
Novel Molecular Mechanism of Lenalidomide in Myeloid Malignancies Independent of Deletion of Chromosome 5q


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Actions of Lenalidomide
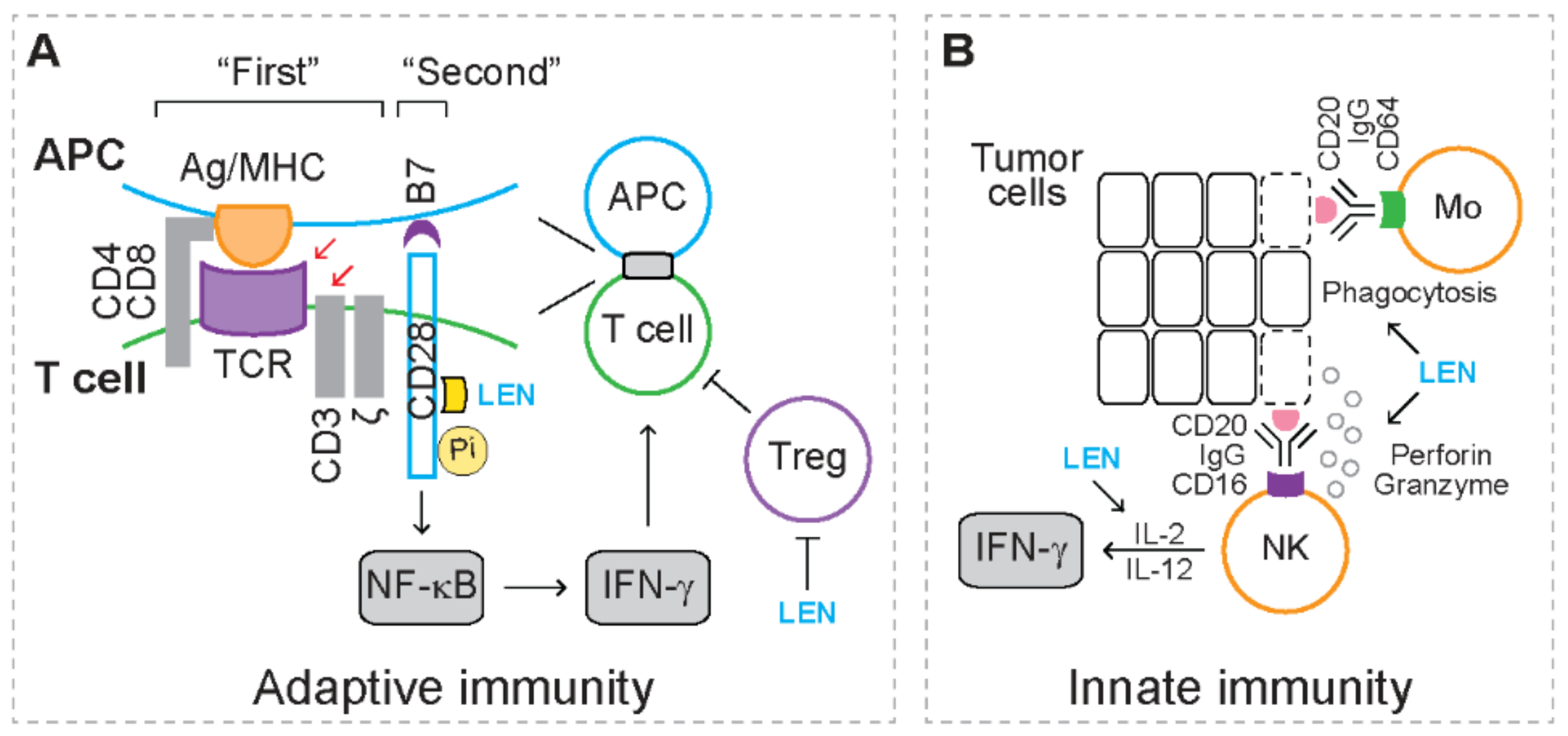
2.1. Anti-Inflammation
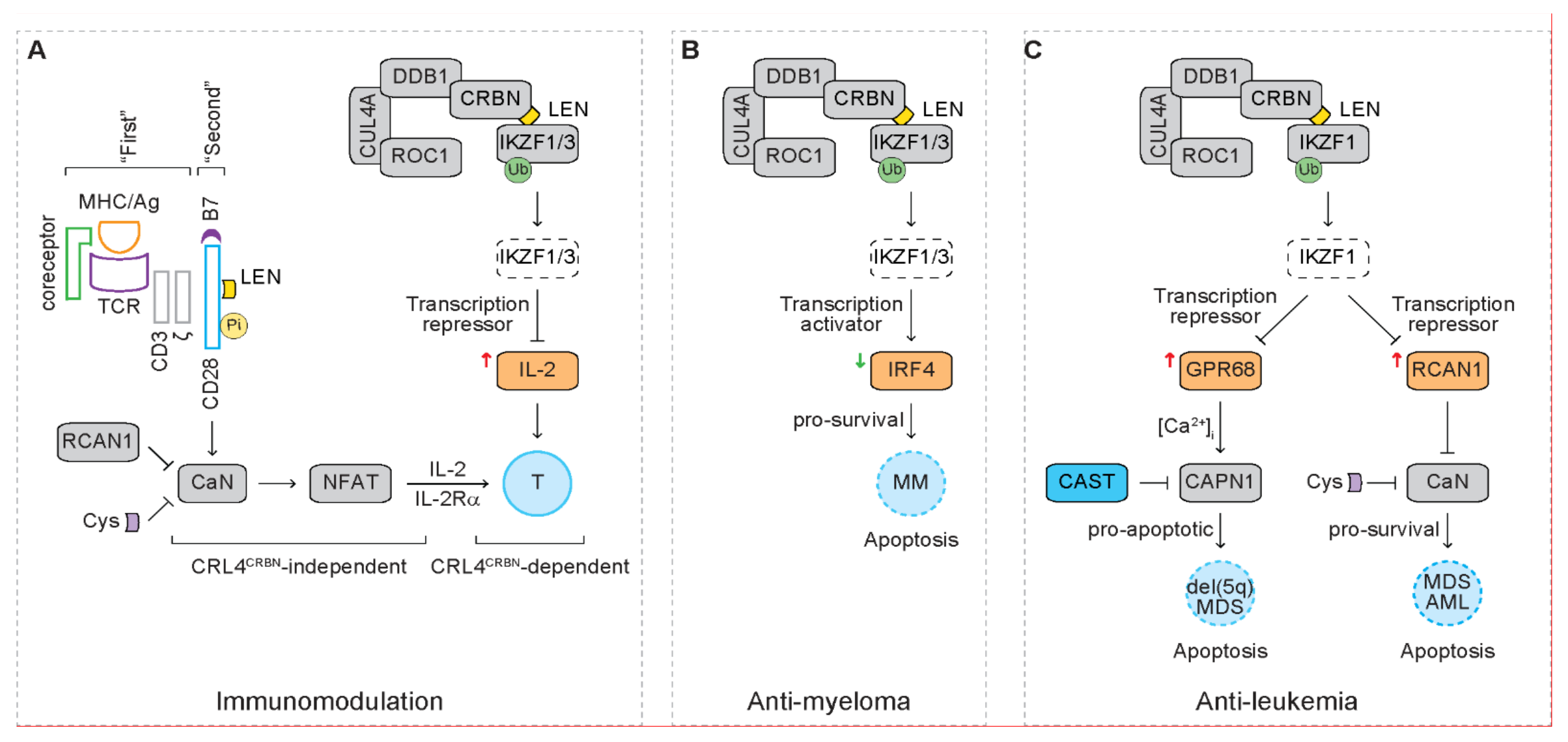
2.2. Immunomodulation
2.3. Anti-Angiogenesis
2.4. Anti-Tumor
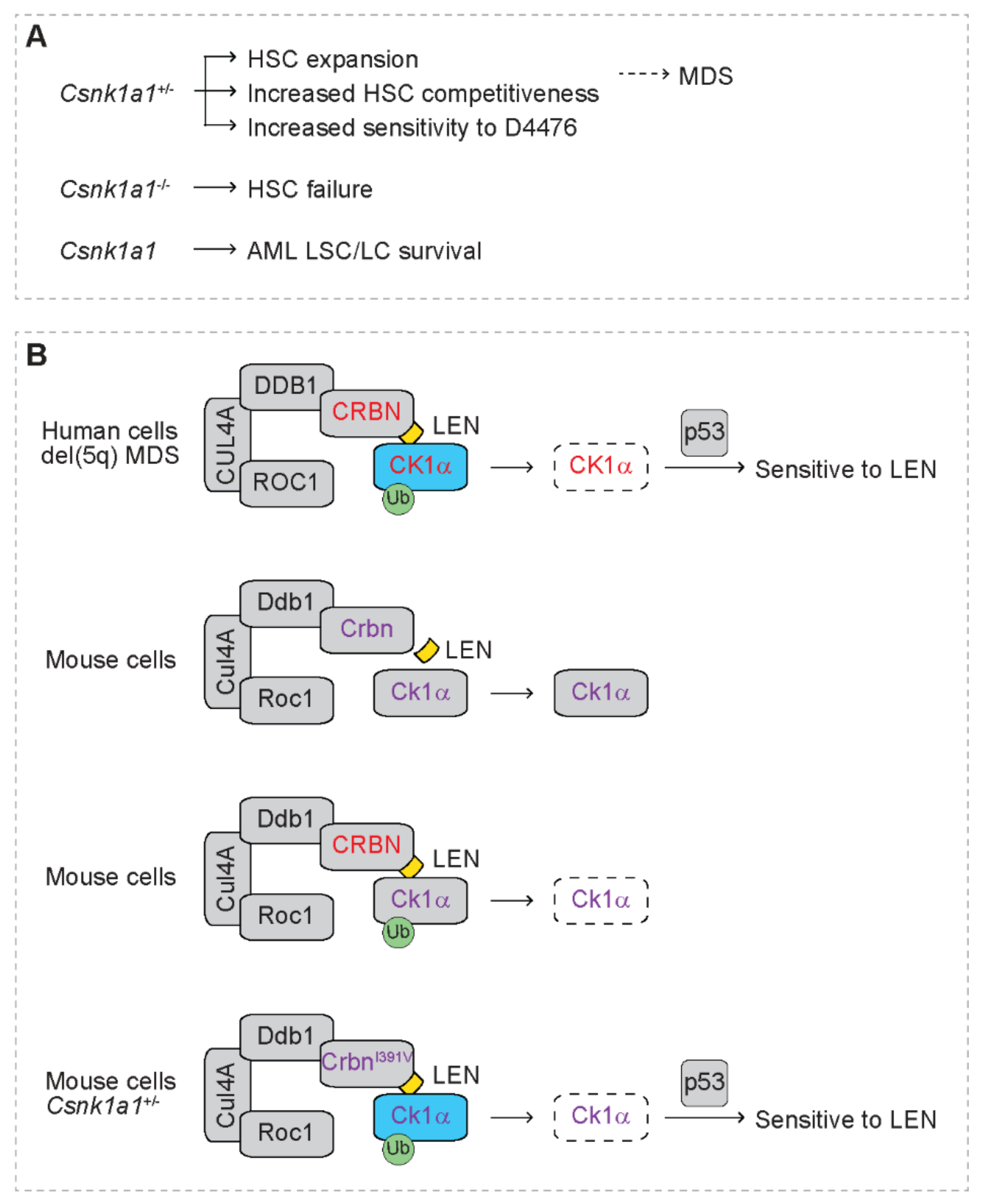
3. Haploinsufficient 5q Genes Confers Therapeutic Vulnerability to Lenalidomide
4. CRBN Is Required for IMiD-Mediated Anti-Tumor Effects
4.1. CRBN Is the Primary Target Protein of IMiDs
4.2. Expression of CRBN Determines Response to Lenalidomide
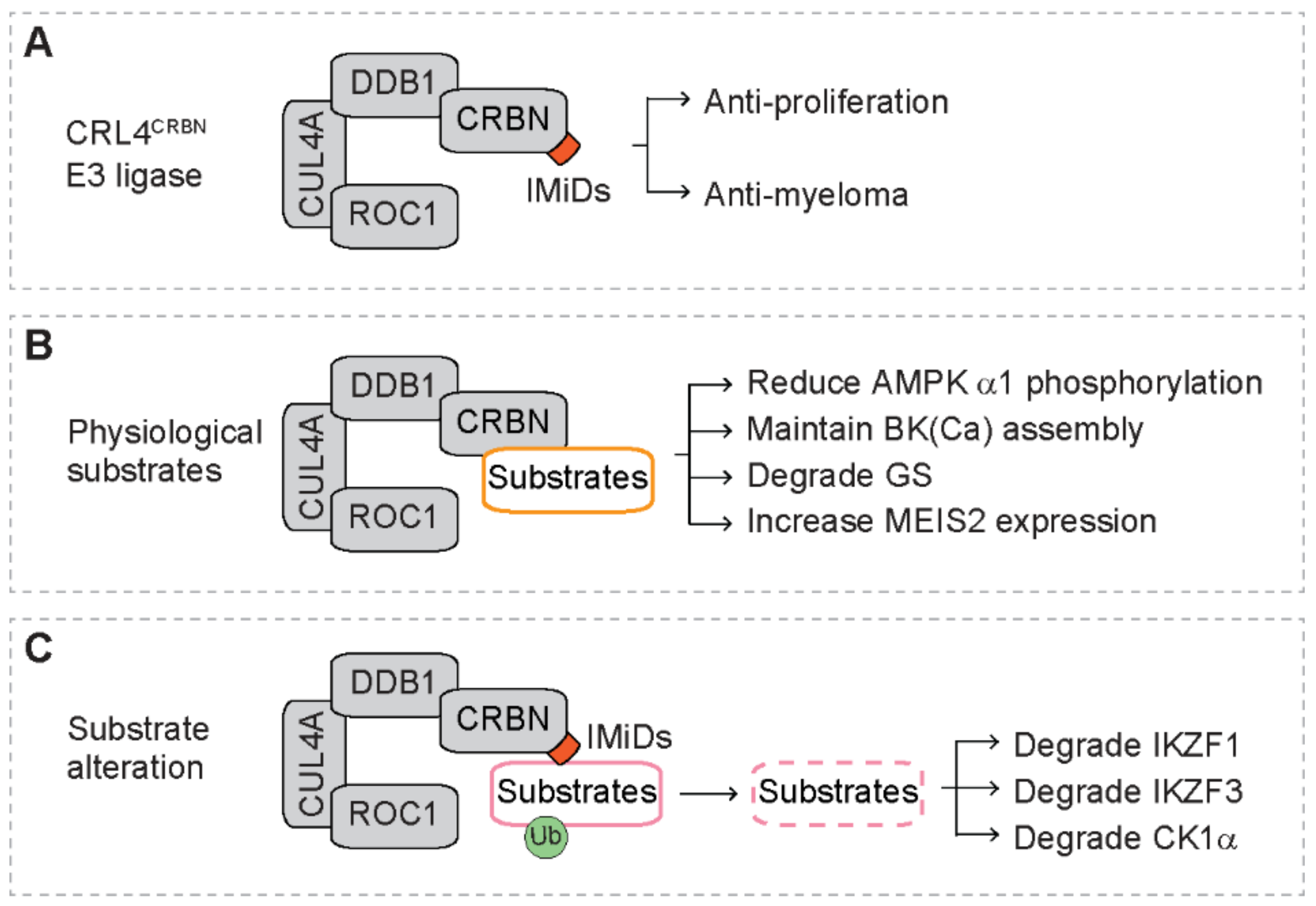
5. Substrates of The CRL4CRBN E3 Ligase Complex
5.1. Physiological Substrates
5.2. Substrate Alteration in the Presence of IMiDs
6. Signaling Pathways Downstream of IKZF1 and IKZF3
6.1. IL-2
6.2. IRF4
6.3. GPR68
6.4. RCAN1
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vargesson, N. Thalidomide-induced teratogenesis: History and mechanisms. Birth Defects Res. Part C Embryo Today Rev. 2015, 105, 140–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, S.; Mehta, J.; Desikan, R.; Ayers, D.; Roberson, P.; Eddlemon, P.; Munshi, N.; Anaissie, E.; Wilson, C.; Dhodapkar, M.; et al. Antitumor Activity of Thalidomide in Refractory Multiple Myeloma. N. Engl. J. Med. 1999, 341, 1565–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, G.W.; Corral, L.G.; Shire, M.G.; Wang, H.; Moreira, A.; Kaplan, A.G.; Stirling, D.I. Structural Modifications of Thalidomide Produce Analogs with Enhanced Tumor Necrosis Factor Inhibitory Activity. J. Med. Chem. 1996, 39, 3238–3240. [Google Scholar] [CrossRef] [PubMed]
- Marriott, J.B.; Muller, G.; Stirling, D.; Dalgleish, A.G. Immunotherapeutic and antitumour potential of thalidomide analogues. Expert Opin. Biol. Ther. 2001, 1, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Raedler, L.A. Revlimid (Lenalidomide) Now FDA Approved as First-Line Therapy for Patients with Multiple Myeloma. Am. Health Drug Benefits 2016, 9, 140–143. [Google Scholar]
- Weber, D.M.; Chen, C.; Niesvizky, R.; Wang, M.; Belch, A.R.; Stadtmauer, E.A.; Siegel, D.; Borrello, I.; Rajkumar, S.V.; Chanan-Khan, A.A.; et al. Lenalidomide plus Dexamethasone for Relapsed Multiple Myeloma in North America. N. Engl. J. Med. 2007, 357, 2133–2142. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Spencer, A.; Attal, M.; Prince, H.M.; Harousseau, J.-L.; Dmoszynska, A.; Miguel, J.S.; Hellmann, A.; Facon, T.; Foà, R.; et al. Lenalidomide plus Dexamethasone for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2007, 357, 2123–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sher, T.; Miller, K.C.; Lawrence, D.; Whitworth, A.; Hernandez-Ilizaliturri, F.; Czuczman, M.S.; Miller, A.; Lawrence, W.; Bilgrami, S.A.; Sood, R.; et al. Efficacy of lenalidomide in patients with chronic lymphocytic leukemia with high-risk cytogenetics. Leuk. Lymphoma 2009, 51, 85–88. [Google Scholar] [CrossRef]
- Byrd, J.C.; Ruppert, A.S.; Heerema, N.A.; Halvorson, A.E.; Hoke, E.; Smith, M.R.; Godwin, J.; Couban, S.; Fehniger, T.A.; Thirman, M.J.; et al. Lenalidomide consolidation benefits patients with CLL receiving chemoimmunotherapy: Results for CALGB 10404 (Alliance). Blood Adv. 2018, 2, 1705–1718. [Google Scholar] [CrossRef]
- Habermann, T.M.; Lossos, I.S.; Justice, G.; Vose, J.M.; Wiernik, P.H.; McBride, K.; Wride, K.; Ervin-Haynes, A.; Takeshita, K.; Pietronigro, D.; et al. Lenalidomide oral monotherapy produces a high response rate in patients with relapsed or refractory mantle cell lymphoma. Br. J. Haematol. 2009, 145, 344–349. [Google Scholar] [CrossRef]
- Wiernik, P.H.; Lossos, I.S.; Tuscano, J.M.; Justice, G.; Vose, J.M.; Cole, C.E.; Lam, W.; McBride, K.; Wride, K.; Pietronigro, D.; et al. Lenalidomide Monotherapy in Relapsed or Refractory Aggressive Non-Hodgkin’s Lymphoma. J. Clin. Oncol. 2008, 26, 4952–4957. [Google Scholar] [CrossRef] [PubMed]
- Witzig, T.E.; Wiernik, P.H.; Moore, T.; Reeder, C.; Cole, C.; Justice, G.; Kaplan, H.; Voralia, M.; Pietronigro, D.; Takeshita, K.; et al. Lenalidomide Oral Monotherapy Produces Durable Responses in Relapsed or Refractory Indolent Non-Hodgkin’s Lymphoma. J. Clin. Oncol. 2009, 27, 5404–5409. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Fowler, N.H.; Feugier, P.; Bouabdallah, R.; Tilly, H.; Palomba, M.L.; Fruchart, C.; Libby, E.N.; Casasnovas, R.-O.; Flinn, I.W.; et al. Rituximab plus Lenalidomide in Advanced Untreated Follicular Lymphoma. N. Engl. J. Med. 2018, 379, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, M.; Ajayi, S.; Reinhardt, H.; Müller, S.J.; Dold, S.M.; Wäsch, R. A Phase 2/3 Multicenter, Randomized, Open-Label Study to Compare the Efficacy and Safety of Le-nalidomide Versus Investigator’s Choice in Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Clin. Cancer Res. 2017, 23, 4127–4137. [Google Scholar]
- Engelhardt, M.; Wäsch, R.; Reinhardt, H.; Kleber, M. Pomalidomide. In Small Molecules in Oncology; Springer: Cham, Switzerland, 2018; pp. 169–185. [Google Scholar]
- Arcuri, L.J.; Americo, A.D. Treatment of relapsed/refractory multiple myeloma in the bortezomib and lenalidomide era: A systematic review and network meta-analysis. Ann. Hematol. 2021, 100, 725–734. [Google Scholar] [CrossRef]
- Raza, S.; Safyan, R.; Suzanne, L. Immunomodulatory Drugs (IMiDs) in Multiple Myeloma. Curr. Cancer Drug Targets 2017, 17, 846–857. [Google Scholar] [CrossRef]
- Holstein, S.A.; McCarthy, P.L. Immunomodulatory Drugs in Multiple Myeloma: Mechanisms of Action and Clinical Ex-perience. Drugs 2017, 77, 505–520. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Kortuem, K.M.; Stewart, A.K. Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leuk. Lymphoma 2012, 54, 683–687. [Google Scholar] [CrossRef]
- Itchaki, G.; Brown, J.R. Lenalidomide in the treatment of chronic lymphocytic leukemia. Expert Opin. Investig. Drugs 2017, 26, 633–650. [Google Scholar] [CrossRef]
- Liang, L.; Zhao, M.; Zhu, Y.-C.; Hu, X.; Yang, L.-P.; Liu, H. Efficacy of lenalidomide in relapsed/refractory chronic lymphocytic leukemia patient: A systematic review and meta-analysis. Ann. Hematol. 2016, 95, 1473–1482. [Google Scholar] [CrossRef]
- Chen, C.I. Lenalidomide Alone and in Combination for Chronic Lymphocytic Leukemia. Curr. Hematol. Malign-Rep. 2012, 8, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Morabito, F.; Skafi, M.; Recchia, A.G.; Kashkeesh, A.; Hindiyeh, M.; Sabatleen, A.; Morabito, L.; Alijanazreh, H.; Hamamreh, Y.; Gentile, M. Lenalidomide for the treatment of mantle cell lymphoma. Expert Opin. Pharmacother. 2019, 20, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Witzig, T.E.; Nowakowski, G.S.; Habermann, T.M.; Goy, A.; Hernandez-Ilizaliturri, F.J.; Chiappella, A.; Vitolo, U.; Fowler, N.; Czuczman, M.S. A comprehensive review of lenalidomide therapy for B-cell non-Hodgkin lymphoma. Ann. Oncol. 2015, 26, 1667–1677. [Google Scholar] [CrossRef] [PubMed]
- Flowers, C.R.; Leonard, J.P.; Fowler, N.H. Lenalidomide in follicular lymphoma. Blood 2020, 135, 2133–2136. [Google Scholar] [CrossRef]
- Salles, G.; Duell, J.; Barca, E.G.; Tournilhac, O.; Jurczak, W.; Liberati, A.M.; Nagy, Z.; Obr, A.; Gaidano, G.; André, M.; et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): A multi-centre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020, 21, 978–988. [Google Scholar] [CrossRef]
- List, A.; Kurtin, S.; Roe, D.J.; Buresh, A.; Mahadevan, D.; Fuchs, D.; Rimsza, L.; Heaton, R.; Knight, R.; Zeldis, J.B. Efficacy of Lenalidomide in Myelodysplastic Syndromes. N. Engl. J. Med. 2005, 352, 549–557. [Google Scholar] [CrossRef] [Green Version]
- List, A.; Dewald, G.; Bennett, J.; Giagounidis, A.; Raza, A.; Feldman, E.; Powell, B.; Greenberg, P.; Thomas, D.; Stone, R.; et al. Lenalidomide in the Myelodysplastic Syndrome with Chromosome 5q Deletion. N. Engl. J. Med. 2006, 355, 1456–1465. [Google Scholar] [CrossRef] [Green Version]
- Fenaux, P.; Giagounidis, A.; Selleslag, D.; Beyne-Rauzy, O.; Mufti, G.; Mittelman, M.; Muus, P.; Boekhorst, P.T.; Sanz, G.; Del Cañizo, C.; et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with Low-/Intermediate-1-risk myelodysplastic syndromes with del5q. Blood 2011, 118, 3765–3776. [Google Scholar] [CrossRef]
- Komrokji, R.S.; Padron, E.; Ebert, B.L.; List, A.F. Deletion 5q MDS: Molecular and therapeutic implications. Best Pract. Res. Clin. Haematol. 2013, 26, 365–375. [Google Scholar] [CrossRef]
- Pellagatti, A.; Jadersten, M.; Forsblom, A.-M.; Cattan, H.; Christensson, B.; Emanuelsson, E.K.; Merup, M.; Nilsson, L.; Samuelsson, J.; Sander, B.; et al. Lenalidomide inhibits the malignant clone and up-regulates the SPARC gene mapping to the commonly deleted region in 5q-syndrome patients. Proc. Natl. Acad. Sci. USA 2007, 104, 11406–11411. [Google Scholar] [CrossRef] [Green Version]
- Boultwood, J.; Fidler, C.; Strickson, A.J.; Watkins, F.; Gama, S.; Kearney, L.; Tosi, S.; Kasprzyk, A.; Cheng, J.-F.; Jaju, R.J.; et al. Narrowing and genomic annotation of the commonly deleted region of the 5q−syndrome. Blood 2002, 99, 4638–4641. [Google Scholar] [CrossRef]
- Wei, S.; Chen, X.; Rocha, K.; Epling-Burnette, P.K.; Djeu, J.Y.; Liu, Q.; Byrd, J.; Sokol, L.; Lawrence, N.; Pireddu, R.; et al. A critical role for phosphatase haplodeficiency in the selective suppression of deletion 5q MDS by lenalidomide. Proc. Natl. Acad. Sci. USA 2009, 106, 12974–12979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, J.; Bolesta, E.; Brautigan, D.L.; Larner, J.M. PP2A regulates ionizing radiation–induced apoptosis through Ser46 phosphorylation of p53. Mol. Cancer Ther. 2009, 8, 135–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.; Chen, X.; McGraw, K.; Zhang, L.; Komrokji, R.; Clark, J.; Caceres, G.; Billingsley, D.; Sokol, L.; Lancet, J.; et al. Lenalidomide promotes p53 degradation by inhibiting MDM2 auto-ubiquitination in myelodysplastic syndrome with chromosome 5q deletion. Oncogene 2012, 32, 1110–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; List, A.; Sallman, D.A. Molecular pathogenesis of myelodysplastic syndromes with deletion 5q. Eur. J. Haematol. 2018, 102, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, R.K.; Schenone, M.; Ferreira, M.S.V.; Kramann, R.; Joyce, C.E.; Hartigan, C.; Beier, F.; Brümmendorf, T.H.; Gehrming, U.; Platzbecker, U.; et al. Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A9. Nat. Med. 2016, 22, 288–297. [Google Scholar] [CrossRef]
- Venugopal, S.; Mascarenhas, J.; Steensma, D.P. Loss of 5q in myeloid malignancies – A gain in understanding of biological and clinical consequences. Blood Rev. 2020, 46, 100735. [Google Scholar] [CrossRef]
- Fink, E.C.; Ebert, B.L. The novel mechanism of lenalidomide activity. Blood 2015, 126, 2366–2369. [Google Scholar] [CrossRef] [Green Version]
- Guirguis, A.; Ebert, B.L. Lenalidomide: Deciphering mechanisms of action in myeloma, myelodysplastic syndrome and beyond. Curr. Opin. Cell Biol. 2015, 37, 61–67. [Google Scholar] [CrossRef]
- Gaballa, M.R.; Besa, E.C. Myelodysplastic syndromes with 5q deletion: Pathophysiology and role of lenalidomide. Ann. Hematol. 2014, 93, 723–733. [Google Scholar] [CrossRef]
- Giagounidis, A.; Mufti, G.J.; Fenaux, P.; Germing, U.; List, A.; Macbeth, K.J. Lenalidomide as a disease-modifying agent in patients with del(5q) myelodysplastic syndromes: Linking mechanism of action to clinical outcomes. Ann. Hematol. 2013, 93, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heise, C.; Carter, T.; Schafer, P.; Chopra, R. Pleiotropic mechanisms of action of lenalidomide efficacy in del(5q) myelodysplastic syndromes. Expert Rev. Anticancer Ther. 2010, 10, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Talati, C.; Sallman, D.; List, A. Lenalidomide: Myelodysplastic syndromes with del(5q) and beyond. Semin. Hematol. 2017, 54, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Komrokji, R.S.; List, A.F. Lenalidomide for Treatment of Myelodysplastic Syndromes: Current Status and Future Directions. Hematol. Clin. N. Am. 2010, 24, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Kelaidi, C.; Eclache, V.; Fenaux, P. The role of lenalidomide in the management of myelodysplasia with del 5q. Br. J. Haematol. 2008, 140, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Melchert, M.; Kale, V.; List, A. The role of lenalidomide in the treatment of patients with chromosome 5q deletion and other myelodysplastic syndromes. Curr. Opin. Hematol. 2007, 14, 123–129. [Google Scholar] [CrossRef] [PubMed]
- List, A.; Ebert, B.L.; Fenaux, P. A decade of progress in myelodysplastic syndrome with chromosome 5q deletion. Leukemia 2018, 32, 1493–1499. [Google Scholar] [CrossRef]
- Jan, M.; Sperling, A.S.; Ebert, B.L. Cancer therapies based on targeted protein degradation—Lessons learned with le-nalidomide. Nat. Rev. Clin. Oncol. 2021, 18, 401–417. [Google Scholar] [CrossRef]
- List, A. Lenalidomide—A Transforming Therapeutic Agent in Myelodysplastic Syndromes. Clin. Lymphoma Myeloma 2009, 9, S302–S304. [Google Scholar] [CrossRef]
- Padron, E.; Komrokji, R.; List, A. Biology and treatment of the 5q- syndrome. Expert Rev. Hematol. 2011, 4, 61–69. [Google Scholar] [CrossRef]
- Sallman, D.A.; Wei, S.; List, A. PP2A: The Achilles Heal in MDS with 5q Deletion. Front. Oncol. 2014, 4, 264. [Google Scholar] [CrossRef] [Green Version]
- Caceres, G.; McGraw, K.; Yip, B.H.; Pellagatti, A.; Johnson, J.; Zhang, L.; Liu, K.; Fulp, W.J.; Lee, J.-H.; Al Ali, N.H.; et al. TP53 suppression promotes erythropoiesis in del(5q) MDS, suggesting a targeted therapeutic strategy in lenalidomide-resistant patients. Proc. Natl. Acad. Sci. USA 2013, 110, 16127–16132. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [Green Version]
- Higgins, J.J.; Pucilowska, J.; Lombardi, R.Q.; Rooney, J. A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology 2004, 63, 1927–1931. [Google Scholar] [CrossRef]
- Sato, T.; Ito, T.; Handa, H. Cereblon-Based Small-Molecule Compounds to Control Neural Stem Cell Proliferation in Re-generative Medicine. Front. Cell. Dev. Biol. 2021, 9, 453. [Google Scholar] [CrossRef]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2013, 343, 301–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2013, 343, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krönke, J.; Fink, E.; Hollenbach, P.W.; Macbeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Boultwood, J.; Pellagatti, A.; Cattan, H.; Lawrie, C.H.; Giagounidis, A.; Malcovati, L.; Della Porta, M.G.; Jädersten, M.; Killick, S.; Fidler, C.; et al. Gene expression profiling of CD34+cells in patients with the 5q− syndrome. Br. J. Haematol. 2007, 139, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Liu, X.; Bolanos, L.; Barker, B.; Rigolino, C.; Cortelezzi, A.; Oliva, E.N.; Cuzzola, M.; Grimes, H.L.; Fontanillo, C.; et al. A calcium- and calpain-dependent pathway determines the response to lenalidomide in myelodysplastic syn-dromes. Nat. Med. 2016, 22, 727–734. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Dou, A.; Feng, S.; Roman-Rivera, A.; Hawkins, C.; Lawley, L.; Zhang, J.; Wunderlich, M.; Mizukawa, B.; Halene, S.; et al. Cyclosporine enhances the sensitivity to lenalidomide in MDS/AML in vitro. Exp. Hematol. 2020, 86, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Dou, A.; Fang, J. Cyclosporine Broadens the Therapeutic Potential of Lenalidomide in Myeloid Malignancies. J. Cell. Immunol. 2020, 2, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Colombo, D.; Ammirati, E. Cyclosporine in transplantation - A history of converging timelines. J. Boil. Regul. Homeost. Agents 2011, 25, 493–504. [Google Scholar]
- Corral, L.G.; A Haslett, P.; Muller, G.W.; Chen, R.; Wong, L.M.; Ocampo, C.J.; Patterson, R.T.; I Stirling, D.; Kaplan, G. Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-alpha. J. Immunol. 1999, 163, 380–386. [Google Scholar] [PubMed]
- Bernard, A.; Lamy, L.; Alberti, I. THE TWO-SIGNAL MODEL OF T-CELL ACTIVATION AFTER 30 YEARS. Transplantation 2002, 73, S31–S35. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, R.; Hideshima, T.; Catley, L.P.; Shringarpure, R.; Burger, R.; Mitsiades, N.; Mitsiades, C.; Cheema, P.; Chauhan, D.; Richardson, P.G.; et al. Immunomodulatory drug costimulates T cells via the B7-CD28 pathway. Blood 2004, 103, 1787–1790. [Google Scholar] [CrossRef] [PubMed]
- Galustian, C.; Meyer, B.; Labarthe, M.C.; Dredge, K.; Klaschka, D.; Henry, J.; Todryk, S.; Chen, R.; Muller, G.; Stirling, D.; et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regu-latory cells. Cancer Immunol. Immunother. 2009, 58, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Nigro, C.L.; Macagno, M.; Sangiolo, D.; Bertolaccini, L.; Aglietta, M.; Merlano, M.C. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: Biological evidence and clinical perspectives. Ann. Transl. Med. 2019, 7, 105. [Google Scholar] [CrossRef] [Green Version]
- Yeap, W.H.; Wong, K.L.; Shimasaki, N.; Teo, E.C.Y.; Quek, J.K.S.; Yong, H.X.; Diong, C.P.; Bertoletti, A.; Linn, Y.C.; Wong, S.C. CD16 is indispensable for antibody-dependent cellular cytotoxicity by human monocytes. Sci. Rep. 2016, 6, 34310. [Google Scholar] [CrossRef]
- Wu, L.; Adams, M.; Carter, T.; Chen, R.; Muller, G.; Stirling, D.; Schafer, P.; Bartlett, J.B. Lenalidomide Enhances Natural Killer Cell and Monocyte-Mediated Antibody-Dependent Cellular Cytotoxicity of Rituximab-Treated CD20+ Tumor Cells. Clin. Cancer Res. 2008, 14, 4650–4657. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Payvandi, F.; Wu, L.; Zhang, L.H.; Hariri, R.J.; Man, H.W.; Chen, R.S.; Muller, G.W.; Hughes, C.C.; Stirling, D.I.; et al. The anti-cancer drug lenalidomide inhibits angiogenesis and metastasis via multiple inhibitory effects on endo-thelial cell function in normoxic and hypoxic conditions. Microvasc. Res. 2009, 77, 78–86. [Google Scholar] [CrossRef]
- Dredge, K.; Horsfall, R.; Robinson, S.P.; Zhang, L.-H.; Lu, L.; Tang, Y.; Shirley, M.A.; Muller, G.; Schafer, P.; Stirling, D.; et al. Orally administered lenalidomide (CC-5013) is anti-angiogenic in vivo and inhibits endothelial cell migration and Akt phosphorylation in vitro. Microvasc. Res. 2005, 69, 56–63. [Google Scholar] [CrossRef]
- Piccolomo, A.; Schifone, C.P.; Strafella, V.; Specchia, G.; Musto, P.; Albano, F. Immunomodulatory Drugs in Acute Myeloid Leukemia Treatment. Cancers 2020, 12, 2528. [Google Scholar] [CrossRef]
- Ximeri, M.; Galanopoulos, A.; Klaus, M.; Parcharidou, A.; Giannikou, K.; Psyllaki, M.; Symeonidis, A.; Pappa, V.; Kartasis, Z.; Liapi, D.; et al. Effect of lenalidomide therapy on hematopoiesis of patients with myelodysplastic syndrome associated with chromosome 5q deletion. Haematologica 2009, 95, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Millan, B.; De La Guardia, R.D.; Roca-Ho, H.; Anguita, E.; Islam, A.B.M.M.K.; Romero-Moya, D.; Prieto, C.; Gutiérrez-Agüera, F.; Bejarano-Garcia, J.A.; Perez-Simon, J.A.; et al. IMiDs mobilize acute myeloid leukemia blasts to peripheral blood through downregulation of CXCR4 but fail to potentiate AraC/Idarubicin activity in preclinical models of non del5q/5q- AML. OncoImmunology 2018, 7, e1477460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, J.S.; Bhatt, S.; Fell, G.; Sperling, A.; Burgess, M.; Keshishian, H.; Yilma, B.; Brunner, A.; Neuberg, D.; Carr, S.A.; et al. Increased mitochondrial apoptotic priming with targeted therapy predicts clinical response to re-induction chemotherapy. Am. J. Hematol. 2019, 95, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Cerca, S.; Pöll, G.; Gleizes, P.-E.; Tschochner, H.; Milkereit, P. Roles of Eukaryotic Ribosomal Proteins in Maturation and Transport of Pre-18S rRNA and Ribosome Function. Mol. Cell 2005, 20, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.L.; Pretz, J.; Bosco, J.; Chang, C.Y.; Tamayo, P.; Galili, N.; Raza, A.; Root, D.E.; Attar, E.; Ellis, S.R.; et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 2008, 451, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.L.; Drynan, L.F.; Hewett, D.R.; Holmes, L.; Lorenzo-Abalde, S.; Lane, A.L.; E Jolin, H.; Pannell, R.; Middleton, A.J.; Wong, S.H.; et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q– syndrome. Nat. Med. 2009, 16, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutt, S.; Narla, A.; Lin, K.; Mullally, A.; Abayasekara, N.; Megerdichian, C.; Wilson, F.H.; Currie, T.; Khanna-Gupta, A.; Berliner, N.; et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 2011, 117, 2567–2576. [Google Scholar] [CrossRef] [Green Version]
- Margolis, S.S.; Perry, J.A.; Forester, C.M.; Nutt, L.K.; Guo, Y.; Jardim, M.J.; Thomenius, M.J.; Freel, C.D.; Darbandi, R.; Ahn, J.H.; et al. Role for the PP2A/B56delta phosphatase in regulating 14–3-3 release from Cdc25 to control mitosis. Cell 2006, 127, 759–773. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Li, H.; Jensen, M.R.; Zhang, T.; Taya, Y.; Thorgeirsson, S.S.; Prives, C. Cyclin G Recruits PP2A to Dephosphorylate Mdm2. Mol. Cell 2002, 9, 761–771. [Google Scholar] [CrossRef]
- Lopez-Girona, A.E.A.; Mendy, D.; Ito, T.; Miller, K.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.; Abbasian, M.; et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalid-omide and pomalidomide. Leukemia 2012, 26, 2326–3235. [Google Scholar] [CrossRef] [PubMed]
- Barankiewicz, J.; Szumera-Ciećkiewicz, A.; Salomon-Perzyński, A.; Wieszczy, P.; Malenda, A.; Garbicz, F.; Prochorec-Sobieszek, M.; Misiewicz-Krzemińska, I.; Juszczyński, P.; Lech-Marańda, E. The CRBN, CUL4A and DDB1 Expression Predicts the Response to Immunomodulatory Drugs and Survival of Multiple Myeloma Patients. J. Clin. Med. 2021, 10, 2683. [Google Scholar] [CrossRef]
- Heintel, D.; Rocci, A.; Ludwig, H.; Bolomsky, A.; Caltagirone, S.; Schreder, M.; Geiselhart, S.; Gisslinger, H.; Zojer, N.; Jäger, U.; et al. High expression of cereblon (CRBN) is associated with improved clinical response in patients with multiple myeloma treated with lenalidomide and dexamethasone. Br. J. Haematol. 2013, 161, 695–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.-Y.; Lin, C.-W.; Lin, H.-H.; Yao, M.; Tang, J.-L.; Wu, S.-J.; Chen, Y.-C.; Lu, H.-Y.; Hou, H.-A.; Chen, C.-Y.; et al. Expression of cereblon protein assessed by immunohistochemicalstaining in myeloma cells is associated with superior response of thalidomide- and lenalidomide-based treatment, but not bortezomib-based treatment, in patients with multiple myeloma. Ann. Hematol. 2014, 93, 1371–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.X.; Braggio, E.; Shi, C.-X.; Kortuem, K.M.; Bruins, L.A.; Schmidt, J.E.; Chang, X.-B.; Langlais, P.; Luo, M.; Jedlowski, P.; et al. Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood 2014, 124, 536–545. [Google Scholar] [CrossRef]
- Haertle, L.; Barrio, S.; Munawar, U.; Han, S.; Zhou, X.; Vogt, C.; Alonso, R.; Bittrich, M.; Ruiz-Heredia, Y.; Da-Via, M.; et al. Cereblon Enhancer Methylation and IMiD Resistance in Multiple Myeloma. Blood 2021, in press. [Google Scholar] [CrossRef]
- Jonasova, A.; Bokorová, R.; Polak, J.; Vostry, M.; Kostecka, A.; Hajkova, H.; Neuwirtova, R.; Siskova, M.; Sponerova, D.; Cermak, J.; et al. High level of full-length cereblon mRNA in lower risk myelodysplastic syndrome with isolated 5q deletion is implicated in the efficacy of lenalidomide. Eur. J. Haematol. 2014, 95, 27–34. [Google Scholar] [CrossRef]
- Lindner, S.; Krönke, J. The molecular mechanism of thalidomide analogs in hematologic malignancies. J. Mol. Med. 2016, 94, 1327–1334. [Google Scholar] [CrossRef]
- Lee, K.M.; Jo, S.; Kim, H.; Lee, J.; Park, C.-S. Functional modulation of AMP-activated protein kinase by cereblon. Biochim. Biophys. Acta (BBA)—Bioenerg. 2011, 1813, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Higgins, J.J.; Hao, J.; Kosofsky, B.E.; Rajadhyaksha, A. M Dysregulation of large-conductance Ca2+-activated K+ channel expression in nonsyndromal mental re-tardation due to a cereblon p.R419X mutation. Neurogenetics 2008, 9, 219–223. [Google Scholar] [CrossRef]
- Van Nguyen, T.; Lee, J.E.; Sweredoski, M.J.; Yang, S.J.; Jeon, S.J.; Harrison, J.S.; Yim, J.H.; Lee, S.G.; Handa, H.; Kuhlman, B.; et al. Glutamine Triggers Acetylation-Dependent Degradation of Glutamine Synthetase via the Thalidomide Receptor Cereblon. Mol. Cell 2016, 61, 809–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, E.S.; Böhm, K.; Lydeard, J.R.; Yang, H.; Stadler, M.B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G.M.; et al. Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamberlain, P.P.; Lopez-Girona, A.; Miller, K.; Carmel, G.; Pagarigan, B.; Chie-Leon, B.; Rychak, E.; Corral, L.G.; Ren, Y.J.; Wang, M.; et al. Structure of the human Cereblon–DDB1–lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat. Struct. Mol. Biol. 2014, 21, 803–809. [Google Scholar] [CrossRef]
- Georgopoulos, K.; Bigby, M.; Wang, J.-H.; Molnar, A.; Wu, P.; Winandy, S.; Sharpe, A. The ikaros gene is required for the development of all lymphoid lineages. Cell 1994, 79, 143–156. [Google Scholar] [CrossRef]
- Morgan, B.; Sun, L.; Avitahl, N.; Andrikopoulos, K.; Ikeda, T.; Gonzales, E.; Wu, P.; Neben, S.; Georgopoulos, K. Aiolos, a lymphoid restricted transcription factor that interacts with Ikaros to regulate lymphocyte differ-entiation. EMBO J. 1997, 16, 2004–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés, M.; Georgopoulos, K. Aiolos Is Required for the Generation of High Affinity Bone Marrow Plasma Cells Responsible for Long-Term Immunity. J. Exp. Med. 2004, 199, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullighan, C.; Goorha, S.; Radtke, I.; Miller, C.B.; Coustan-Smith, E.; Dalton, J.D.; Girtman, K.; Mathew, S.; Ma, J.; Pounds, S.; et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007, 446, 758–764. [Google Scholar] [CrossRef]
- Nückel, H.; Frey, U.H.; Sellmann, L.; Collins, C.H.; Dührsen, U.; Siffert, W. The IKZF3 (Aiolos) transcription factor is highly upregulated and inversely correlated with clinical pro-gression in chronic lymphocytic leukaemia. Br. J. Haematol. 2009, 144, 268–270. [Google Scholar] [CrossRef]
- Schneider, R.K.; Ademà, V.; Heckl, D.; Järås, M.; Mallo, M.; Lord, A.M.; Chu, L.P.; McConkey, M.E.; Kramann, R.; Mullally, A.; et al. Role of Casein Kinase 1A1 in the Biology and Targeted Therapy of del(5q) MDS. Cancer Cell 2014, 26, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Järås, M.; Miller, P.; Chu, L.P.; Puram, R.V.; Fink, E.C.; Schneider, R.K.; Al-Shahrour, F.; Peña-Martínez, P.; Breyfogle, L.J.; Hartwell, K.A.; et al. Csnk1a1 inhibition has p53-dependent therapeutic efficacy in acute myeloid leukemia. J. Exp. Med. 2014, 211, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.-Y.; Zhang, Z.-H.; Deng, Z.-Q.; He, P.-F.; Yao, D.-M.; Xu, Z.-J.; Wen, X.-M.; Yang, L.; Lin, J.; Qian, J. Efficacy and Safety of Lenalidomide for Treatment of Low-/Intermediate-1-Risk Myelodysplastic Syndromes with or without 5q Deletion: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0165948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komrokji, R.S.; List, A. Short- and long-term benefits of lenalidomide treatment in patients with lower-risk del(5q) myelodysplastic syndromes. Ann. Oncol. 2015, 27, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Möllgård, L.; Saft, L.; Treppendahl, M.B.; Dybedal, I.; Nørgaard, J.M.; Astermark, J.; Ejerblad, E.; Garelius, H.; Dufva, I.H.; Jansson, M.; et al. Clinical effect of increasing doses of lenalidomide in high-risk myelodysplastic syndrome and acute myeloid leukemia with chromosome 5 abnormalities. Haematologica 2011, 96, 963–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintana, F.J.; Jin, H.; Burns, E.J.; Nadeau, M.; Yeste, A.; Kumar, D.; Rangachari, M.; Zhu, C.; Xiao, S.; Seavitt, J.; et al. Aiolos promotes TH17 differentiation by directly silencing Il2 expression. Nat. Immunol. 2012, 13, 770–777. [Google Scholar] [CrossRef]
- Klein, U.; Casola, S.; Cattoretti, G.; Shen, Q.; Lia, M.; Mo, T.; Ludwig, T.; Rajewsky, K.; Dalla-Favera, R. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat. Immunol. 2006, 7, 773–782. [Google Scholar] [CrossRef]
- Sciammas, R.; Shaffer, A.L.; Schatz, J.H.; Zhao, H.; Staudt, L.M.; Singh, H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell dif-ferentiation. Immunity 2006, 25, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, A.L.; Emre, T.; Lamy, L.; Ngo, V.; Wright, G.; Xiao, W.; Powell, J.; Dave, S.; Yu, X.; Zhao, H.; et al. IRF4 addiction in multiple myeloma. Nature 2008, 454, 226–231. [Google Scholar] [CrossRef]
- Matsuoka, A.J.; Tochigi, A.; Kishimoto, M.; Nakahara, T.; Kondo, T.; Tsujioka, T.; Tasaka, T.; Tohyama, Y.; Tohyama, K. Lenalidomide induces cell death in an MDS-derived cell line with deletion of chromosome 5q by inhibition of cytokinesis. Leukemia 2010, 24, 748–755. [Google Scholar] [CrossRef] [Green Version]
- Tohyama, K.; Tohyama, Y.; Nakayama, T.; Ueda, T.; Nakamura, T.; Yoshida, Y. A novel factor-dependent human myelodysplastic cell line, MDS92, contains haemopoietic cells of several lineages. Br. J. Haematol. 1995, 91, 795–799. [Google Scholar] [CrossRef]
- Ludwig, M.-G.; Vanek, M.; Guerini, D.; Gasser, J.A.; Jones, C.E.; Junker, U.; Hofstetter, H.; Wolf, R.M.; Seuwen, K. Proton-sensing G-protein-coupled receptors. Nature 2003, 425, 93–98. [Google Scholar] [CrossRef]
- Wang, J.-Q.; Kon, J.; Mogi, C.; Tobo, M.; Damirin, A.; Sato, K.; Komachi, M.; Malchinkhuu, E.; Murata, N.; Kimura, T.; et al. TDAG8 Is a Proton-sensing and Psychosine-sensitive G-protein-coupled Receptor. J. Biol. Chem. 2004, 279, 45626–45633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seuwen, K.; Ludwig, M.G.; Wolf, R.M. Wolf, Receptors for protons or lipid messengers or both? J. Recept. Signal Transduct. Res. 2006, 26, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Radu, C.; Nijagal, A.; McLaughlin, J.; Wang, L.; Witte, O.N. Differential proton sensitivity of related G protein-coupled receptors T cell death-associated gene 8 and G2A expressed in immune cells. Proc. Natl. Acad. Sci. USA 2005, 102, 1632–1637. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.Y.; Dai, S.P.; Chang, Y.C.; Sun, W.H. Acidosis Mediates the Switching of Gs-PKA and Gi-PKCepsilon Dependence in Prolonged Hyperalgesia Induced by Inflammation. PLoS ONE 2015, 10, e0125022. [Google Scholar]
- Ligt, R.A.F.D.; Kourounakis, A.P.; Ijzerman, A.P. Inverse agonism at G protein-coupled receptors: (patho)physiological relevance and implications for drug discovery. Br. J. Pharmacol. 2000, 130, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Justus, C.R.; Dong, L.; Yang, L.V. Acidic tumor microenvironment and pH-sensing G protein-coupled receptors. Front. Physiol. 2013, 4, 354. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.-W.; Lee, S.Y.; Choi, Y. Identification of a Putative G Protein-Coupled Receptor Induced during Activation-Induced Apoptosis of T Cells. Cell. Immunol. 1996, 168, 78–84. [Google Scholar] [CrossRef]
- Mogi, C.; Tobo, M.; Tomura, H.; Murata, N.; He, X.-D.; Sato, K.; Kimura, T.; Ishizuka, T.; Sasaki, T.; Sato, T.; et al. Involvement of Proton-Sensing TDAG8 in Extracellular Acidification-Induced Inhibition of Proinflammatory Cytokine Production in Peritoneal Macrophages. J. Immunol. 2009, 182, 3243–3251. [Google Scholar] [CrossRef]
- Justus, C.R.; Sanderlin, E.J.; Dong, L.; Sun, T.; Chi, J.T.; Lertpiriyapong, K.; Yang, L.V. Contextual tumor suppressor function of T cell death-associated gene 8 (TDAG8) in hematological malig-nancies. J. Transl. Med. 2017, 15, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nii, T.; Prabhu, V.V.; Ruvolo, V.; Madhukar, N.; Zhao, R.; Mu, H.; Heese, L.; Nishida, Y.; Kojima, K.; Garnett, M.J.; et al. Imipridone ONC212 activates orphan G protein-coupled receptor GPR132 and integrated stress response in acute myeloid leukemia. Leukemia 2019, 33, 2805–2816. [Google Scholar] [CrossRef]
- He, X.; Feng, S.; Hawkins, C.; Lawley, L.; Fan, W.; Xu, Y.; Zha, X.; Fang, J. G protein-coupled receptor 68 increases the number of B lymphocytes. Am. J. Blood Res. 2020, 10, 15–21. [Google Scholar] [PubMed]
- He, X.; Hawkins, C.; Lawley, L.; Freeman, K.; Phan, T.M.; Zhang, J.; Xu, Y.; Fang, J. Whole body deletion of Gpr68 does not change hematopoietic stem cell function. Stem Cell Res. 2020, 47, 101869. [Google Scholar] [CrossRef] [PubMed]
- Arron, J.; Winslow, M.M.; Polleri, A.; Chang, C.-P.; Wu, H.; Gao, X.; Neilson, J.R.; Chen, L.; Heit, J.J.; Kim, S.K.; et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 2006, 441, 595–600. [Google Scholar] [CrossRef]
- Rusnak, F.; Mertz, P. Calcineurin: Form and Function. Physiol. Rev. 2000, 80, 1483–1521. [Google Scholar] [CrossRef]
- Ding, Y.; Zhang, B.; Payne, J.; Song, C.; Ge, Z.; Gowda, C.; Iyer, S.; Dhanyamraju, P.K.; Dorsam, G.; Reeves, M.E.; et al. Ikaros tumor suppressor function includes induction of active enhancers and super-enhancers along with pioneering activity. Leukemia 2019, 33, 2720–2731. [Google Scholar] [CrossRef] [Green Version]
- Flores, C.; Fouquet, G.; Moura, I.C.; Maciel, T.T.; Hermine, O. Lessons to Learn From Low-Dose Cyclosporin-A: A New Approach for Unexpected Clinical Applications. Front. Immunol. 2019, 10, 588. [Google Scholar] [CrossRef] [Green Version]
- Wiederrecht, G.; Lam, E.; Hung, S.; Martin, M.; Sigal, N. The Mechanism of Action of FK-506 and Cyclosporin A. Ann. N. Y. Acad. Sci. 2006, 696, 9–19. [Google Scholar] [CrossRef]
- Brandt, C.; Liman, P.; Bendfeldt, H.; Mueller, K.; Reinke, P.; Radbruch, A.; Worm, M.; Baumgrass, R. Whole blood flow cytometric measurement of NFATc1 and IL-2 expression to analyze cyclosporine A-mediated effects in T cells. Cytom. Part A 2010, 77, 607–613. [Google Scholar] [CrossRef]
- Kronke, M.; Leonard, W.J.; Depper, J.M.; Arya, S.K.; Wong-Staal, F.; Gallo, R.C.; Waldmann, T.A.; Greene, W.C. Cyclosporin A inhibits T-cell growth factor gene expression at the level of mRNA transcription. Proc. Natl. Acad. Sci. USA 1984, 81, 5214–5218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brini, A.T.; Harel-Bellan, A.; Farrar, W.L. Cyclosporin A inhibits induction of IL-2 receptor alpha chain expression by affecting activation of NF-kB-like factor(s) in cultured human T lymphocytes. Eur. Cytokine Netw. 1990, 1, 131–139. [Google Scholar] [PubMed]
- Kasaian, M.T.; A Biron, C. Effects of cyclosporin A on IL-2 production and lymphocyte proliferation during infection of mice with lymphocytic choriomeningitis virus. J. Immunol. 1990, 144, 299–306. [Google Scholar] [PubMed]
- Kolata, G. FDA speeds approval of cyclosporin. Science 1983, 221, 1273. [Google Scholar] [CrossRef]
- Bretscher, P.A.; Havele, C. Cyclosporin A can switch the immune response induced by antigen from a humoral to a cell-mediated mode. Eur. J. Immunol. 1992, 22, 349–355. [Google Scholar] [CrossRef]
- Prud’Homme, G.J.; A Parfrey, N.; E Vanier, L. Cyclosporine-induced autoimmunity and immune hyperreactivity. Autoimmunity 1991, 9, 345–356. [Google Scholar] [CrossRef]
- Mustafa, M.; Diener, P.; Sun, J.-B.; Link, H.; Olsson, T. Immunopharmacologic Modulation of Experimental Allergic Encephalomyelitis: Low-Dose Cyclosporin-A Treatment Causes Disease Relapse and Increased Systemic T and B Cell-Mediated Myelin-Directed Autoimmunity. Scand. J. Immunol. 1993, 38, 499–507. [Google Scholar] [CrossRef]
- Miroux, C.; Morales, O.; Carpentier, A.; Dharancy, S.; Conti, F.; Boleslowski, E.; Podevin, P.; Auriault, C.; Pancré, V.; Delhem, N. Inhibitory Effects of Cyclosporine on Human Regulatory T Cells In Vitro. Transplant. Proc. 2009, 41, 3371–3374. [Google Scholar] [CrossRef]
- López-Flores, R.; Bojalil, R.; Benítez, J.C.; Ledesma-Soto, Y.; Terrazas, C.A.; Rodríguez-Sosa, M.; Terrazas, L.I. Consecutive Low Doses of Cyclosporine A Induce Pro-Inflammatory Cytokines and Accelerate Allograft Skin Rejection. Molecules 2011, 16, 3969. [Google Scholar] [CrossRef]
- Lowe, N.J.; Wieder, J.M.; Rosenbach, A.; Johnson, K.; Kunkel, R.; Bainbridge, C.; Bourget, T.; Dimov, I.; Simpson, K.; Glass, E.; et al. Long-term low-dose cyclosporine therapy for severe psoriasis: Effects on renal function and structure. J. Am. Acad. Dermatol. 1996, 35, 710–719. [Google Scholar] [CrossRef]
- Bagnis, C.I.; Du Montcel, S.T.; Beaufils, H.; Jouanneau, C.; Jaudon, M.C.; Maksud, P.; Mallet, A.; LeHoang, P.; Deray, G. Long-term renal effects of low-dose cyclosporine in uveitis-treated patients: Follow-up study. J. Am. Soc. Nephrol. 2002, 13, 2962–2968. [Google Scholar] [CrossRef] [Green Version]
- Kessel, A.; Toubi, E. Low-dose cyclosporine is a good option for severe chronic urticaria. J. Allergy Clin. Immunol. 2009, 123, 970–971. [Google Scholar] [CrossRef]
- Ross, H.J.; Cho, J.; Osann, K.; Wong, S.-F.; Ramsinghani, N.; Williams, J.; Downey-Hurtado, N.; Slater, L.M. Phase I/II trial of low dose cyclosporin A with EP for advanced non-small cell lung cancer1. Lung Cancer 1997, 18, 189–198. [Google Scholar] [CrossRef]
- Cooper, D.L.; Braverman, I.M.; Sarris, A.H.; Durivage, H.J.; Saidman, B.H.; Davis, C.A.; Hait, W.N. Cyclosporine treatment of refractory T-cell lymphomas. Cancer 1993, 71, 2335–2341. [Google Scholar] [CrossRef]
- Isshiki, Y.; Tanaka, H.; Suzuki, Y.; Yoshida, Y. Cyclosporine is a potential curative treatment option for advanced thymoma. Exp. Hematol. Oncol. 2017, 6, 13. [Google Scholar] [CrossRef]
- Yamada, T.; Tsurumi, H.; Kasahara, S.; Hara, T.; Sawada, M.; Moriwaki, H. Immunosuppressive therapy for myelodysplastic syndrome: Efficacy of methylprednisolone pulse therapy with or without cyclosporin A. J. Cancer Res. Clin. Oncol. 2003, 129, 485–491. [Google Scholar] [CrossRef]
- Ishikawa, T.; Tohyama, K.; Nakao, S.; Yoshida, Y.; Teramura, M.; Motoji, T.; Takatoku, M.; Kurokawa, M.; Mitani, K.; Uchiyama, T.; et al. A Prospective Study of Cyclosporine A Treatment of Patients with Low-Risk Myelodysplastic Syndrome: Presence of CD55-CD59- Blood Cells Predicts Platelet Response. Int. J. Hematol. 2007, 86, 150–157. [Google Scholar] [CrossRef]
- Ogata, M.; Ohtsuka, E.; Imamura, T.; Ikewaki, J.; Ogata, Y.; Kohno, K.; Nakayama, T.; Ono, K.; Saburi, Y.; Kikuchi, H.; et al. Response to Cyclosporine Therapy in Patients with Myelodysplastic Syndrome: A Clinical Study of 12 Cases and Literature Review. Int. J. Hematol. 2004, 80, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Deveaux, M.; De Witte, T.; Neukirchen, J.; Sekeres, M.A.; Brunner, A.M.; Roboz, G.J.; Steensma, D.P.; Bhatt, V.R.; Platzbecker, U.; et al. The use of immunosuppressive therapy in MDS: Clinical outcomes and their predictors in a large international patient cohort. Blood Adv. 2018, 2, 1765–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haider, M.; Al Ali, N.; Padron, E.; Epling-Burnette, P.; Lancet, J.; List, A.; Komrokji, R. Immunosuppressive Therapy: Exploring an Underutilized Treatment Option for Myelodysplastic Syndrome. Clin. Lymphoma Myeloma Leuk. 2016, 16, S44–S48. [Google Scholar] [CrossRef] [PubMed]
- Gafter-Gvili, A.; Ram, R.; Gurion, R.; Paul, M.; Yeshurun, M.; Raanani, P.; Shpilberg, O. ATG plus Cyclosporine Reduces All-Cause Mortality in Patients with Severe Aplastic Anemia – Systematic Review and Meta-Analysis. Acta Haematol. 2008, 120, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Reeves, J.A.; Feldman, E.J.; Dewald, G.W.; Bennett, J.M.; Deeg, H.J.; Dreisbach, L.; Schiffer, C.A.; Stone, R.M.; Greenberg, P.L.; et al. Phase 2 study of lenalidomide in transfusion-dependent, low-risk, and intermediate-1 risk myelodysplastic syndromes with karyotypes other than deletion 5q. Blood 2008, 111, 86–93. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, I.; Phan, T.M.; Fang, J. Novel Molecular Mechanism of Lenalidomide in Myeloid Malignancies Independent of Deletion of Chromosome 5q. Cancers 2021, 13, 5084. https://doi.org/10.3390/cancers13205084
Park I, Phan TM, Fang J. Novel Molecular Mechanism of Lenalidomide in Myeloid Malignancies Independent of Deletion of Chromosome 5q. Cancers. 2021; 13(20):5084. https://doi.org/10.3390/cancers13205084
Chicago/Turabian StylePark, Isaac, Tra Mi Phan, and Jing Fang. 2021. "Novel Molecular Mechanism of Lenalidomide in Myeloid Malignancies Independent of Deletion of Chromosome 5q" Cancers 13, no. 20: 5084. https://doi.org/10.3390/cancers13205084
APA StylePark, I., Phan, T. M., & Fang, J. (2021). Novel Molecular Mechanism of Lenalidomide in Myeloid Malignancies Independent of Deletion of Chromosome 5q. Cancers, 13(20), 5084. https://doi.org/10.3390/cancers13205084