.M.; Gravina, L.; Salvatorelli, L.; Musumeci, G.; Vasquez, E.; Magro, G. Immunolocalization of Wilms’ Tumor protein (WT1) in developing human peripheral sympathetic and gastroenteric nervous system. Acta Histochem. 2014, 116, 48–54. [Google Scholar] [CrossRef]
- Al Dhaybi, R.; Powell, J.; McCuaig, C.; Kokta, V. Differentiation of vascular tumors from vascular malformations by expression of Wilms tumor 1 gene: Evaluation of 126 cases. J. Am. Acad. Dermatol. 2010, 63, 1052–1057. [Google Scholar] [CrossRef]
- Trindade, F.; Tellechea, O.; Torrelo, A.; Requena, L.; Colmenero, I. Wilms tumor 1 expression in vascular neoplasms and vascular malformations. Am. J. Dermatopathol. 2011, 33, 569–572. [Google Scholar] [CrossRef]
- Carpentieri, D.F.; Nichols, K.; Chou, P.M.; Matthews, M.; Pawel, B.; Huff, D. The expression of WT1 in the differentiation of rhabdomyosarcoma from other pediatric small round blue cell tumors. Mod. Pathol. 2002, 15, 1080–1086. [Google Scholar] [CrossRef] [Green Version]
- Magro, G.; Salvatorelli, L.; Puzzo, L.; Musumeci, G.; Bisceglia, M.; Parenti, R. Oncofetal expression of Wilms’ tumor 1 (WT1) protein in human fetal, adult and neoplastic skeletal muscle tissues. Acta Histochem. 2015, 117, 492–504. [Google Scholar] [CrossRef]
- Magro, G.; Salvatorelli, L.; Vecchio, G.M.; Musumeci, G.; Rita, A.; Parenti, R. Cytoplasmic expression of Wilms tumor transcription factor-1 (WT1): A useful immunomarker for young-type fibromatoses and infantile fibrosarcoma. Acta Histochem. 2014, 116, 1134–1140. [Google Scholar] [CrossRef]
- Magro, G.; Spadola, S.; Motta, F.; Palazzo, J.; Catalano, F.; Vecchio, G.M.; Salvatorelli, L. STAT6 expression in spindle cell lesions of the breast: An immunohistochemical study of 48 cases. Pathol. Res. Pract. 2018, 214, 1544–1549. [Google Scholar] [CrossRef]
- Cammarata, F.P.; Forte, G.I.; Broggi, G.; Bravatà, V.; Minafra, L.; Pisciotta, P.; Calvaruso, M.; Tringali, R.; Tomasello, B.; Torrisi, F.; et al. Molecular Investigation on a Triple Negative Breast Cancer Xenograft Model Exposed to Proton Beams. Int. J. Mol. Sci. 2020, 21, 6337. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnosis/Number of Cases | Positive Cases (%) | Staining Extension Cases (%) | Staining Intensity Cases (%) |
|---|---|---|---|
| Dermatofibrosarcoma protuberans (n = 57) | 54/57 (95%) | Diffuse: 42/57 (75%) Heterogeneous: 9/57 (15%) Focal: 6/57 (6%) | Strong: 53/57 (93%) Weak: 4/57 (7%) |
| Dermatofibroma (n = 15) | 0/15 (0%) | No staining | No staining |
| Deep fibrous histiocytoma (n = 5) | 0/5 (0%) | No staining | No staining |
| Dermal scars (n = 8) | 0/8 (0%) | No staining | No staining |
| Spindle cell lipoma (n = 5) | 0/5 (0%) | No staining | No staining |
| Nodular fasciitis (n = 6) | 0/6 (0%) | No staining | No staining |
| Cutaneous leiomyomas (n = 5) | 0/5 (0%) | No staining | No staining |
| Neurofibroma (n = 8) | 8/8 (100%) | Heterogeneous 8/8 (100%) | Weak/moderate 8/8 (100%) |
| Solitary fibrous tumor (n =5) | 0/5 (0%) | No staining | No staining |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piombino, E.; Broggi, G.; Barbareschi, M.; Castorina, S.; Parenti, R.; Bartoloni, G.; Salvatorelli, L.; Magro, G. Wilms’ Tumor 1 (WT1): A Novel Immunomarker of Dermatofibrosarcoma Protuberans—An Immunohistochemical Study on a Series of 114 Cases of Bland-Looking Mesenchymal Spindle Cell Lesions of the Dermis/Subcutaneous Tissues. Cancers 2021, 13, 252. https://doi.org/10.3390/cancers13020252
Piombino E, Broggi G, Barbareschi M, Castorina S, Parenti R, Bartoloni G, Salvatorelli L, Magro G. Wilms’ Tumor 1 (WT1): A Novel Immunomarker of Dermatofibrosarcoma Protuberans—An Immunohistochemical Study on a Series of 114 Cases of Bland-Looking Mesenchymal Spindle Cell Lesions of the Dermis/Subcutaneous Tissues. Cancers. 2021; 13(2):252. https://doi.org/10.3390/cancers13020252
Chicago/Turabian StylePiombino, Eliana, Giuseppe Broggi, Mattia Barbareschi, Sergio Castorina, Rosalba Parenti, Giovanni Bartoloni, Lucia Salvatorelli, and Gaetano Magro. 2021. "Wilms’ Tumor 1 (WT1): A Novel Immunomarker of Dermatofibrosarcoma Protuberans—An Immunohistochemical Study on a Series of 114 Cases of Bland-Looking Mesenchymal Spindle Cell Lesions of the Dermis/Subcutaneous Tissues" Cancers 13, no. 2: 252. https://doi.org/10.3390/cancers13020252
APA StylePiombino, E., Broggi, G., Barbareschi, M., Castorina, S., Parenti, R., Bartoloni, G., Salvatorelli, L., & Magro, G. (2021). Wilms’ Tumor 1 (WT1): A Novel Immunomarker of Dermatofibrosarcoma Protuberans—An Immunohistochemical Study on a Series of 114 Cases of Bland-Looking Mesenchymal Spindle Cell Lesions of the Dermis/Subcutaneous Tissues. Cancers, 13(2), 252. https://doi.org/10.3390/cancers13020252