The Giant HECT E3 Ubiquitin Ligase HERC1 Is Aberrantly Expressed in Myeloid Related Disorders and It Is a Novel BCR-ABL1 Binding Partner

 ,
, 
 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
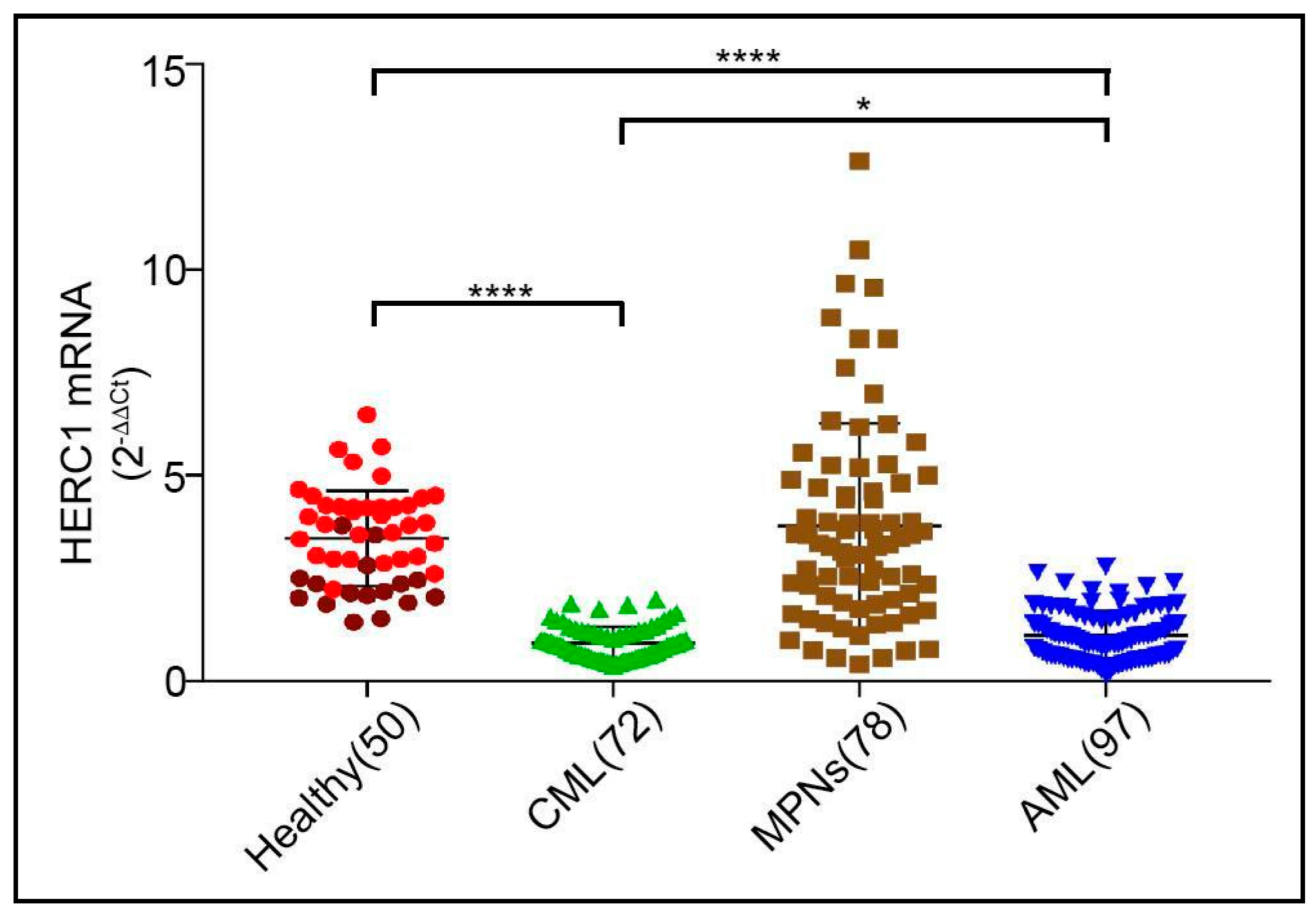
2.1. HERC1 Gene Expression Is Aberrantly Regulated in Myeloid Related Disorders and Leukemias
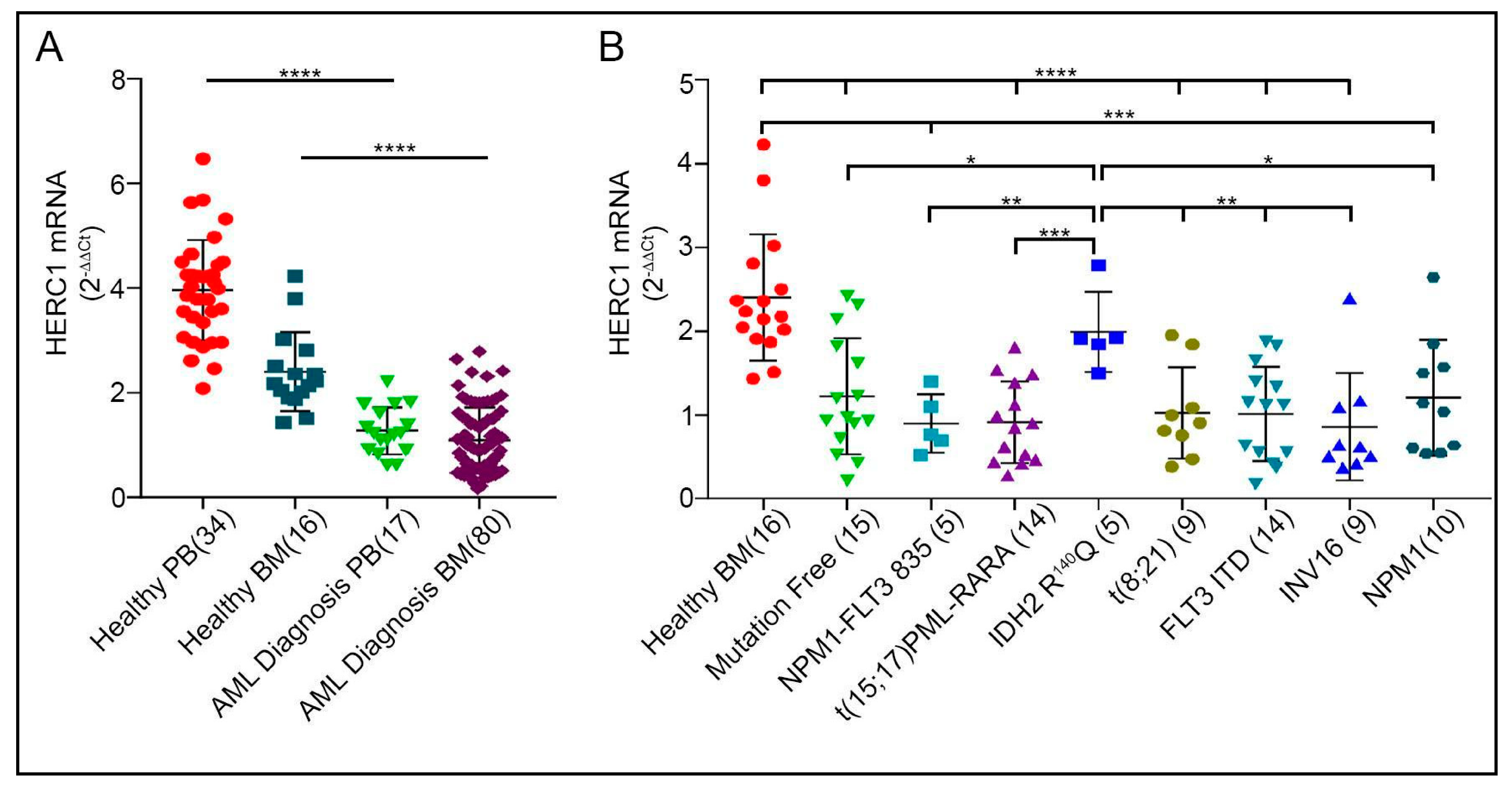
2.2. Newly Diagnosed AML Display HERC1 Down-Regulation Independently from the Genetic Alterations
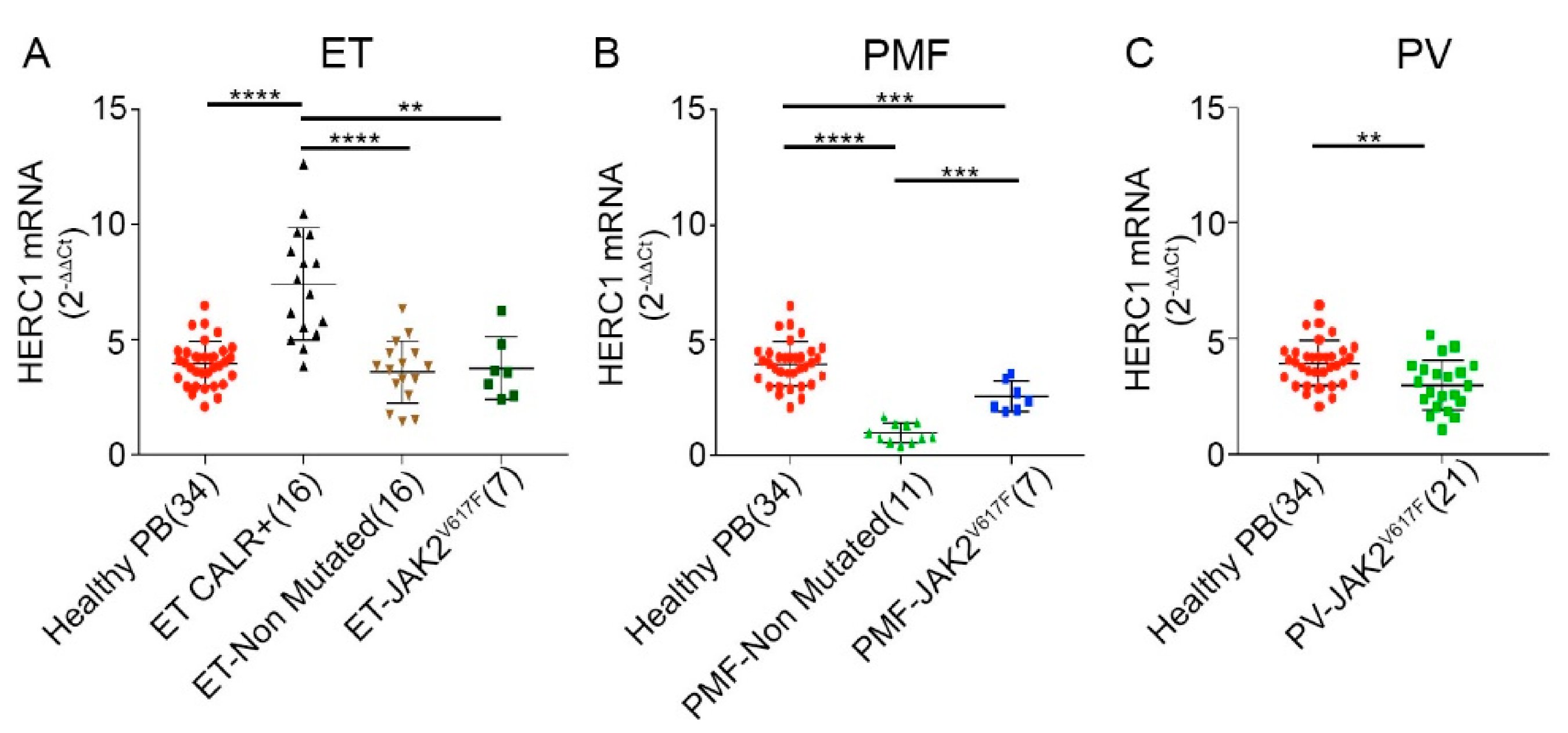
2.3. MPNs Display a Peculiar Pattern of HERC1 mRNA Abundance
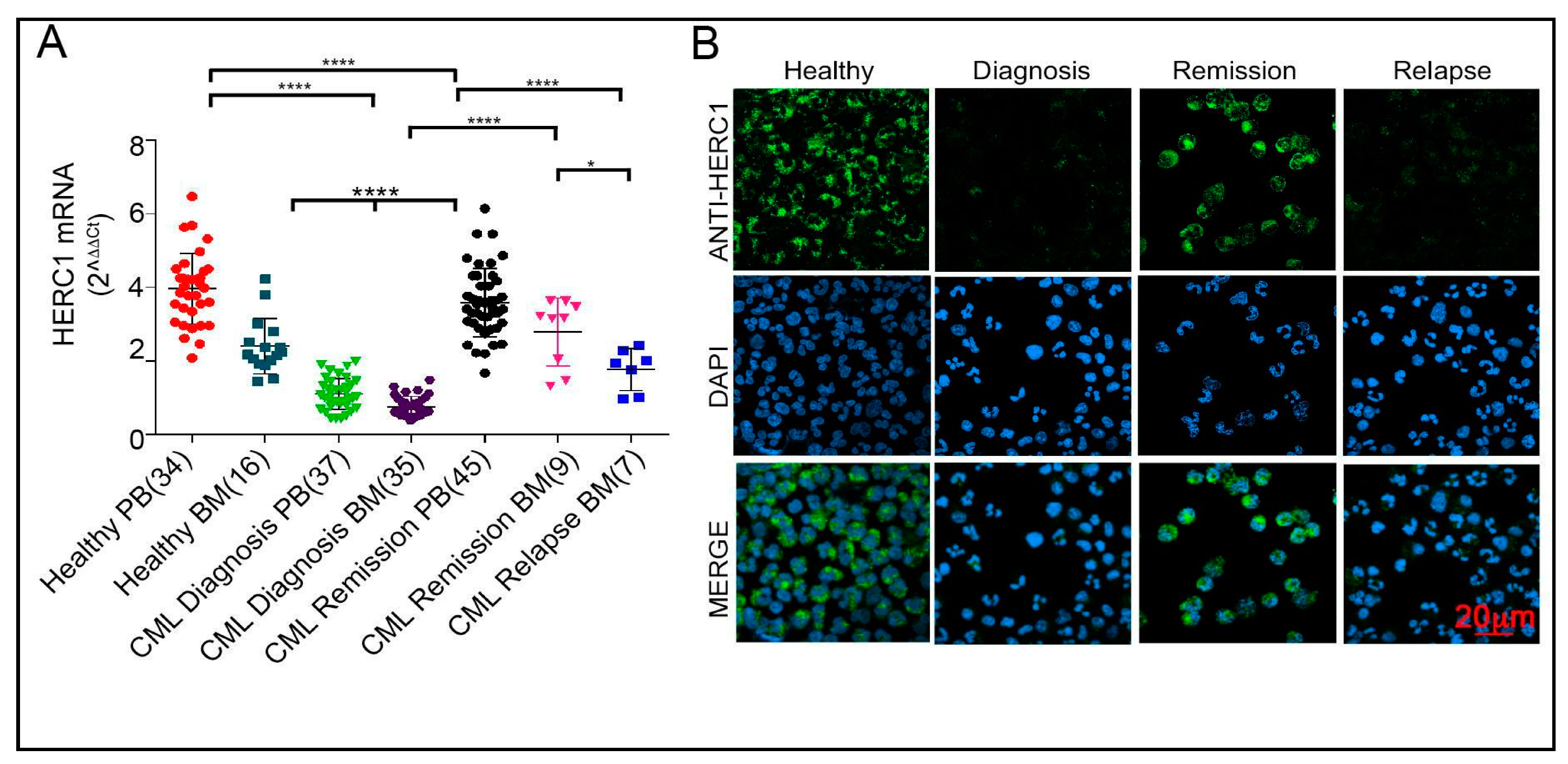
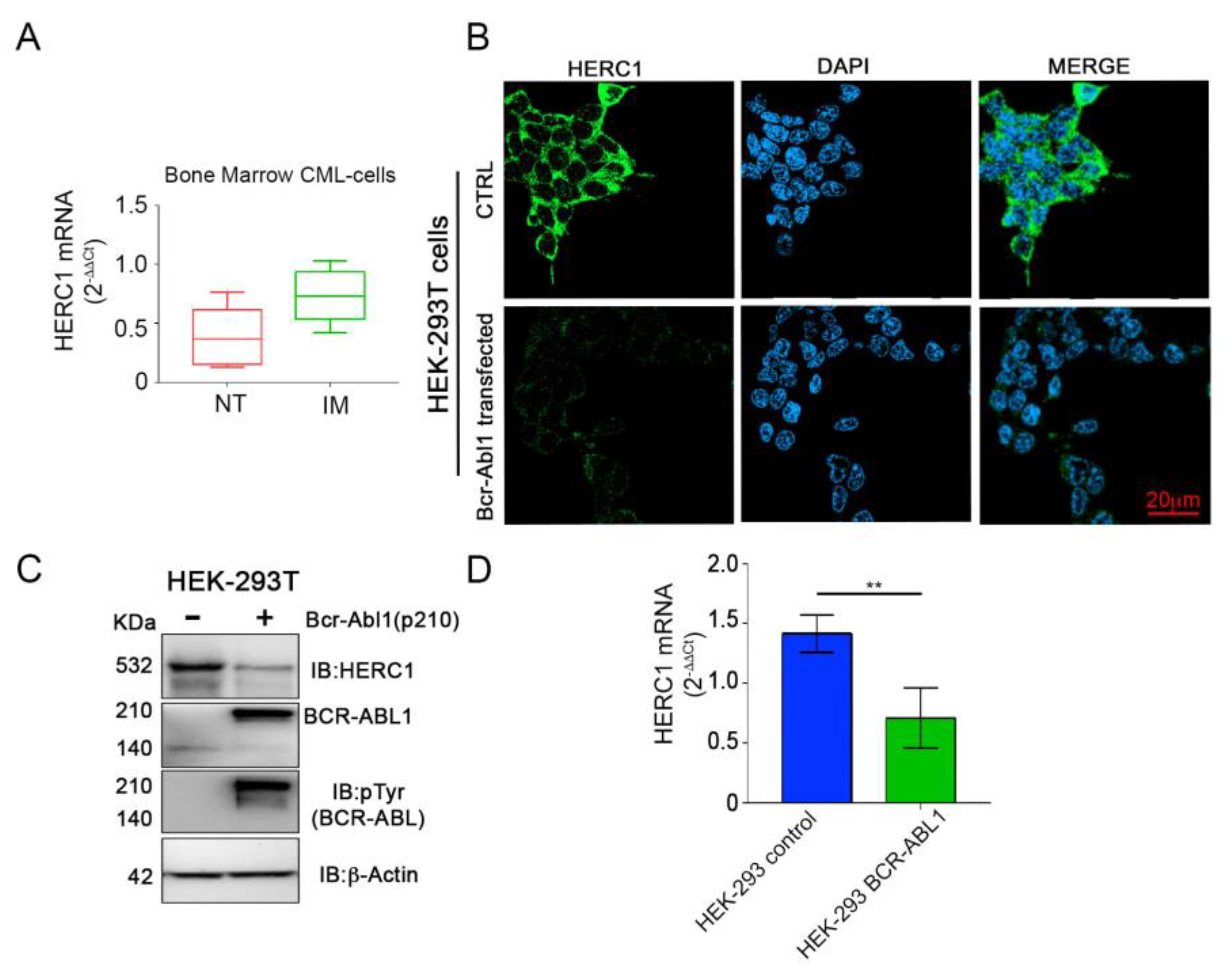
2.4. The HERC1 Expression Is Sensitive to the Bcr-Abl1 Tyrosine Kinase Activity
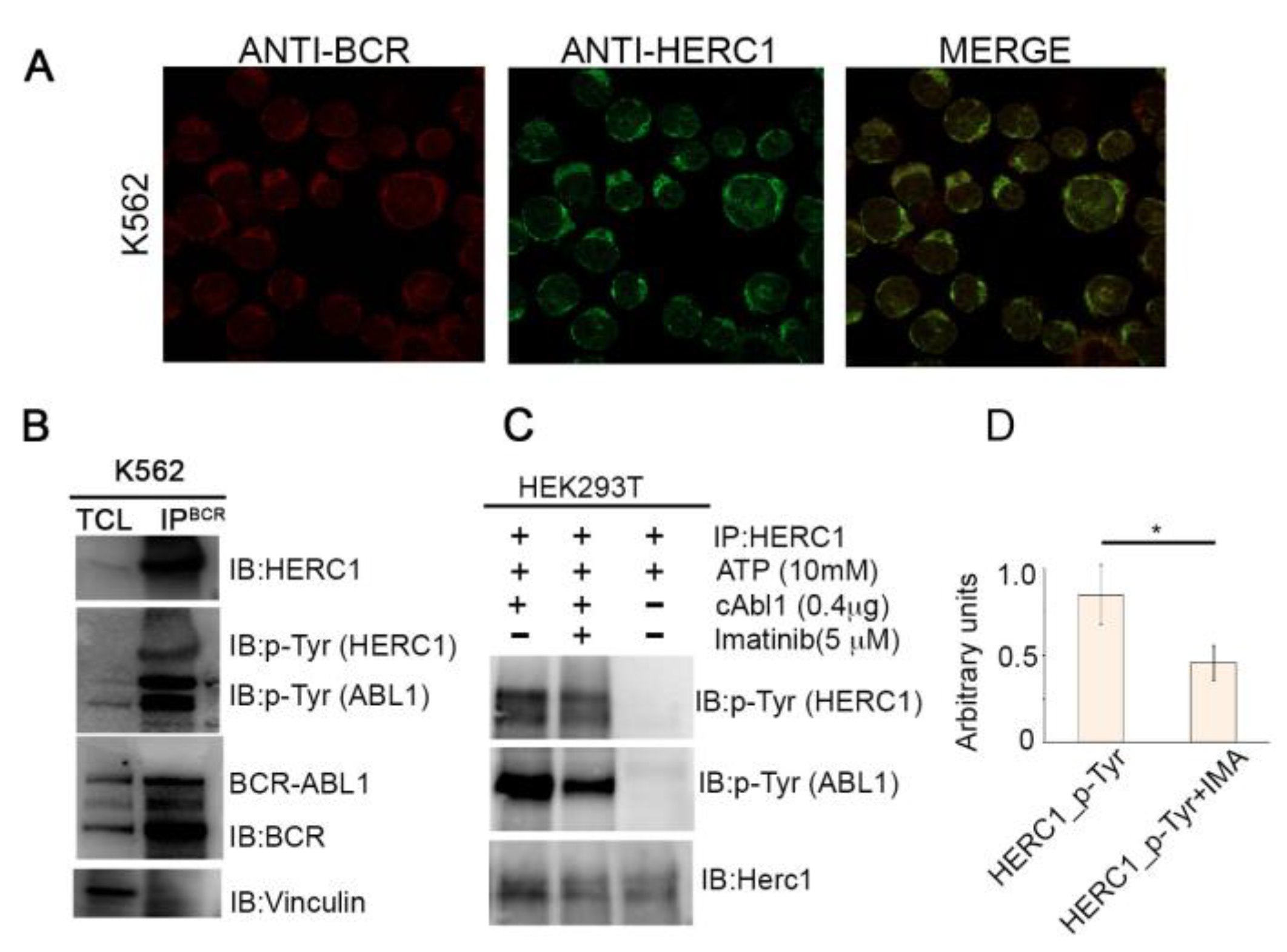
2.5. HERC1 Is a Novel Binding Partner and a Substrate of BCR-ABL1
3. Discussion
4. Materials and Methods
4.1. Patients and Cell Lines
4.2. Quantitative Real-Time PCR
4.3. Cell Lysis and Immunoprecipitation
4.4. Western Blotting
4.5. Immunofluorescence Staining
4.6. Plasmids and Transfection
4.7. In-Vitro Kinase Assay
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nielsen, C.P.; MacGurn, J.A. Coupling Conjugation and Deconjugation Activities to Achieve Cellular Ubiquitin Dynamics. Trends Biochem. Sci. 2020, 45, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Pergolizzi, B.; Bozzaro, S.; Bracco, E. Dictyostelium as model for studying ubiquitination and deubiquitination. Int. J. Dev. Biol. 2019, 63, 529–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, J.; Lerman, L.O.; Lerman, A. Ubiquitin and ubiquitin-like proteins in protein regulation. Circ. Res. 2007, 100, 1276–1291. [Google Scholar] [CrossRef] [Green Version]
- Wright, J.D.; Mace, P.D.; Day, C.L. Noncovalent Ubiquitin Interactions Regulate the Catalytic Activity of Ubiquitin Writers. Trends Biochem. Sci. 2016, 41, 924–937. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Finley, D.; Ciechanover, A.; Varshavsky, A. Ubiquitin as a central cellular regulator. Cell 2004, 116, S29–S32. [Google Scholar] [CrossRef] [Green Version]
- Groothuis, T.A.M.; Dantuma, N.P.; Neefjes, J.; Salomons, F.A. Ubiquitin crosstalk connecting cellular processes. Cell Div. 2006, 1, 1–7. [Google Scholar] [CrossRef]
- Chaugule, V.K.; Walden, H. Specificity and disease in the ubiquitin system. Biochem. Soc. Trans. 2016, 44, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Marteijn, J.A.F.; Jansen, J.H.; van der Reijden, B.A. Ubiquitylation in normal and malignant hematopoiesis: Novel therapeutic targets. Leukemia 2006, 20, 1511–1518. [Google Scholar] [CrossRef] [Green Version]
- Diouf, B.; Cheng, Q.; Krynetskaia, N.F.; Yang, W.; Cheok, M.; Pei, D.; Fan, Y.; Cheng, C.; Krynetskiy, E.Y.; Geng, H.; et al. Somatic deletions of genes regulating MSH2 protein stability cause DNA mismatch repair deficiency and drug resistance in human leukemia cells. Nat. Med. 2011, 17, 1298–1303. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Vosberg, S.; Schlee, C.; Heesch, S.; Schwartz, S.; Gökbuget, N.; Hoelzer, D.; Graf, A.; Krebs, S.; Bartram, I.; et al. Mutational spectrum of adult T-ALL. Oncotarget 2015, 6, 2754–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opatz, S.; Bamopoulos, S.A.; Metzeler, K.H.; Herold, T.; Ksienzyk, B.; Bräundl, K.; Tschuri, S.; Vosberg, S.; Konstandin, N.P.; Wang, C. The clinical mutatome of core binding factor leukemia. Leukemia 2020, 34, 1553–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, P.; Klein-Hitpass, L.; Choidas, A.; Habenberger, P.; Mahboubi, B.; Kim, B.; Bergmann, A.; Scholtysik, R.; Brauser, M.; Lollies, A.; et al. SAMHD1 is recurrently mutated in T-cell prolymphocytic leukemia. Blood Cancer J. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Walz, C.; Grimwade, D.; Saussele, S.; Lengfelder, E.; Haferlach, C.; Schnittger, S.; Lafage-Pochitaloff, M.; Hochhaus, A.; Cross, N.C.P.; Reiter, A. Atypical mRNA fusions in PML-RARA positive, RARA-PML negative acute promyelocytic leukemia. Genes Chromosom. Cancer 2010, 49. [Google Scholar] [CrossRef]
- Pedrazza, L.; Schneider, T.; Bartrons, R.; Ventura, F.; Rosa, J.L. The ubiquitin ligase HERC1 regulates cell migration via RAF-dependent regulation of MKK3/p38 signaling. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Pergolizzi, B.; Bracco, E.; Bozzaro, S. A new HECT ubiquitin ligase regulating chemotaxis and development in Dictyostelium discoideum. J. Cell Sci. 2017, 130, 551–562. [Google Scholar] [CrossRef] [Green Version]
- Panuzzo, C.; Signorino, E.; Calabrese, C.; Ali, M.S.; Petiti, J.; Bracco, E.; Cilloni, D. Landscape of Tumor Suppressor Mutations in Acute Myeloid Leukemia. J. Clin. Med. 2020, 9, 802. [Google Scholar] [CrossRef] [Green Version]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- Spivak, J.L. Myeloproliferative neoplasms. N. Engl. J. Med. 2017, 376, 2168–2181. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Tena, S.; Cubillos-Rojas, M.; Schneider, T.; Rosa, J.L. Functional and pathological relevance of HERC family proteins: A decade later. Cell. Mol. Life Sci. 2016, 73, 1955–1968. [Google Scholar] [CrossRef] [PubMed]
- Sala-Gaston, J.; Martinez-Martinez, A.; Pedrazza, L.; Lorenzo-Martín, L.F.; Caloto, R.; Bustelo, X.R.; Ventura, F.; Rosa, J.L. Herc ubiquitin ligases in cancer. Cancers 2020, 12, 1653. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Argiles-Castillo, D.; Kane, E.I.; Zhou, A.; Spratt, D.E. HECT E3 ubiquitin ligases—Emerging insights into their biological roles and disease relevance. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Galligan, J.T.; Martinez-Noël, G.; Arndt, V.; Hayes, S.; Chittenden, T.W.; Harper, J.W.; Howley, P.M. Proteomic analysis and identification of cellular interactors of the giant ubiquitin ligase HERC2. J. Proteome Res. 2015, 14, 953–966. [Google Scholar] [CrossRef] [Green Version]
- Preisinger, C.; Schwarz, J.P.; Bleijerveld, O.B.; Corradini, E.; Müller, P.J.; Anderson, K.I.; Kolch, W.; Scholten, A.; Heck, A.J.R. Imatinib-dependent tyrosine phosphorylation profiling of Bcr-Abl-positive chronic myeloid leukemia cells. Leukemia 2013, 27, 743–746. [Google Scholar] [CrossRef]
- Park, C.W.; Ryu, K.Y. Cellular ubiquitin pool dynamics and homeostasis. BMB Rep. 2014, 47, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Bernassola, F.; Chillemi, G.; Melino, G. HECT-Type E3 Ubiquitin Ligases in Cancer. Trends Biochem. Sci. 2019, 44, 1057–1075. [Google Scholar] [CrossRef]
- Senft, D.; Qi, J.; Ronaiev, A. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Publ. Gr. 2017, 18. [Google Scholar] [CrossRef]
- Marteijn, J.A.F.; Van Emst, L.; Erpelinck-Verschueren, C.A.J.; Nikoloski, G.; Menke, A.; De Witte, T.; Löwenberg, B.; Jansen, J.H.; Van Der Reijden, B.A. The E3 ubiquitin-protein ligase Triad1 inhibits clonogenic growth of primary myeloid progenitor cells. Blood 2005, 106, 4114–4123. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Bei, L.; Shah, C.A.; Huang, W.; Platanias, L.C.; Eklund, E.A. The E3 ubiquitin ligase Triad1 influences development of Mll-Ell-induced acute myeloid leukemia. Oncogene 2018, 37, 2532–2544. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Bei, L.; Shah, C.A.; Horvath, E.; Eklund, E.A. HoxA10 influences protein ubiquitination by activating transcription of ARIH2, the gene encoding Triad1. J. Biol. Chem. 2011, 286, 16832–16845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanarico, A.G.; Ronchini, C.; Croce, A.; Memmi, E.M.; Cammarata, U.A.; De Antoni, A.; Lavorgna, S.; DIvona, M.; Giacò, L.; Melloni, G.E.M.; et al. The E3 ubiquitin ligase WWP1 sustains the growth of acute myeloid leukaemia. Leukemia 2018, 32, 911–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molenaar, R.J.; Maciejewski, J.P.; Wilmink, J.W.; Van Noorden, C.J.F. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 2018, 37, 1949–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uvarov, A.V.; Mesaeli, N. Enhanced ubiquitin-proteasome activity in calreticulin deficient cells: A compensatory mechanism for cell survival. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 1237–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deegan, S.; Koryga, I.; Glynn, S.A.; Gupta, S.; Gorman, A.M.; Samali, A. A close connection between the PERK and IRE arms of the UPR and the transcriptional regulation of autophagy. Biochem. Biophys. Res. Commun. 2015, 456, 305–311. [Google Scholar] [CrossRef]
- Schneider, T.; Martinez-Martinez, A.; Cubillos-Rojas, M.; Bartrons, R.; Ventura, F.; Rosa, J.L. The E3 ubiquitin ligase HERC1 controls the ERK signaling pathway targeting C-RAF for degradation. Oncotarget 2018, 9, 31531–31548. [Google Scholar] [CrossRef]
- Reckel, S.; Hamelin, R.; Georgeon, S.; Armand, F.; Jolliet, Q.; Chiappe, D.; Moniatte, M.; Hantschel, O. Differential signaling networks of Bcr-Abl p210 and p190 kinases in leukemia cells defined by functional proteomics. Leukemia 2017, 31, 1502–1512. [Google Scholar] [CrossRef]
- Antonenko, S.V.; Telegeev, G.D. Inhibition of USP1, a new partner of Bcr-Abl, results in decrease of Bcr-Abl level in K562 cells. Exp. Oncol. 2020, 42, 109–114. [Google Scholar] [CrossRef]
- Mao, J.H.; Sun, X.Y.; Liu, J.X.; Zhang, Q.Y.; Liu, P.; Huang, Q.H.; Li, K.K.; Chen, Q.; Chen, Z.; Chen, S.J. As4S4 targets RING-type E3 ligase c-CBL to induce degradation of BCR-ABL in chronic myelogenous leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 21683–21688. [Google Scholar] [CrossRef] [Green Version]
- Persaud, A.; Alberts, P.; Mari, S.; Tong, J.; Murchie, R.; Maspero, E.; Safi, F.; Moran, M.F.; Polo, S.; Rotin, D. Tyrosine phosphorylation of NEDD4 activates its ubiquitin ligase activity. Sci. Signal. 2014, 7, ra95. [Google Scholar] [CrossRef]
- Rosso, V.; Panuzzo, C.; Petiti, J.; Carturan, S.; Dragani, M.; Andreani, G.; Fava, C.; Saglio, G.; Bracco, E.; Cilloni, D. Reduced Expression of Sprouty1 Contributes to the Aberrant Proliferation and Impaired Apoptosis of Acute Myeloid Leukemia Cells. J. Clin. Med. 2019, 8, 972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubillos-Rojas, M.; Amair-Pinedo, F.; Tato, I.; Bartrons, R.; Ventura, F.; Rosa, J.L. Tris-acetate polyacrylamide gradient gels for the simultaneous electrophoretic analysis of proteins of very high and low molecular mass. Methods Mol. Biol. 2012, 869, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.S.; Gill, K.S.; Saglio, G.; Cilloni, D.; Soden, D.M.; Forde, P.F. Expressional changes in stemness markers post electrochemotherapy in pancreatic cancer cells. Bioelectrochemistry 2018, 122, 84–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morotti, A.; Panuzzo, C.; Crivellaro, S.; Pergolizzi, B.; Familiari, U.; Berger, A.H.; Saglio, G.; Pandolfi, P.P. BCR-ABL disrupts PTEN nuclear-cytoplasmic shuttling through phosphorylation-dependent activation of HAUSP. Leukemia 2014, 28, 1326–1333. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.S.; Panuzzo, C.; Calabrese, C.; Maglione, A.; Piazza, R.; Cilloni, D.; Saglio, G.; Pergolizzi, B.; Bracco, E. The Giant HECT E3 Ubiquitin Ligase HERC1 Is Aberrantly Expressed in Myeloid Related Disorders and It Is a Novel BCR-ABL1 Binding Partner. Cancers 2021, 13, 341. https://doi.org/10.3390/cancers13020341
Ali MS, Panuzzo C, Calabrese C, Maglione A, Piazza R, Cilloni D, Saglio G, Pergolizzi B, Bracco E. The Giant HECT E3 Ubiquitin Ligase HERC1 Is Aberrantly Expressed in Myeloid Related Disorders and It Is a Novel BCR-ABL1 Binding Partner. Cancers. 2021; 13(2):341. https://doi.org/10.3390/cancers13020341
Chicago/Turabian StyleAli, Muhammad Shahzad, Cristina Panuzzo, Chiara Calabrese, Alessandro Maglione, Rocco Piazza, Daniela Cilloni, Giuseppe Saglio, Barbara Pergolizzi, and Enrico Bracco. 2021. "The Giant HECT E3 Ubiquitin Ligase HERC1 Is Aberrantly Expressed in Myeloid Related Disorders and It Is a Novel BCR-ABL1 Binding Partner" Cancers 13, no. 2: 341. https://doi.org/10.3390/cancers13020341
APA StyleAli, M. S., Panuzzo, C., Calabrese, C., Maglione, A., Piazza, R., Cilloni, D., Saglio, G., Pergolizzi, B., & Bracco, E. (2021). The Giant HECT E3 Ubiquitin Ligase HERC1 Is Aberrantly Expressed in Myeloid Related Disorders and It Is a Novel BCR-ABL1 Binding Partner. Cancers, 13(2), 341. https://doi.org/10.3390/cancers13020341