
Microenvironmental Reactive Oxygen Species in Colorectal Cancer: Involved Processes and Therapeutic Opportunities

 , ,
, ,
Abstract
:Simple Summary
Abstract
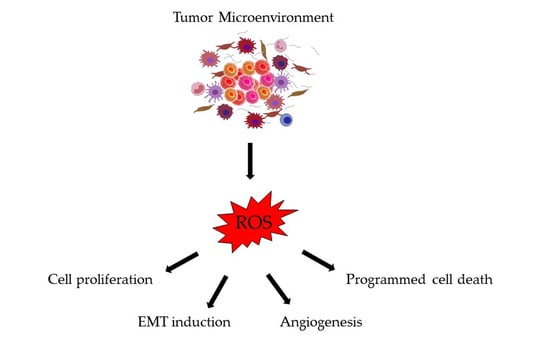
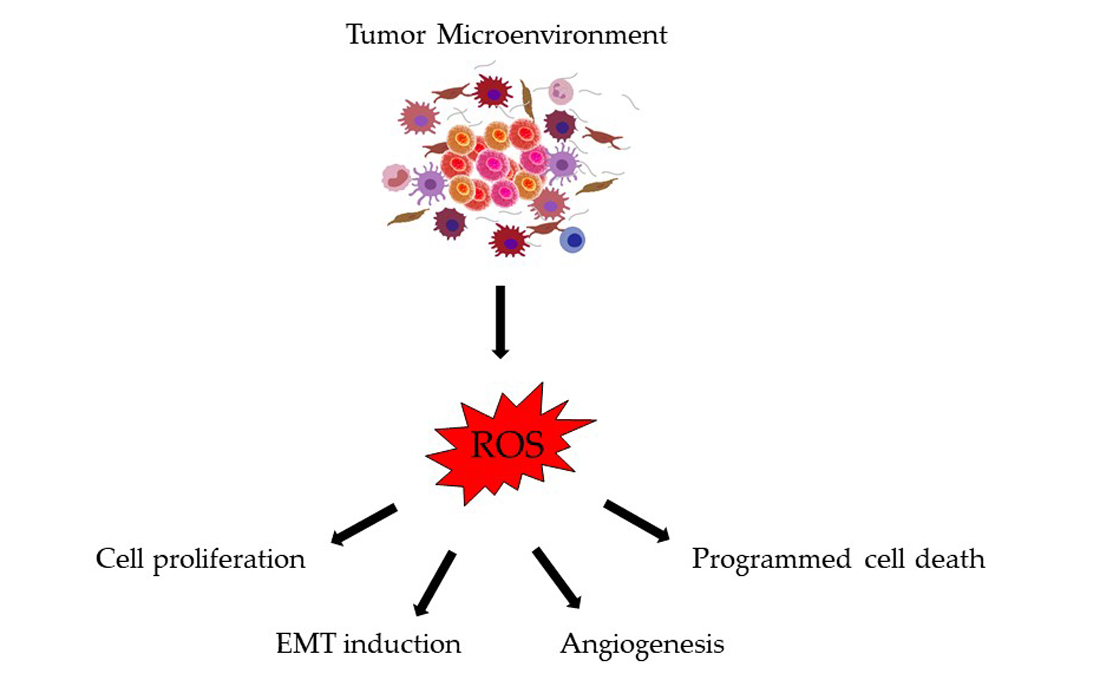
1. Introduction
2. Oxidative Stress in Tumorigenesis
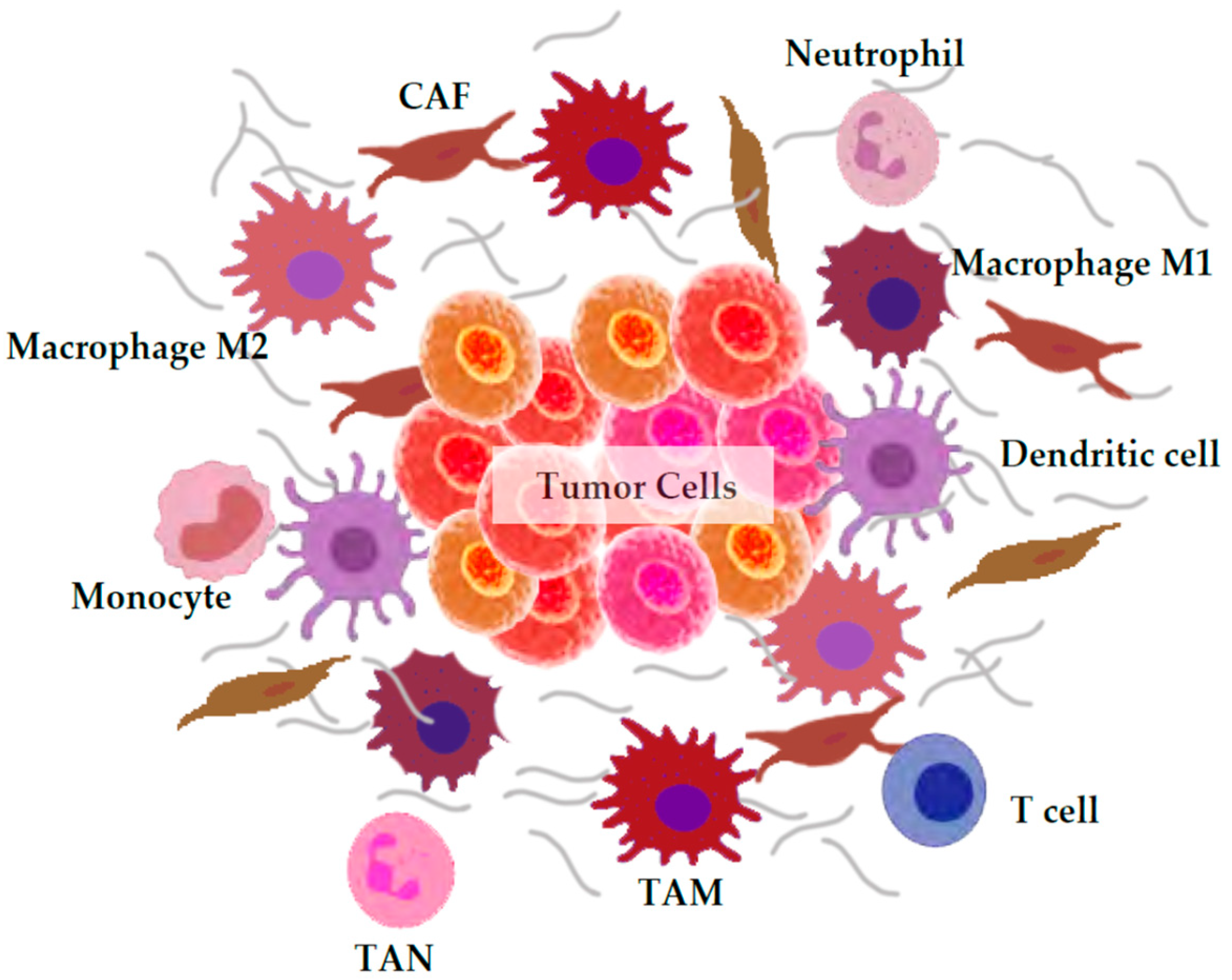
3. ROS Generation by the Tumor Microenvironment
3.1. Macrophages
3.2. Neutrophils
3.3. Cancer-Associated Fibroblasts (CAFs)
3.4. Others
4. Consequences of Oxidative Stress in Colorectal Cancer Progression and Metastasis
4.1. Cell Proliferation
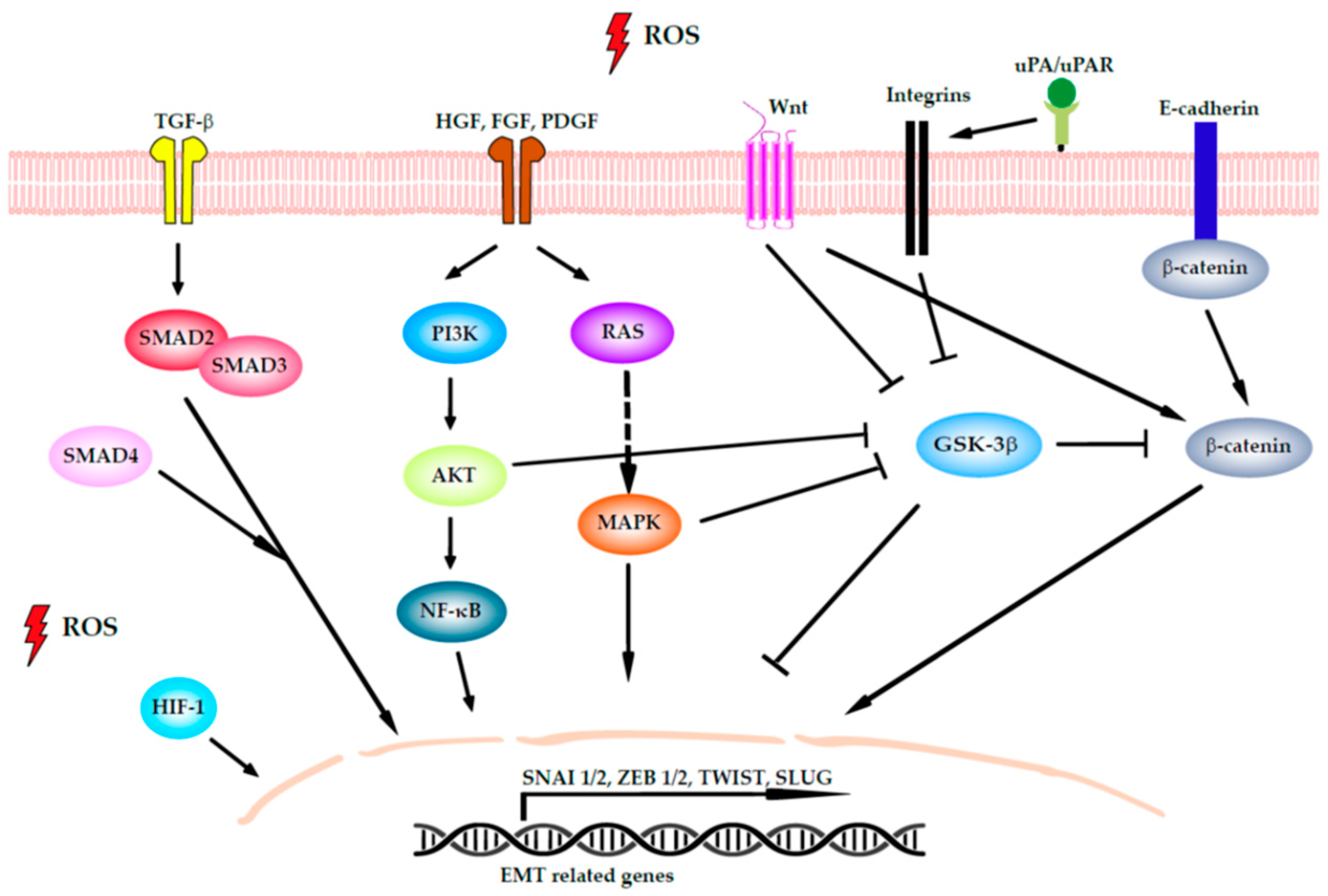
4.2. Induction of EMT
4.3. Angiogenesis
4.4. Apoptosis, Autophagy, and Anoikis
5. Pro-Oxidants and Antioxidants in Colorectal Cancer Therapeutics
5.1. Pro-Oxidants
5.1.1. Oxaliplatin
5.1.2. Arsenic Trioxide (AT)
5.1.3. Tyrosine Kinase Inhibitors (TKIs)
5.1.4. Endoplasmic Reticulum (ER) Stress Inductors
5.1.5. Novel Anthracyclines: AVA6000
5.1.6. Poly (ADP-Ribose) Polymerase (PARP) Inhibitors
5.2. Antioxidant Treatments or Interventions
5.2.1. Dietary Supplementation of Vitamins
5.2.2. Trace Element Supplementation
5.2.3. SOD Mimetics
5.2.4. Polyphenols
5.2.5. Organosulfur Compounds
5.3. Future Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Kennel, K.B.; Greten, F.R. Immune cell-produced ROS and their impact on tumor growth and metastasis. Redox Biol. 2021, 42, 101891. [Google Scholar] [CrossRef]
- Del Idelchik, M.P.S.; Begley, U.; Begley, T.J.; Melendez, J.A. Mitochondrial ROS control of cancer. Semin. Cancer Biol. 2017, 47, 57–66. [Google Scholar] [CrossRef]
- Saikolappan, S.; Kumar, B.; Shishodia, G.; Koul, S.; Koul, H.K. Reactive oxygen species and cancer: A complex interaction. Cancer Lett. 2019, 452, 132–143. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [Green Version]
- Aboelella, N.S.; Brandle, C.; Kim, T.; Ding, Z.C.; Zhou, G. Oxidative stress in the tumor microenvironment and its relevance to cancer immunotherapy. Cancers 2021, 13, 986. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 287. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Bone 2013, 23, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Akella, N.M.; Ciraku, L.; Reginato, M.J. Fueling the fire: Emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biol. 2019, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Locasale, J.W. Serine, Glycine and the one-carbon cycle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [Green Version]
- Baenke, F.; Peck, B.; Miess, H.; Schulze, A. Hooked on fat: The role of lipid synthesis in cancer metabolism and tumour development. DMM Dis. Model. Mech. 2013, 6, 1353–1363. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2008, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, B.; Huang, T.; Sun, Y.; Jin, Z.; Li, X.F. Revisit 18F-fluorodeoxyglucose oncology positron emission tomography: “systems molecular imaging” of glucose metabolism. Oncotarget 2017, 8, 43536–43542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, S.; Badana, A.K.; Murali Mohan, G.; Shailender, G.; Malla, R.R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS generation and antioxidant defense systems in normal and malignant cells. Oxid. Med. Cell. Longev. 2019. [Google Scholar] [CrossRef]
- Martínez, M.C.; Andriantsitohaina, R. Reactive nitrogen species: Molecular mechanisms and potential significance in health and disease. Antioxid. Redox Signal. 2009, 11, 669–702. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive Oxygen Species in Cancer. Free Radic Res 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-clermont, P.J. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997, 14, 1649–1652. [Google Scholar] [CrossRef]
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 1999, 274, 7936–7940. [Google Scholar] [CrossRef] [Green Version]
- Son, Y.; Cheong, Y.-K.; Kim, N.-H.; Chung, H.-T.; Kang, D.G.; Pae, H.-O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduct. 2011, 2011, 1–6. [Google Scholar] [CrossRef]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C.P. Redox regulation of PI 3-kinase signalling via PTEN. EMBO J 2003, 22, 5501–5510. [Google Scholar] [CrossRef]
- Kodama, R.; Kato, M.; Furuta, S.; Ueno, S.; Zhang, Y.; Matsuno, K.; Yabe-Nishimura, C.; Tanaka, E.; Kamata, T. ROS-generating oxidases Nox1 and Nox4 contribute to oncogenic Ras-induced premature senescence. Genes Cells 2013, 18, 32–41. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Chandra, J.; Samali, A.; Orrenius, S. Triggering and modulation of apoptosis by oxidative stress. Free Radic. Biol. Med. 2000, 29, 323–333. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- López-Lázaro, M. Dual role of hydrogen peroxide in cancer: Possible relevance to cancer chemoprevention and therapy. Cancer Lett. 2007, 252, 1–8. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef] [PubMed]
- Jarosz, M.; Sekuła, W.; Rychlik, E. Trends in dietary patterns, alcohol intake, tobacco smoking, and colorectal cancer in polish population in 1960–2008. Biomed Res. Int. 2013, 2013, 183204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappelman, M.D.; Farkas, D.K.; Long, M.D.; Erichsen, R.; Sandler, R.S.; Sørensen, H.T.; Baron, J.A. Risk of cancer in patients with inflammatory bowel diseases: A nationwide population-based cohort study with 30 years of follow-up evaluation. Clin. Gastroenterol. Hepatol. 2014, 176, 100–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinu, D.; Dobre, M.; Panaitescu, E.; Bîrlă, R.; Iosif, C.; Hoara, P.; Caragui, A.; Boeriu, M.; Constantinoiu, S.; Ardeleanu, C. Prognostic significance of KRAS gene mutations in colorectal cancer-preliminary study. J. Med. Life 2014, 7, 581–587. [Google Scholar]
- Lim, J.K.M.; Leprivier, G. The impact of oncogenic RAS on redox balance and implications for cancer development. Cell Death Dis. 2019, 10, 955. [Google Scholar] [CrossRef]
- Mitsushita, J.; Lambeth, J.D.; Kamata, T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Res. 2004, 64, 3580–3585. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Li, C.; Cheng, N.; Cui, X.; Xu, X.; Zhou, G. Redox regulation in cancer stem cells. Oxid. Med. Cell. Longev. 2015, 2015, 750798. [Google Scholar] [CrossRef] [Green Version]
- Dando, I.; Cordani, M.; Pozza, E.D.; Biondani, G.; Donadelli, M.; Palmieri, M. Antioxidant mechanisms and ROS-related MicroRNAs in cancer stem cells. Oxid. Med. Cell. Longev. 2015, 2015, 425708. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Tseng, H.; Chen, Y.; Tanzih, A.; Haq, A.; Hwang, P.; Hsu, H. Oligo-Fucoidan Prevents M2 Macrophage Differentiation and HCT116 Tumor Progression. Cancers 2020, 12, 421. [Google Scholar] [CrossRef] [Green Version]
- Mariani, F.; Sena, P.; Roncucci, L. Inflammatory pathways in the early steps of colorectal cancer development. World, J. Gastroenterol. 2014, 20, 9716–9731. [Google Scholar] [CrossRef]
- Nishikawa, M. Reactive oxygen species in tumor metastasis. Cancer Lett. 2008, 266, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Mytar, B.; Woloszyn, M.; Macura-Biegun, A.; Hajto, B.; Ruggiero, I.; Piekarska, B.; Zembala, M. Involvement of pattern recognition receptors in the induction of cytokines and reactive oxygen intermediates production by human monocytes/macrophages stimulated with tumour cells. Anticancer Res. 2004, 24, 2287–2293. [Google Scholar] [PubMed]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, R.; Kawada, K.; Itatani, Y.; Ogawa, R.; Kiyasu, Y.; Sakai, Y. The role of tumor-associated neutrophils in colorectal cancer. Int. J. Mol. Sci. 2019, 20, 529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winterbourn, C.C.; Kettle, A.J.; Hampton, M.B. Reactive Oxygen Species and Neutrophil Function. Annu. Rev. Biochem. 2016, 85, 765–792. [Google Scholar] [CrossRef]
- Canli, Ö.; Nicolas, A.M.; Gupta, J.; Finkelmeier, F.; Goncharova, O.; Pesic, M.; Neumann, T.; Horst, D.; Löwer, M.; Sahin, U.; et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell 2017, 32, 869–883. [Google Scholar] [CrossRef] [Green Version]
- Rao, H.L.; Chen, J.W.; Li, M.; Xiao, Y.B.; Fu, J.; Zeng, Y.X.; Cai, M.Y.; Xie, D. Increased intratumoral neutrophil in colorectal carcinomas correlates closely with malignant phenotype and predicts patients’ adverse prognosis. PLoS ONE 2012, 7, e30806. [Google Scholar] [CrossRef] [Green Version]
- Berry, R.S.; Xiong, M.J.; Greenbaum, A.; Mortaji, P.; Nofchissey, R.A.; Schultz, F.; Martinez, C.; Luo, L.; Morris, K.T.; Hanson, J.A. High levels of tumor-associated neutrophils are associated with improved overall survival in patients with stage II colorectal cancer. PLoS ONE 2017, 12, e0188799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.S.K.; Tan, M.J.; Sng, M.K.; Teo, Z.; Phua, T.; Choo, C.C.; Li, L.; Zhu, P.; Tan, N.S. Cancer-associated fibroblasts enact field cancerization by promoting extratumoral oxidative stress. Cell Death Dis. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Onfroy-Roy, L.; Hamel, D.; Malaquin, L.; Ferrand, A. Colon fibroblasts and inflammation: Sparring partners in colorectal cancer initiation? Cancers 2021, 13, 1749. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010, 9, 3256–3276. [Google Scholar] [CrossRef] [Green Version]
- Policastro, L.L.; Ibañez, I.L.; Notcovich, C.; Duran, H.A.; Podhajcer, O.L. The tumor microenvironment: Characterization, redox considerations, and novel approaches for reactive oxygen species-targeted gene therapy. Antioxid. Redox Signal. 2013, 19, 854–895. [Google Scholar] [CrossRef]
- Qu, P.; Boelte, K.C.; Lin, P.C. Negative regulation of myeloid-derived suppressor cells in cancer. Immunol. Investig. 2012, 41, 562–580. [Google Scholar] [CrossRef]
- Talmadge, J.E. Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clin. Cancer Res. 2007, 13, 5243–5248. [Google Scholar] [CrossRef] [Green Version]
- Toyokuni, S.; Okamoto, K.; Yodoi, J.; Hiai, H. Persistent oxidative stress in cancer. FEBS Lett. 1995, 358, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Savina, A.; Jancic, C.; Hugues, S.; Guermonprez, P.; Vargas, P.; Moura, I.C.; Lennon-Duménil, A.M.; Seabra, M.C.; Raposo, G.; Amigorena, S. NOX2 Controls Phagosomal pH to Regulate Antigen Processing during Crosspresentation by Dendritic Cells. Cell 2006, 126, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Aceto, G.M.; Catalano, T.; Curia, M.C.; Tong, Q. Molecular Aspects of Colorectal Adenomas: The Interplay among Microenvironment, Oxidative Stress, and Predisposition. Biomed. Res. Int. 2020, 2020, 1726309. [Google Scholar] [CrossRef] [PubMed]
- Vodicka, P.; Urbanova, M.; Makovicky, P.; Tomasova, K.; Kroupa, M.; Stetina, R.; Opattova, A.; Kostovcikova, K.; Siskova, A.; Schneiderova, M.; et al. Oxidative damage in sporadic colorectal cancer: Molecular mapping of base excision repair glycosylases in colorectal cancer patients. Int. J. Mol. Sci. 2020, 21, 2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, R.S.; Shi, J.; Murad, E.; Whalen, A.M.; Sun, C.Q.; Polavarapu, R.; Parthasarathy, S.; Petros, J.A.; Lambeth, J.D. Hydrogen peroxide mediates the cell growth and transformation caused by the mitogenic oxidase Nox1. Proc. Natl. Acad. Sci. USA 2001, 98, 5550–5555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szatrowski, T.P.; Nathan, C.F. Production of Large Amounts of Hydrogen Peroxide by Human Tumor Cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Policastro, L.; Molinari, B.; Larcher, F.; Blanco, P.; Podhajcer, O.L.; Costa, C.S.; Rojas, P.; Durán, H. Imbalance of Antioxidant Enzymes in Tumor Cells and Inhibition of Proliferation and Malignant Features by Scavenging Hydrogen Peroxide. Mol. Carcinog. 2004, 39, 103–113. [Google Scholar] [CrossRef]
- De Oliveira, V.A.; da Motta, L.L.; De Bastiani, M.A.; Lopes, F.M.; Müller, C.B.; Gabiatti, B.P.; França, F.S.; Castro, M.A.A.; Klamt, F. In vitro evaluation of antitumoral efficacy of catalase in combination with traditional chemotherapeutic drugs against human lung adenocarcinoma cells. Tumor Biol. 2016, 37, 10775–10784. [Google Scholar] [CrossRef]
- Yang, J.Q.; Buettner, G.R.; Domann, F.E.; Li, Q.; Engelhardt, J.F.; Weydert, C.D.; Oberley, L.W. v-Ha-ras mitogenic signaling through superoxide and derived reactive oxygen species. Mol. Carcinog. 2002, 33, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Cullen, J.J.; Weydert, C.; Hinkhouse, M.M.; Ritchie, J.; Domann, F.E.; Spitz, D.; Oberley, L.W. The Role of Manganese Superoxide Dismutase in the Growth of Pancreatic Adenocarcinoma. Cancer Res. 2003, 63, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Juhasz, A.; Markel, S.; Gaur, S.; Liu, H.; Lu, J.; Jiang, G.; Wu, X.; Antony, S.; Wu, Y.; Melillo, G.; et al. NADPH oxidase 1 supports proliferation of colon cancer cells by modulating reactive oxygen species-dependent signal transduction. J. Biol. Chem. 2017, 292, 7866–7887. [Google Scholar] [CrossRef] [Green Version]
- Sithara, T.; Arun, K.B.; Syama, H.P.; Reshmitha, T.R.; Nisha, P. Morin inhibits proliferation of SW480 colorectal cancer cells by inducing apoptosis mediated by reactive oxygen species formation and uncoupling of warburg effect. Front. Pharmacol. 2017, 8, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Obeed, O.; El-Obeid, A.S.; Matou-Nasri, S.; Vaali-Mohammed, M.A.; Alhaidan, Y.; Elwatidy, M.; Al Dosary, H.; Alehaideb, Z.; Alkhayal, K.; Haseeb, A.; et al. Herbal melanin inhibits colorectal cancer cell proliferation by altering redox balance, inducing apoptosis, and modulating MAPK signaling. Cancer Cell Int. 2020, 20, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Al-Khayal, K.; Alafeefy, A.; Vaali-Mohammed, M.A.; Mahmood, A.; Zubaidi, A.; Al-Obeed, O.; Khan, Z.; Abdulla, M.; Ahmad, R. Novel derivative of aminobenzenesulfonamide (3c) induces apoptosis in colorectal cancer cells through ROS generation and inhibits cell migration. BMC Cancer 2017, 17, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, S.; Wang, P.; Toh, A.; Thompson, E.W. New Insights Into the Role of Phenotypic Plasticity and EMT in Driving Cancer Progression. Front. Mol. Biosci. 2020, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Basak, D.; Uddin, M.N.; Hancock, J. The Role of Oxidative Stress and Its Counteractive Utility in Colorectal Cancer (CRC). Cancers 2020, 12, 3336. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, K.; Chen, Y.; Chen, H.; Nice, E.C.; Huang, C. Redox regulation in tumor cell epithelial–mesenchymal transition: Molecular basis and therapeutic strategy. Signal Transduct. Target. Ther. 2017, 2, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Cannito, S.; Novo, E.; Di Bonzo, L.V.; Busletta, C.; Colombatto, S.; Parola, M. Epithelial-mesenchymal transition: From molecular mechanisms, redox regulation to implications in human health and disease. Antioxid. Redox Signal. 2010, 12, 1383–1430. [Google Scholar] [CrossRef] [PubMed]
- Parisi, E.; Sorolla, A.; Montal, R.; González-Resina, R.; Novell, A.; Salud, A.; Sorolla, M.A. Prognostic factors involved in the epithelial–mesenchymal transition process in colorectal cancer have a preponderant role in oxidative stress: A systematic review and meta-analysis. Cancers 2020, 12, 3330. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Xu, E.; Liu, H.; Wan, L.; Lai, M. Epithelial-mesenchymal transition in colorectal cancer metastasis: A system review. Pathol. Res. Pract. 2015, 211, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Goitre, L.; Pergolizzi, B.; Ferro, E.; Trabalzini, L.; Retta, S.F. Molecular Crosstalk between Integrins and Cadherins: Do Reactive Oxygen Species Set the Talk? J. Signal Transduct. 2012, 2012, 1–12. [Google Scholar] [CrossRef]
- Lee, K.H.; Kim, S.W.; Kim, J.R. Reactive oxygen species regulate urokinase plasminogen activator expression and cell invasion via mitogen-activated protein kinase pathways after treatment with hepatocyte growth factor in stomach cancer cells. J. Exp. Clin. Cancer Res. 2009, 28, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, H.; Maurer, F.; Nagamine, Y. Stabilization of Urokinase and Urokinase Receptor mRNAs by HuR Is Linked to Its Cytoplasmic Accumulation Induced by Activated Mitogen-Activated Protein Kinase-Activated Protein Kinase 2. Mol. Cell. Biol. 2003, 23, 7177–7188. [Google Scholar] [CrossRef] [Green Version]
- Tochhawng, L.; Deng, S.; Pugalenthi, G.; Kumar, A.P.; Lim, K.H.; Yang, H.; Hooi, S.C.; Goh, Y.C.; Maciver, S.K.; Pervaiz, S.; et al. Gelsolin-Cu/ZnSOD interaction alters intracellular reactive oxygen species levels to promote cancer cell invasion. Oncotarget 2016, 7, 52832–52848. [Google Scholar] [CrossRef]
- Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers 2019, 16, 1191. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; McNiven, M.A. Invasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK-p130Cas complex. J. Cell Biol. 2012, 196, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, E.; Chiarugi, P. Redox circuitries driving Src regulation. Antioxid. Redox Signal. 2014, 20, 2011–2025. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M. Redox signaling in angiogenesis: Role of NADPH oxidase. Cardiovasc. Res. 2006, 71, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Zimna, A.; Kurpisz, M. Hypoxia-Inducible factor-1 in physiological and pathophysiological angiogenesis: Applications and therapies. Biomed Res. Int. 2015, 2015, 549412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, C.C.; Hamdy, R.C.; Chua, B.H.L. Upregulation of vascular endothelial growth factor by H2O2 in rat heart endothelial cells. Free Radic. Biol. Med. 1998, 25, 891–897. [Google Scholar] [CrossRef]
- Kuroki, M.; Voest, E.E.; Amano, S.; Beerepoot, L.V.; Takashima, S.; Tolentino, M.; Kim, R.Y.; Rohan, R.M.; Colby, K.A.; Yeo, K.T.; et al. Reactive oxygen intermediates increase vascular endothelial growth factor expression in vitro and in vivo. J. Clin. Investig. 1996, 98, 1667–1675. [Google Scholar] [CrossRef] [Green Version]
- Fukai, M.U.; Nakamura, Y. Reactive Oxygen Species and Angiogenesis NADPH Oxidase as. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Khromova, N.V.; Kopnin, P.B.; Stepanova, E.V.; Agapova, L.S.; Kopnin, B.P. p53 hot-spot mutants increase tumor vascularization via ROS-mediated activation of the HIF1/VEGF-A pathway. Cancer Lett. 2009, 276, 143–151. [Google Scholar] [CrossRef]
- Larsen, A.K.; Ouaret, D.; El Ouadrani, K.; Petitprez, A. Targeting EGFR and VEGF(R) pathway cross-talk in tumor survival and angiogenesis. Pharmacol. Ther. 2011, 131, 80–90. [Google Scholar] [CrossRef]
- Cao, D.; Hou, M.; Guan, Y.S.; Jiang, M.; Yang, Y.; Gou, H.F. Expression of HIF-1alpha and VEGF in colorectal cancer: Association with clinical outcomes and prognostic implications. BMC Cancer 2009, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Fan, F.; Wey, J.S.; McCarty, M.F.; Belcheva, A.; Liu, W.; Bauer, T.W.; Somcio, R.J.; Wu, Y.; Hooper, A.; Hicklin, D.J.; et al. Expression and function of vascular endothelial growth factor receptor-1 on human colorectal cancer cells. Oncogene 2005, 24, 2647–2653. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, R.; Fan, F.; Wang, R.; Ye, X.; Xia, L.; Boulbes, D.; Ellis, L.M. Intracrine VEGF signalling mediates colorectal cancer cell migration and invasion. Br. J. Cancer 2017, 117, 848–855. [Google Scholar] [CrossRef] [Green Version]
- Battaglin, F.; Puccini, A.; Intini, R.; Schirripa, M.; Ferro, A.; Bergamo, F.; Lonardi, S.; Zagonel, V.; Lenz, H.J.; Loupakis, F. The role of tumor angiogenesis as a therapeutic target in colorectal cancer. Expert Rev. Anticancer Ther. 2018, 18, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative stress in cancer cell metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Chung, W.-G.; Mirandaa, C.L.; Stevensb, J.F.; Maiera, C.S. Hop proanthocyanidins induce apoptosis, protein carbonylation, and cytoskeleton disorganization in human colorectal adenocarcinoma cells via reactive oxygen species. Food Chem. Toxicol. 2009, 47, 827–836. [Google Scholar] [CrossRef] [Green Version]
- Juan, M.E.; Wenzel, U.; Daniel, H.; Planas, J.M. Resveratrol induces apoptosis through ROS-dependent mitochondria pathway in HT-29 human colorectal carcinoma cells. J. Agric. Food Chem. 2008, 56, 4813–4818. [Google Scholar] [CrossRef]
- Miki, H.; Uehara, N.; Kimura, A.; Sasaki, T.; Yuri, T.; Yoshizawa, K.; Tsubura, A. Resveratrol induces apoptosis via ROS-triggered autophagy in human colon cancer cells. Int. J. Oncol. 2012, 40, 1020–1028. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Kim, N.Y.; Suh, Y.A.; Lee, C.H. Involvement of ROS in curcumin-induced autophagic cell death. Korean J. Physiol. Pharmacol. 2011, 15, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Kim, Y.S.; Kim, D.E.; Lee, J.S.; Song, J.H.; Kim, H.G.; Cho, D.H.; Jeong, S.Y.; Jin, D.H.; Jang, S.J.; et al. BIX-01294 induces autophagy-associated cell death via EHMT2/G9a dysfunction and intracellular reactive oxygen species production. Autophagy 2013, 9, 2126–2139. [Google Scholar] [CrossRef]
- Song, X.; Kim, S.Y.; Zhang, L.; Tang, D.; Bartlett, D.L.; Kwon, Y.T.; Lee, Y.J. Role of AMP-activated protein kinase in cross-talk between apoptosis and autophagy in human colon cancer. Cell Death Dis. 2014, 5, e1504–e1513. [Google Scholar] [CrossRef] [Green Version]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [Green Version]
- Hawk, M.A.; Schafer, Z.T. Mechanisms of redox metabolism and cancer cell survival during extracellular matrix detachment. J. Biol. Chem. 2018, 293, 7531–7537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, B.H.; Masson, O.; Li, Y.; Khan, I.A.; Gowda, P.S.; Rosen, K.V. Anoikis of colon carcinoma cells triggered by β-catenin loss can be enhanced by tumor necrosis factor receptor 1 antagonists. Oncogene 2015, 34, 4939–4951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Windham, T.C.; Parikh, N.U.; Siwak, D.R.; Summy, J.M.; McConkey, D.J.; Kraker, A.J.; Gallick, G.E. Src activation regulates anoikis in human colon tumor cell lines. Oncogene 2002, 21, 7797–7807. [Google Scholar] [CrossRef] [Green Version]
- Stone, W.L.; Krishnan, K.; Campbell, S.E.; Palau, V.E. The role of antioxidants and pro-oxidants in colon cancer. World J. Gastrointest. Oncol. 2014, 6, 55. [Google Scholar] [CrossRef]
- De Gramont, A.; Figer, A.; Seymour, M.; Homerin, M.; Hmissi, A.; Cassidy, J.; Boni, C.; Cortes-Funes, H.; Cervantes, A.; Freyer, G.; et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J. Clin. Oncol. 2000, 18, 2938–2947. [Google Scholar] [CrossRef]
- Tolan, D.; Gandin, V.; Morrison, L.; El-Nahas, A.; Marzano, C.; Montagner, D.; Erxleben, A. Oxidative Stress Induced by Pt(IV) Pro-drugs Based on the Cisplatin Scaffold and Indole Carboxylic Acids in Axial Position. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Al-Fahdawi, M.Q.; Al-Doghachi, F.A.J.; Abdullah, Q.K.; Hammad, R.T.; Rasedee, A.; Ibrahim, W.N.; Alshwyeh, H.A.; Alosaimi, A.A.; Aldosary, S.K.; Eid, E.E.M.; et al. Oxidative stress cytotoxicity induced by platinum-doped magnesia nanoparticles in cancer cells. Biomed. Pharmacother. 2021, 138, 111483. [Google Scholar] [CrossRef]
- Davison, K.; Mann, K.K.; Miller, W.H. Arsenic trioxide: Mechanisms of action. Semin. Hematol. 2002, 39, 3–7. [Google Scholar] [CrossRef]
- Zhang, P. On arsenic trioxide in the clinical treatment of acute promyelocytic leukemia. Leuk. Res. Rep. 2017, 7, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Hernández, M.A.; de la Cruz-Ojeda, P.; López-Grueso, M.J.; Navarro-Villarán, E.; Requejo-Aguilar, R.; Castejón-Vega, B.; Negrete, M.; Gallego, P.; Vega-Ochoa, Á.; Victor, V.M.; et al. Integrated molecular signaling involving mitochondrial dysfunction and alteration of cell metabolism induced by tyrosine kinase inhibitors in cancer. Redox Biol. 2020, 36, 101510. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.L.; Lee, D.H.; Jeong, S.; Kim, B.R.; Na, Y.J.; Park, S.H.; Jo, M.J.; Jeong, Y.A.; Oh, S.C. Imatinib-induced apoptosis of gastric cancer cells is mediated by endoplasmic reticulum stress. Oncol. Rep. 2019, 41, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Hoehler, T.; Von Wichert, G.; Schimanski, C.; Kanzler, S.; Moehler, M.H.; Hinke, A.; Seufferlein, T.; Siebler, J.; Hochhaus, A.; Arnold, D.; et al. Phase I/II trial of capecitabine and oxaliplatin in combination with bevacizumab and imatinib in patients with metastatic colorectal cancer: AIO KRK 0205. Br. J. Cancer 2013, 109, 1408–1413. [Google Scholar] [CrossRef] [PubMed]
- Orcutt, K.P.; Parsons, A.D.; Sibenaller, Z.A.; Scarbrough, P.M.; Zhu, Y.; Sobhakumari, A.; Wilke, W.W.; Kalen, A.L.; Goswami, P.; Francis, J.; et al. Erlotinib-mediated Inhibition of EGFR Signaling Induces Metabolic Oxidative Stress through NOX4. Cancer Res. 2011, 71, 3932–3940. [Google Scholar] [CrossRef] [Green Version]
- Corazao-Rozas, P.; Guerreschi, P.; Jendoubi, M.; André, F.; Jonneaux, A.; Scalbert, C.; Garçon, G.; Malet-Martino, M.; Balayssac, S.; Rocchi, S.; et al. Mitochondrial oxidative stress is the achille’s heel of melanoma cells resistant to Braf-mutant inhibitor. Oncotarget 2013, 4, 1986–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Lee, Y.L.; Kim, S.; Lee, H.; Chung, H. PCGF2 negatively regulates arsenic trioxide-induced PML-RARA protein degradation via UBE2I inhibition in NB4 cells. Biochim. Biophys. Acta 2016, 1986, 1499–1509. [Google Scholar] [CrossRef]
- Takenokuchi, M.; Miyamoto, K.; Saigo, K.; Taniguchi, T. Bortezomib Causes ER Stress-related Death of Acute Promyelocytic Leukemia Cells Through Excessive Accumulation of PML–RARA. Anticancer Res. 2015, 35, 3307–3316. [Google Scholar] [PubMed]
- Mackay, H.; Hedley, D.; Major, P.; Townsley, C.; Mackenzie, M.; Vincent, M.; Degendorfer, P.; Tsao, M.S.; Nicklee, T.; Birle, D.; et al. A phase II trial with pharmacodynamic endpoints of the proteasome inhibitor bortezomib in patients with metastatic colorectal cancer. Clin. Cancer Res. 2005, 11, 5526–5533. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Thorn, C.F.; Bertagnolli, M.M.; Grosser, T.; Altman, R.B.; Klein, T.E. Celecoxib pathways: Pharmacokinetics and pharmacodynamics. Pharmacogenet. Genom. 2012, 22, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar] [CrossRef]
- Morales, J.C.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Hou, D.; Liu, Z.; Xu, X.; Liu, Q.; Zhang, X.; Kong, B.; Wei, J.J.; Gong, Y.; Shao, C. Increased oxidative stress mediates the antitumor effect of PARP inhibition in ovarian cancer. Redox Biol. 2018, 17, 99–111. [Google Scholar] [CrossRef]
- Zimmer, A.S.; Gillard, M.; Lipkowitz, S.; Lee, J.M. Update on PARP Inhibitors in Breast Cancer. Curr. Treat. Options Oncol. 2018, 19, 21. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.X.; Jonker, D.J.; Siu, L.L.; McKeever, K.; Keller, D.; Wells, J.; Hagerman, L.; Seymour, L. A Phase I study of olaparib and irinotecan in patients with colorectal cancer: Canadian Cancer Trials Group IND 187. Investig. New Drugs 2016, 34, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, V.; Beck, J.T.; Hofheinz, R.D.; Garcia-Alfonso, P.; Nechaeva, M.; Gracian, A.C.; Mangel, L.; Fernandez, E.E.; Deming, D.A.; Ramanathan, R.K.; et al. Correction: A phase 2 randomised study of veliparib plus FOLFIRI±bevacizumab versus placebo plus FOLFIRI±bevacizumab in metastatic colorectal cancer (British Journal of Cancer, (2019), 120, 2, (183-189), 10.1038/s41416-018-0343-z). Br. J. Cancer 2019, 121, 429–430. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Rodríguez, E.; Hernández-García, S.; Sanz, E.; Pandiella, A. Antitumoral effect of Ocoxin on acute myeloid leukemia. Oncotarget 2016, 7, 6231–6242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-García, S.; González, V.; Sanz, E.; Pandiella, A. Effect of Oncoxin Oral Solution in HER2-Overexpressing Breast Cancer. Nutr. Cancer 2015, 67, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Rodríguez, E.; Sanz, E.; Pandiella, A. Antitumoral effect of Ocoxin, a natural compound-containing nutritional supplement, in small cell lung cancer. Int. J. Oncol. 2018, 53, 113–123. [Google Scholar] [CrossRef]
- Das Gupta, S.; Suh, N. Tocopherols in cancer: An update. Mol. Nutr. Food Res. 2016, 60, 1354–1363. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.S.; Suh, N.; Kong, A.N.T. Does vitamin E prevent or promote cancer? Cancer Prev. Res. 2012, 5, 701–705. [Google Scholar] [CrossRef] [Green Version]
- Roohani, N.; Hurrell, R.; Kelishadi, R.; Schulin, R. Zinc and its importance for human health: An integrative review. J. Res. Med. Sci. 2013, 18, 144–157. [Google Scholar]
- De Ribeiro, S.M.F.; Braga, C.B.M.; Peria, F.M.; Domenici, F.A.; Martinez, E.Z.; Feres, O.; da Rocha, J.J.R.; de Cunha, S.F. Effect of Zinc Supplementation on Antioxidant Defenses and Oxidative Stress Markers in Patients Undergoing Chemotherapy for Colorectal Cancer: A Placebo-Controlled, Prospective Randomized Trial. Biol. Trace Elem. Res. 2016, 169, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Glimelius, B.; Manojlovic, N.; Pfeiffer, P.; Mosidze, B.; Kurteva, G.; Karlberg, M.; Mahalingam, D.; Buhl Jensen, P.; Kowalski, J.; Bengtson, M.; et al. Persistent prevention of oxaliplatin-induced peripheral neuropathy using calmangafodipir (PledOx®): A placebo-controlled randomised phase II study (PLIANT). Acta Oncol. 2018, 57, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, J.O.G.; Ignarro, L.J.; Lundström, I.; Jynge, P.; Almén, T. Calmangafodipir [Ca4Mn(DPDP)5], mangafodipir (MnDPDP) and MnPLED with special reference to their SOD mimetic and therapeutic properties. Drug Discov. Today 2015, 20, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Kurz, T.; Grant, D.; Andersson, R.G.G.; Towart, R.; De Cesare, M.; Karlsson, J.O.G. Effects of mndpdp and icrf-187 on doxorubicin-induced cardiotoxicity and anticancer activity. Transl. Oncol. 2012, 5, 252–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cory, H.; Passarelli, S.; Szeto, J.; Tamez, M.; Mattei, J. The Role of Polyphenols in Human Health and Food Systems: A Mini-Review. Front. Nutr. 2018, 5, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.K.; Nanda, P.K.; Chowdhury, N.R.; Dandapat, P.; Gagaoua, M.; Chauhan, P.; Pateiro, M.; Lorenzo, J.M. Application of pomegranate by-products in muscle foods: Oxidative indices, colour stability, shelf life and health benefits. Molecules 2021, 26, 467. [Google Scholar] [CrossRef] [PubMed]
- Nuñez-Sánchez, M.A.; González-Sarrías, A.; García-Villalba, R.; Monedero-Saiz, T.; García-Talavera, N.V.; Gómez-Sánchez, M.B.; Sánchez-Álvarez, C.; García-Albert, A.M.; Rodríguez-Gil, F.J.; Ruiz-Marín, M.; et al. Gene expression changes in colon tissues from colorectal cancer patients following the intake of an ellagitannin-containing pomegranate extract: A randomized clinical trial. J. Nutr. Biochem. 2017, 42, 126–133. [Google Scholar] [CrossRef]
- Shaito, A.; Posadino, A.M.; Younes, N.; Hasan, H.; Halabi, S.; Alhababi, D.; Al-Mohannadi, A.; Abdel-Rahman, W.M.; Eid, A.H.; Nasrallah, G.K.; et al. Potential adverse effects of resveratrol: A literature review. Int. J. Mol. Sci. 2020, 21, 2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.V.; Martinez, M.; Stamos, M.J.; Moyer, M.P.; Planutis, K.; Hope, C.; Holcombe, R.F. Results of a phase I pilot clinical trial examining the effect of plant-derived resveratrol and grape powder on Wnt pathway target gene expression in colonic mucosa and colon cancer. Cancer Manag. Res. 2009, I, 25–37. [Google Scholar] [CrossRef] [Green Version]
- Isemura, M. Catechin in human health and disease. Molecules 2019, 24, 528. [Google Scholar] [CrossRef] [Green Version]
- Bernatoniene, J.; Kopustinskiene, D.M. The Role of Catechins in Cellular Responses to Oxidative Stress. Molecules 2018, 23, 965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinicrope, F.A.; Viggiano, T.R.; Buttar, N.S.; Wong Kee Song, L.M.; Schroeder, K.W.; Kraichely, R.E.; Larson, M.V.; Sedlack, R.E.; Kisiel, J.B.; Gostout, C.J.; et al. Randomized phase II trial of polyphenon E versus placebo in patients at high risk of recurrent colonic neoplasia. Cancer Prev. Res. 2021, 14, 573–580. [Google Scholar] [CrossRef]
- Salehi, B.; Machin, L.; Monzote, L.; Sharifi-Rad, J.; Ezzat, S.M.; Salem, M.A.; Merghany, R.M.; El Mahdy, N.M.; Klllç, C.S.; Sytar, O.; et al. Therapeutic Potential of Quercetin: New Insights and Perspectives for Human Health. ACS Omega 2020, 5, 11849–11872. [Google Scholar] [CrossRef]
- Maurya, A.K.; Vinayak, M. Modulation of PKC signaling and induction of apoptosis through suppression of reactive oxygen species and tumor necrosis factor receptor 1 (TNFR1): Key role of quercetin in cancer prevention. Tumor Biol. 2015, 36, 8913–8924. [Google Scholar] [CrossRef]
- Hewlings, S.J.; Kalman, D.S. Curcumin: A review of its effects on human health. Foods 2017, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Houghton, C.A. Sulforaphane: Its “Coming of Age” as a Clinically Relevant Nutraceutical in the Prevention and Treatment of Chronic Disease. Oxid. Med. Cell. Longev. 2019, 2019, 2716870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Sorolla, A.; Sorolla, M.A.; Wang, E.; Ceña, V. Peptides, proteins and nanotechnology: A promising synergy for breast cancer targeting and treatment. Expert Opin. Drug Deliv. 2020, 17, 1597–1613. [Google Scholar] [CrossRef] [PubMed]
- Kretzmann, J.A.; Evans, C.W.; Moses, C.; Sorolla, A.; Kretzmann, A.L.; Wang, E.; Ho, D.; Hackett, M.J.; Dessauvagie, B.F.; Smith, N.M.; et al. Tumour suppression by targeted intravenous non-viral CRISPRa using dendritic polymers. Chem. Sci. 2019, 10, 7718–7727. [Google Scholar] [CrossRef] [Green Version]
- Sorolla, A.; Wang, E.; Clemons, T.D.; Evans, C.W.; Plani-Lam, J.H.; Golden, E.; Dessauvagie, B.; Redfern, A.D.; Swaminathan-Iyer, K.; Blancafort, P. Triple-hit therapeutic approach for triple negative breast cancers using docetaxel nanoparticles, EN1-iPeps and RGD peptides. Nanomed. Nanotechnol. Biol. Med. 2019, 20, 102003. [Google Scholar] [CrossRef] [PubMed]
- Cisterna, B.A.; Kamaly, N.; Choi, W.I.L.; Tavakkoli, A.; Farokhzad, O.C.; Vilos, C. Targeted nanoparticles for colorectal cancer. Nanomedicine 2016, 11, 2443–2456. [Google Scholar] [CrossRef] [Green Version]
- Datta, A.; Mishra, S.; Manna, K.; Saha, K.D.; Mukherjee, S.; Roy, S. Pro-Oxidant Therapeutic Activities of Cerium Oxide Nanoparticles in Colorectal Carcinoma Cells. ACS Omega 2020, 5, 9714–9723. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, A.; Sutaria, D.; Huang, Y.; Wang, J.; Prabhu, S. Chemoprevention of Colon Cancer in a Rat Carcinogenesis Model Using a Novel Nanotechnology-based Combined Treatment System. Cancer Prev. Res. 2011, 4, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhao, L.; Lau, Y.S.; Zhang, C.; Han, R. Genome-wide CRISPR screen identifies LGALS2 as an oxidative stress-responsive gene with an inhibitory function on colon tumor growth. Oncogene 2021, 40, 177–188. [Google Scholar] [CrossRef] [PubMed]
- House, N.C.M.; Parasuram, R.; Layer, J.V.; Price, B.D. Site-specific targeting of a light activated dCas9-KillerRed fusion protein generates transient, localized regions of oxidative DNA damage. PLoS ONE 2020, 15, e0237759. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Drug or Treatment | Mechanism of Action | Interventional Arms | Inclusion Criteria | Endpoints | Phase | Number of Subjects | Status | Results | Start Date | Identifier |
|---|---|---|---|---|---|---|---|---|---|---|
| Picoplatin | A platinum-based drug that increases intracellular ROS levels | Arm 1: FOLPI: leucovorin + 5-FU + picoplatin Arm 2: FOLFOX: leucovorin + 5-FU + oxaliplatin | Metastatic colorectal cancer | Primary: dose-limiting toxicity, maximum tolerated dose Secondary: Safety and efficacy | Phase I/II | 43 | Not specified | No | 2007 | NCT00478946 |
| Arsenic trioxide | Potent oxidant and apoptosis inductor | Single Arm: Arsenic trioxide + 5-FU + leucovorin | Refractory/relapsed metastatic colorectal cancer | Primary: maximum tolerated dose and thymidylate synthase expression | Phase I | 13 | Completed | Yes PMID: 20950119 | 2007 | NCT00449137 |
| Imatinib | TK inhibitor of BCR-ABL | Single arm: XELOX (capecitabine + oxaliplatin) + bevacizumab + imatinib | Metastatic colorectal cancer | Primary: Dose-limiting toxicity Secondary: ORR and PFS | Phase I/II | 51 | Completed | Yes PMID: 23963139 | 2008 | NCT00784446 |
| Erlotinib | TK inhibitor of EGFR | Single arm: pemetrexed and erlotinib | Metastatic and refractory colorectal cancer | Primary: OS and PFS Secondary: OS, disease control rate and treatment-related adverse events | Phase II | 50 | Completed | Yes DOI: https://doi.org/10.1093/annonc/mdy281.114 | 2016 | NCT02723578 |
| Vemurafenib | TK inhibitor of BRAF | Single arm: FOLFIRI (leucovorin + 5-FU + irinotecan) + vemurafenib + cetuximab | BRAF V600E mutated advanced colorectal cancer | Primary: ORR Secondary: early tumor shrinkage and disease control rate | Phase II | 30 | Recruiting | No | 2018 | NCT03727763 |
| Bortezomib | Proteasome inhibitor that promotes endoplasmic reticulum stress | Single arm: bortezomib | Metastatic or recurrent colorectal cancer | Primary: efficacy | Phase II | 19 | Completed | Yes PMID: 16061869 | 2003 | NCT00052507 |
| Celecoxib | Cyclooxygenase 2 inhibitor that causes endoplasmic reticulum stress | Arm 1: FOLFIRI. Arm 2: FOLFIRI + celecoxib | Metastatic colorectal cancer | Primary: number of patients with improved radiology | Phase IV | 50 | Recruiting | No | 2018 | NCT03645187 |
| AVA6000 or Pro-doxorubicin | Targets topoisomerase in DNA replication and promotes apoptosis by oxidative stress | Arm 1: AVA6000 standard 3 + 3 scheme Arm 2: AVA6000 dose-expansion phase | Locally advanced and/or metastatic solid tumors including colorectal cancer | Primary: Dose limiting toxicities, maximum tolerated dose, adverse events Secondary: maximum drug concentration, elimination half-time, renal clearance, ORR, duration of response, PFS and OS | Phase I | 80 | Recruiting | No | 2021 | NCT04969835 |
| Olaparib | PARP inhibitor; promotes DNA damage and oxidative stress | Single arm: olaparib + irinotecan | Locally advanced or metastatic incurable colorectal cancer | Primary: recommended phase II dose, safety, tolerability, dose-limiting toxicities and pharmacokinetic profile Secondary: efficacy and pharmacodynamic outcomes | Phase I | 26 | Completed | Yes PMID: 27075016 | 2007 | NCT00535353 |
| Veliparib | PARP inhibitor; promotes DNA damage and oxidative stress | Arm 1: FOLFIRI ± bevacizumab + veliparib Arm 2: FOLFIRI ± bevacizumab + placebo | Untreated metastatic colorectal cancer | Primary: PFS. Secondary: OS and ORR | Phase II | 130 | Completed | Yes PMID: 30531832 | 2018 | NCT02305758 |
| Drug or Treatment | Mechanism of Action | Interventional Arms | Inclusion Criteria | Endpoints | Phase | Number of Subjects | Status | Results | Start Date | Identifier |
|---|---|---|---|---|---|---|---|---|---|---|
| Ocoxin®-Viusid® (vitamin B6, C, and cinnamic acid) | Nutritional and vitamin supplement with anticancer and antioxidant activity | Single arm: Ocoxin-Viusid® | Metastatic colorectal adenocarcinoma | Primary: quality of life, tolerance of chemotherapy, and nutritional status | Phase II | 40 | Recruiting | No | 2018 | NCT03559543 |
| Vitamin C, B6, and folic acid | Vitamin supplement with anticancer and antioxidant activity | Control group: vitamin C. Arm 1: vitamin B6 Arm 2: folic acid Arm 3: vitamin B6 + folic acid | Confirmed colorectal cancer | Primary: measurement of oxidative stress (TBARS), antioxidant activities, and DNA methylation status | Phase II/III | 300 | Not specified | No | 2011 | NCT01426490 |
| Vitamin E | Vitamin supplement with anticancer and antioxidant activity | Arm 1: no intervention Arm 2: high γ-tocopherol vitamin E mixture (1 week before surgery) Arm 3: high γ-tocopherol vitamin E mixture (2 week before surgery) | Pre-surgical patients with colorectal cancer | Primary: measurement of plasma and urine levels of tocopherols and prostaglandin E2; measurement of plasma levels of F2-isoprostane, C-reactive protein, 3-NT, and urinary levels of 8-OHdG; and measurement of tocopherols, cell proliferation and apoptosis indicators, β-catenin localization, RXR expression, cyclooxygenase-2, 8-OHdG, and 3-NT levels in colon tissue | Phase I | 14 | Completed | No | 2009 | NCT00905918 |
| Zinc | Trace element cofactor of endogenous antioxidant enzymes | Experimental: zinc + chemotherapy (CRC patients) Placebo comparator: placebo + chemotherapy (CRC patients) Zinc control: zinc (healthy volunteers). Placebo control: placebo (healthy volunteers) | Stage II–IV colorectal patients | Primary: oxidative stress markers (SOD, GPx, MDA, isoprostane, vitamin C and E; Secondary: FACIT-F and CTCAE | Not applicable | 55 | Completed | Yes PMID: 26066525 | 2014 | NCT02106806 |
| Calmangafodipir | MnSOD mimetic activity | Arm 1: FOLFOX6 + calmangafodipir 2 µmol/kg Arm 2: FOLFOX6 + calmangafodipir 5 µmol/kg Arm 3: FOLFOX6 + calmangafodipir 10 µmol/kg Placebo arm: FOLFOX6 + 0.9% NaCl | Advanced metastatic colorectal cancer | Primary: number of patients with neuropathy grade 2 or higher | Phase I/II | 186 | Completed | Yes | 186 | NCT01619423 |
| UrolithinA (pomegranate formulation) | Anti-inflammatory and anti-cancer activity | Arm 1: standard pomegranate extract formulation Arm 2: new pomegranate extract formulation-1 Arm 3: new pomegranate extract formulation-2 | Pre-surgical colorectal cancer patients | Primary: measurement of phenolics and their metabolites in colon tissues, plasma, and urine samples; analysis of the gene expression profile in colon tissue Secondary: evaluation of the number of patients with adverse events, measurement of circulating IGF-1 and CEA levels and microRNA expression in tumoral and colon tissues | Phase I/II | 60 | Completed | Yes PMID: 28183047 | 2013 | NCT01916239 |
| Resveratrol (grape extract) | It is suggested that modulates Wnt signaling, with anti-oxidant and pro-apoptotic effects | Single arm: resveratrol | Pre-surgical colorectal cancer patients | Primary: evaluation of the modulation of Wnt signalling in vivo in colon cancer and normal colonic mucosa | Phase I | 11 | Completed | Yes PMID: 21188121 | 2005 | NCT00256334 |
| Polyphenon E (green tea catechin extract) | Reduces free radicals, chelates metal ions, upregulates antioxidant enzymes, and inhibits prooxidant enzymes | Arm I: polyphenon Arm 2: placebo | Stage I–III high-risk colorectal cancer patients | Primary: measurement of the percent change in rectal ACF before and after the intervention Secondary: study of the tolerability dose of catechin extract | Phase II | 39 | Terminated | Yes PMID: 33648940 | 2012 | NCT01606124 |
| Sulindac + rutin + quercetin + curcumin | Antioxidant effect | Arm 1: control diet Arm 2: control diet + sulindac Arms 3–5: control diet + rutin Arms 5–7: control diet + quercetin Arm 8–10: control diet + curcumin | Individuals with average or above risk for development of colon cancer | To determine and compare the response of colon epithelium to different dietary treatments and to sulindac, and to identify the lowest optimal dose of the dietary supplementation to modulate biomarkers of colon epithelial | Not applicable | 130 | Terminated | No | 2004 | NCT00003365 |
| Curcumin | Antioxidant activity | Single arm: curcumin + 5-FU | 5-FU resistant metastatic colon cancer | Early phase I | 13 | Active | No | 2016 | NCT02724202 | |
| Sulforaphane (cruciferous vegetables) | Nrf2 enhancer which promotes antioxidant gene expression | Single arm: cruciferous vegetable intake | Patients scheduled for a screening colonoscopy | Primary: to determine the correlation of sulforaphane and indole-3-carbinol urinary levels with cruciferous intake Secondary: p21 and acetylated histone expression, and HDAC activity in PBMCs and colon tissue | Not applicable | 108 | Completed | No | 2011 | NCT01344330 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorolla, M.A.; Hidalgo, I.; Sorolla, A.; Montal, R.; Pallisé, O.; Salud, A.; Parisi, E. Microenvironmental Reactive Oxygen Species in Colorectal Cancer: Involved Processes and Therapeutic Opportunities. Cancers 2021, 13, 5037. https://doi.org/10.3390/cancers13205037
Sorolla MA, Hidalgo I, Sorolla A, Montal R, Pallisé O, Salud A, Parisi E. Microenvironmental Reactive Oxygen Species in Colorectal Cancer: Involved Processes and Therapeutic Opportunities. Cancers. 2021; 13(20):5037. https://doi.org/10.3390/cancers13205037
Chicago/Turabian StyleSorolla, Maria Alba, Ivan Hidalgo, Anabel Sorolla, Robert Montal, Ona Pallisé, Antonieta Salud, and Eva Parisi. 2021. "Microenvironmental Reactive Oxygen Species in Colorectal Cancer: Involved Processes and Therapeutic Opportunities" Cancers 13, no. 20: 5037. https://doi.org/10.3390/cancers13205037
APA StyleSorolla, M. A., Hidalgo, I., Sorolla, A., Montal, R., Pallisé, O., Salud, A., & Parisi, E. (2021). Microenvironmental Reactive Oxygen Species in Colorectal Cancer: Involved Processes and Therapeutic Opportunities. Cancers, 13(20), 5037. https://doi.org/10.3390/cancers13205037