
Peutz–Jeghers Syndrome and the Role of Imaging: Pathophysiology, Diagnosis, and Associated Cancers

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Diagnostic Criteria
- Two or more pathologically confirmed Peutz-Jeghers polyps;
- Any Peutz-Jeghers polyps and family history of Peutz-Jeghers;
- Characteristic mucocutaneous pigmentations involving the mouth, lips, nose, eyes, genitalia or fingers with a family history of PJS;
- Any Peutz-Jeghers polyps in patients with characteristic mucocutaneous pigmentations.
3. Genomics and Pathophysiology
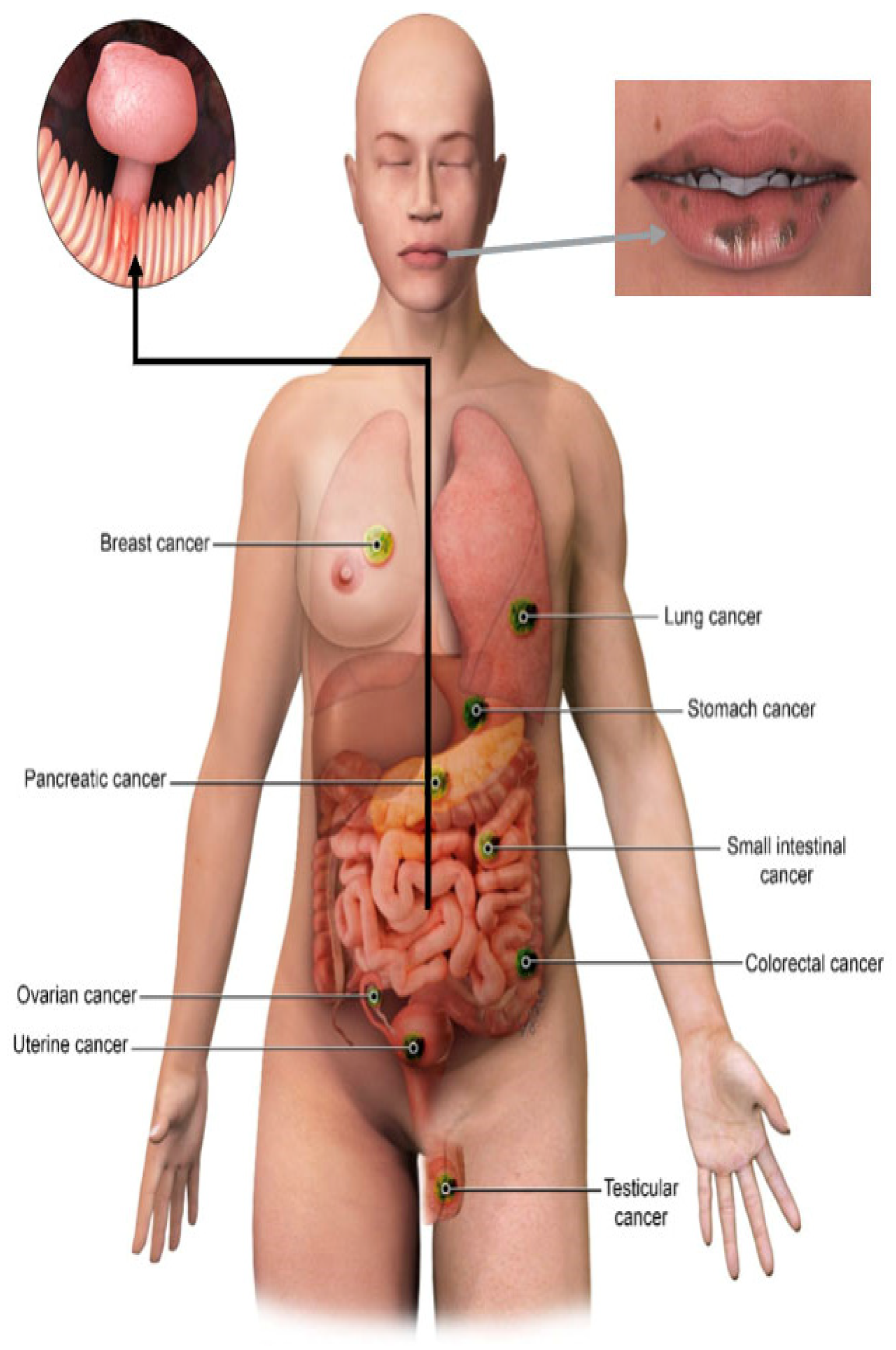
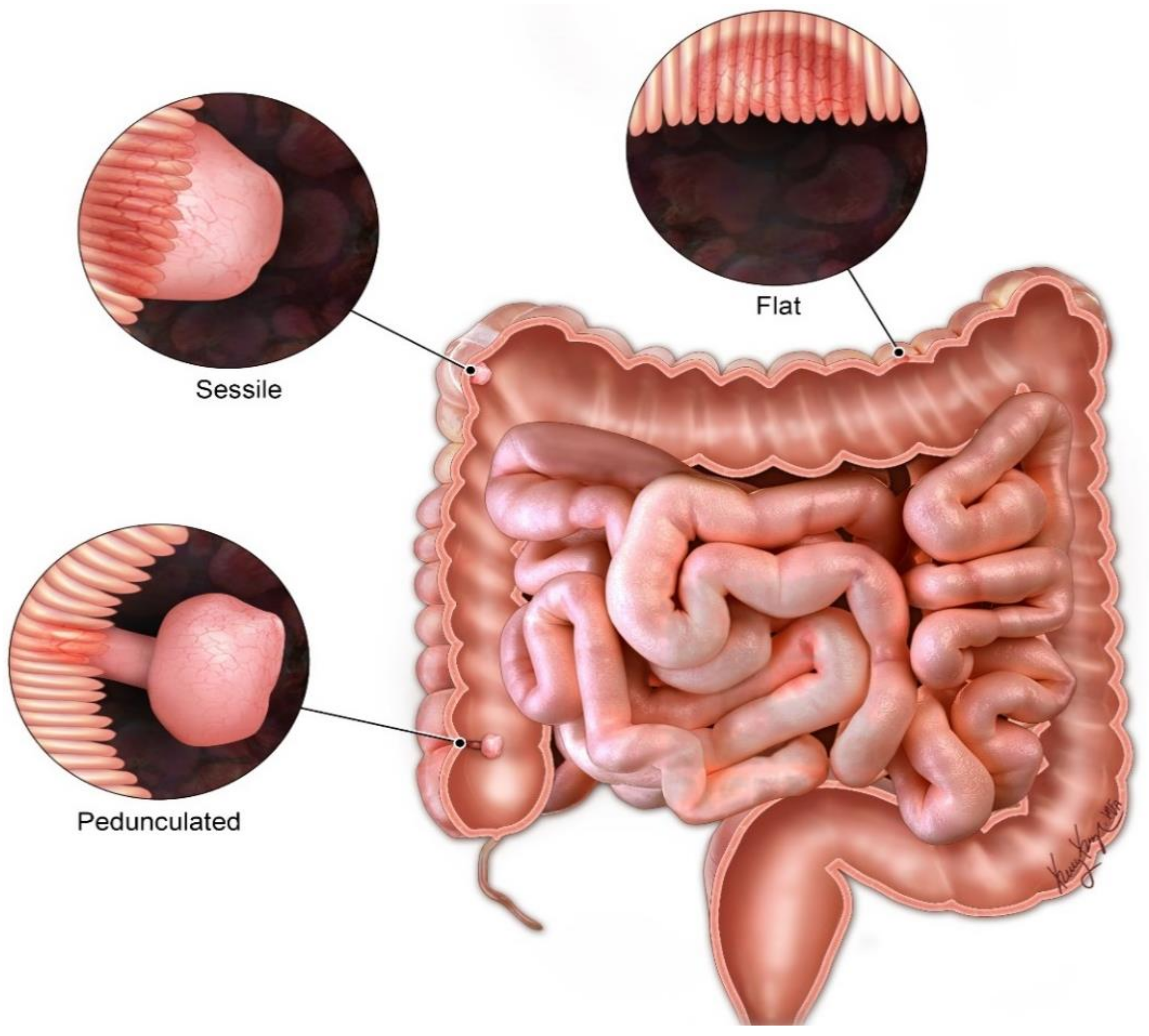
4. Clinical Manifestations





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Usual Intussusception | PJS-Associated Intussusception | |
|---|---|---|
| Cause | ldiopathic, viralinfection, Meckel’s diverticulum | Hamartomatous Polyp (leading point) |
| Age | First 2 years of life | Usually > 10 years old |
| Site | Usually ileocecal | Usually jejuno-jejunal or ileo-ileal |
| Treatment | Usually air or saline enema is enough | Usually surgery or enteroscopy |
| Prognosis | Excellent | Responsible for up to 30% of PJS-associated mortality |
5. Diagnostic Challenges and Differential Considerations
6. Common Malignancies in PJS and Imaging Screening Protocols
6.1. Gastrointestinal Malignancies
6.2. Pancreatic Cancer
6.3. Gynecologic Cancers
6.4. Breast Cancer
6.5. Testicular Cancer
6.6. Lung Cancer
7. Treatment Approach
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Beggs, A.D.; Latchford, A.R.; Vasen, H.F.; Moslein, G.; Alonso, A.; Aretz, S.; Bertario, L.; Blanco, I.; Bulow, S.; Burn, J.; et al. Peutz-Jeghers syndrome: A systematic review and recommendations for management. Gut 2010, 59, 975–986. [Google Scholar] [CrossRef] [Green Version]
- Jeghers, H.; Mc, K.V.; Katz, K.H. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N. Engl. J. Med. 1949, 241, 1031–1036. [Google Scholar] [CrossRef]
- Utsunomiya, J.; Gocho, H.; Miyanaga, T.; Hamaguchi, E.; Kashimure, A. Peutz-Jeghers syndrome: Its natural course and management. Johns Hopkins Med. J. 1975, 136, 71–82. [Google Scholar]
- Burt, R.W. Polyposis Syndrome. Clin. Perspectives Gastro 2002, 51–59. [Google Scholar]
- Amos, C.I.; Keitheri-Cheteri, M.B.; Sabripour, M.; Wei, C.; McGarrity, T.J.; Seldin, M.F.; Nations, L.; Lynch, P.M.; Fidder, H.H.; Friedman, E.; et al. Genotype-phenotype correlations in Peutz-Jeghers syndrome. J. Med. Genet. 2004, 41, 327–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehenni, H.; Resta, N.; Guanti, G.; Mota-Vieira, L.; Lerner, A.; Peyman, M.; Chong, K.A.; Aissa, L.; Ince, A.; Cosme, A.; et al. Molecular and clinical characteristics in 46 families affected with Peutz-Jeghers syndrome. Dig. Dis. Sci. 2007, 52, 1924–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aretz, S.; Stienen, D.; Uhlhaas, S.; Loff, S.; Back, W.; Pagenstecher, C.; McLeod, D.R.; Graham, G.E.; Mangold, E.; Santer, R.; et al. High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome. Hum. Mutat. 2005, 26, 513–519. [Google Scholar] [CrossRef]
- Boardman, L.A.; Couch, F.J.; Burgart, L.J.; Schwartz, D.; Berry, R.; McDonnell, S.K.; Schaid, D.J.; Hartmann, L.C.; Schroeder, J.J.; Stratakis, C.A.; et al. Genetic heterogeneity in Peutz-Jeghers syndrome. Hum. Mutat. 2000, 16, 23–30. [Google Scholar] [CrossRef]
- Hearle, N.; Schumacher, V.; Menko, F.H.; Olschwang, S.; Boardman, L.A.; Gille, J.J.; Keller, J.J.; Westerman, A.M.; Scott, R.J.; Lim, W.; et al. STK11 status and intussusception risk in Peutz-Jeghers syndrome. J. Med. Genet. 2006, 43, e41. [Google Scholar] [CrossRef] [Green Version]
- Hearle, N.; Schumacher, V.; Menko, F.H.; Olschwang, S.; Boardman, L.A.; Gille, J.J.; Keller, J.J.; Westerman, A.M.; Scott, R.J.; Lim, W.; et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin. Cancer Res. 2006, 12, 3209–3215. [Google Scholar] [CrossRef] [Green Version]
- Giardiello, F.M.; Brensinger, J.D.; Tersmette, A.C.; Goodman, S.N.; Petersen, G.M.; Booker, S.V.; Cruz-Correa, M.; Offerhaus, J.A. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000, 119, 1447–1453. [Google Scholar] [CrossRef] [Green Version]
- van Lier, M.G.F.; Wagner, A.; Mathus-Vliegen, E.M.H.; Kuipers, E.J.; Steyerberg, E.W.; van Leerdam, M.E. High Cancer Risk in Peutz-Jeghers Syndrome: A Systematic Review and Surveillance Recommendations. Am. J. Gastroenterol. 2010, 105, 1258–1264. [Google Scholar] [CrossRef]
- Foley, T.R.; McGarrity, T.J.; Abt, A.B. Peutz-Jeghers syndrome: A clinicopathologic survey of the “Harrisburg family” with a 49-year follow-up. Gastroenterology 1988, 95, 1535–1540. [Google Scholar] [CrossRef]
- Perzin, K.H.; Bridge, M.F. Adenomatous and carcinomatous changes in hamartomatous polyps of the small intestine (Peutz-Jeghers syndrome): Report of a case and review of the literature. Cancer 1982, 49, 971–983. [Google Scholar] [CrossRef] [Green Version]
- Hizawa, K.; Iida, M.; Matsumoto, T.; Kohrogi, N.; Yao, T.; Fujishima, M. Neoplastic transformation arising in Peutz-Jeghers polyposis. Dis. Colon. Rectum. 1993, 36, 953–957. [Google Scholar] [CrossRef]
- Narita, T.; Eto, T.; Ito, T. Peutz-Jeghers syndrome with adenomas and adenocarcinomas in colonic polyps. Am. J. Surg. Pathol. 1987, 11, 76–81. [Google Scholar] [CrossRef]
- Miller, L.J.; Bartholomew, L.G.; Dozois, R.R.; Dahlin, D.C. Adenocarcinoma of the rectum arising in a hamartomatous polyp in a patient with Peutz-Jeghers syndrome. Dig. Dis. Sci. 1983, 28, 1047–1051. [Google Scholar] [CrossRef]
- Spigelman, A.D.; Murday, V.; Phillips, R.K. Cancer and the Peutz-Jeghers syndrome. Gut 1989, 30, 1588–1590. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.; de Leng, W.W.; Baas, A.F.; Myoshi, H.; Mathus-Vliegen, L.; Taketo, M.M.; Clevers, H.; Giardiello, F.M.; Offerhaus, G.J. Mucosal prolapse in the pathogenesis of Peutz-Jeghers polyposis. Gut 2006, 55, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGarrity, T.J.; Kulin, H.E.; Zaino, R.J. Peutz-Jeghers syndrome. Am. J. Gastroenterol. 2000, 95, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Lindor, N.M.; McMaster, M.L.; Lindor, C.J.; Greene, M.H. Prevention Trials Research Concise handbook of familial cancer susceptibility syndromes-second edition. J. Natl. Cancer Inst. Monogr. 2008, 38, 1–93. [Google Scholar]
- Gammon, A.; Jasperson, K.; Kohlmann, W.; Burt, R.W. Hamartomatous polyposis syndromes. Best Pract. Res. Clin. Gastroenterol. 2009, 23, 219–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartholomew, L.G.; Dahlin, D.C.; Waugh, J.M. Intestinal polyposis associated with mucocutaneous melanin pigmentation (Peutz-Jeghers syndrome). Proc. Staff Meet Mayo Clin. 1957, 32, 675–680. [Google Scholar] [CrossRef]
- Dippolito, A.D.; Aburano, A.; Bezouska, C.A.; Happ, R.A. Enteritis cystica profunda in Peutz-Jeghers syndrome. Report of a case and review of the literature. Dis. Colon. Rectum. 1987, 30, 192–198. [Google Scholar] [CrossRef]
- Harned, R.K.; Buck, J.L.; Sobin, L.H. The hamartomatous polyposis syndromes: Clinical and radiologic features. AJR Am. J. Roentgenol. 1995, 164, 565–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arber, N.; Moshkowitz, M. Small bowel polyposis syndromes. Curr. Gastroenterol. Rep. 2011, 13, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Calva, D.; Howe, J.R. Hamartomatous polyposis syndromes. Surg. Clin. N. Am. 2008, 88, 779–817. [Google Scholar] [CrossRef] [Green Version]
- Duan, N.; Zhang, Y.H.; Wang, W.M.; Wang, X. Mystery behind labial and oral melanotic macules: Clinical, dermoscopic and pathological aspects of Laugier-Hunziker syndrome. World J. Clin. Cases 2018, 6, 322–334. [Google Scholar] [CrossRef]
- Nayak, R.S.; Kotrashetti, V.S.; Hosmani, J.V. Laugier-Hunziker syndrome. J. Oral Maxillofac. Pathol. 2012, 16, 245–250. [Google Scholar] [CrossRef]
- Kurugoglu, S.; Aksoy, H.; Kantarci, F.; Cetinkaya, S.; Mihmanli, I.; Korman, U. Radiological work-up in Peutz-Jeghers syndrome. Pediatr. Radiol. 2003, 33, 766–771. [Google Scholar] [CrossRef]
- Gupta, A.; Postgate, A.J.; Burling, D.; Ilangovan, R.; Marshall, M.; Phillips, R.K.; Clark, S.K.; Fraser, C.H. A prospective study of MR enterography versus capsule endoscopy for the surveillance of adult patients with Peutz-Jeghers syndrome. AJR Am. J. Roentgenol. 2010, 195, 108–116. [Google Scholar] [CrossRef]
- Burke, C.A.; Santisi, J.; Church, J.; Levinthal, G. The utility of capsule endoscopy small bowel surveillance in patients with polyposis. Am. J. Gastroenterol. 2005, 100, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.; Lopes, L.; Boas, G.V.; Pinho, C. Wireless capsule endoscopy for evaluation of phenotypic expression of small-bowel polyps in patients with Peutz-Jeghers syndrome and in symptomatic first-degree relatives. Endoscopy 2004, 36, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Syngal, S.; Brand, R.E.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W.; American College of Gastroenterology. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am. J. Gastroenterol. 2015, 110, 223–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Signoretti, M.; Bruno, M.J.; Zerboni, G.; Poley, J.W.; Fave, G.D.; Capurso, G. Results of surveillance in individuals at high-risk of pancreatic cancer: A systematic review and meta-analysis. United Eur. Gastroenterol. J. 2018, 6, 489–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, T.; Abe, T.; Amano, H.; Hanada, K.; Minami, T.; Kobayashi, T.; Yonehara, S.; Nakahara, M.; Ohdan, H.; Noriyuki, T. Metachronous triple cancer associated with Peutz-Jeghers syndrome treated with curative surgery: A case report. Surg. Case Rep. 2018, 4, 84. [Google Scholar] [CrossRef] [Green Version]
- Hruban, R.H.; Canto, M.I.; Yeo, C.J. Prevention of pancreatic cancer and strategies for management of familial pancreatic cancer. Dig. Dis. 2001, 19, 76–84. [Google Scholar] [CrossRef]
- Ki, E.Y.; Byun, S.W.; Park, J.S.; Lee, S.J.; Hur, S.Y. Adenoma malignum of the uterine cervix: Report of four cases. World J. Surg. Oncol. 2013, 11, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, M.C.; Marks, J.H.; Mandell, J.B.; New York Breast Cancer Study Group. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef]
- Begg, C.B. On the use of familial aggregation in population-based case probands for calculating penetrance. J. Natl. Cancer Inst. 2002, 94, 1221–1226. [Google Scholar] [CrossRef]
- Ford, D.; Easton, D.F.; Stratton, M.; Narod, S.; Goldgar, D.; Devilee, P.; Bishop, D.T.; Weber, B.; Lenoir, G.; Chang-Claude, J.; et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. Am. J. Hum. Genet. 1998, 62, 676–689. [Google Scholar] [CrossRef] [Green Version]
- Lim, W.; Olschwang, S.; Keller, J.J.; Westerman, A.M.; Menko, F.H.; Boardman, L.A.; Scott, R.J.; Trimbath, J.; Giardiello, F.M.; Gruber, S.B.; et al. Relative frequency and morphology of cancers in STK11 mutation carriers. Gastroenterology 2004, 126, 1788–1794. [Google Scholar] [CrossRef]
- van Lier, M.G.; Mathus-Vliegen, E.M.; Wagner, A.; van Leerdam, M.E.; Kuipers, E.J. High cumulative risk of intussusception in patients with Peutz-Jeghers syndrome: Time to update surveillance guidelines? Am. J. Gastroenterol. 2011, 106, 940–945. [Google Scholar] [CrossRef]
- Oncel, M.; Remzi, F.H.; Church, J.M.; Connor, J.T.; Fazio, V.W. Benefits of ‘clean sweep’ in Peutz-Jeghers patients. Colorectal. Dis. 2004, 6, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Kopacova, M.; Tacheci, I.; Rejchrt, S.; Bures, J. Peutz-Jeghers syndrome: Diagnostic and therapeutic approach. World J. Gastroenterol. 2009, 15, 5397–5408. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Amos, C.I.; Zhang, N.; Wang, X.; Rashid, A.; Walker, C.L.; Behringer, R.R.; Frazier, M.L. Suppression of Peutz-Jeghers polyposis by targeting mammalian target of rapamycin signaling. Clin. Cancer Res. 2008, 14, 1167–1171. [Google Scholar] [CrossRef] [Green Version]
- de Brabander, J.; Eskens, F.; Korsse, S.E.; Dekker, E.; Dewint, P.; van Leerdam, M.E.; van Eeden, S.; Klumpen, H.J. Chemoprevention in Patients with Peutz-Jeghers Syndrome: Lessons Learned. Oncologist 2018, 23, 399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udd, L.; Katajisto, P.; Rossi, D.J.; Lepisto, A.; Lahesmaa, A.M.; Ylikorkala, A.; Jarvinen, H.J.; Ristimaki, A.P.; Makela, T.P. Suppression of Peutz-Jeghers polyposis by inhibition of cyclooxygenase-2. Gastroenterology 2004, 127, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Sosnicki, S.; Kapral, M.; Weglarz, L. Molecular targets of metformin antitumor action. Pharmacol. Rep. 2016, 68, 918–925. [Google Scholar] [CrossRef]
- Huang, X.; Wullschleger, S.; Shpiro, N.; McGuire, V.A.; Sakamoto, K.; Woods, Y.L.; McBurnie, W.; Fleming, S.; Alessi, D.R. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem. J. 2008, 412, 211–221. [Google Scholar] [CrossRef]
- Lefevre, H.; Bouvattier, C.; Lahlou, N.; Adamsbaum, C.; Bougneres, P.; Carel, J.C. Prepubertal gynecomastia in Peutz-Jeghers syndrome: Incomplete penetrance in a familial case and management with an aromatase inhibitor. Eur. J. Endocrinol. 2006, 154, 221–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S.; Takeyama, J.; Tanita, Y.; Ebina, K. Ruby laser therapy for labial lentigines in Peutz-Jeghers syndrome. Eur. J. Pediatr. 1998, 157, 622–624. [Google Scholar] [CrossRef] [PubMed]
- Benedict, L.M.; Cohen, B. Treatment of Peutz-Jeghers lentigines with the carbon dioxide laser. J. Dermatol. Surg. Oncol. 1991, 17, 954–955. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Nelson, J.S. Q-switched ruby laser treatment of mucocutaneous melanosis associated with Peutz-Jeghers syndrome. Ann. Plast Surg. 1996, 36, 394–397. [Google Scholar] [CrossRef]
- Remington, B.K.; Remington, T.K. Treatment of facial lentigines in Peutz-Jeghers syndrome with an intense pulsed light source. Dermatol. Surg. 2002, 28, 1079–1081. [Google Scholar] [CrossRef] [PubMed]





| Location | Lifetime Risk of Developing Malignancy, % | Surveillance Method | Interval of Surveillance | Age to Initiate Surveillance, Years |
|---|---|---|---|---|
| Breast | 32–54 | Mammogram Breast MRI Clinical breast exam | Yearly Yearly Every 6 months | 30 |
| Colon | 39 | Colonoscopy | Every 2–3 years | 18 |
| Stomach | 29 | Endoscopy | Every 2–3 years | 18 |
| Small Intestine | 13 | Video capsule endoscopy or CT/MRI enterography | Every 2–3 years | 18 |
| Pancreas | 11–36 | Endoscopic US or MRI/MRCP | Yearly | 30–35 |
| Cervix | 10 | Pelvic exam/ Pap smear | Yearly | 18–20 |
| Uterus | 9 | Pelvic exam/ Pap smear | Yearly | 18–20 |
| Ovary | 18–21 | Pelvic exam/ Pap smear | Yearly | 18–20 |
| Lung | 7–17 | NA |
| Location | Screening Targets | Surveillance Method | Interval of Surveillance | Age to Initiate Surveillance, Years |
|---|---|---|---|---|
| Colon Cancer Stomach | Bleeding Iron deficiency anemia | Upper endoscopy and colonoscopy | If polyps found then repeat every 2–3 years. If no polyps found, then resume at 18 y Yearly | 8–10 |
| Small Intestine | Intussusception Bleeding Iron deficiency anemia | Video capsule endoscopy or CT/MRI enterography | Every 2–3 years | 8–10 Can start earlier or image more frequently if findings and symptoms warrant |
| Ovary | Sex cord tumor with annular tubules | Physical exam and close observation for precocious puberty | Annually | 8 |
| Testes | Sertoli cell tumors | Physical exam and close observation for feminizing changes | Annually | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klimkowski, S.; Ibrahim, M.; Ibarra Rovira, J.J.; Elshikh, M.; Javadi, S.; Klekers, A.R.; Abusaif, A.A.; Moawad, A.W.; Ali, K.; Elsayes, K.M. Peutz–Jeghers Syndrome and the Role of Imaging: Pathophysiology, Diagnosis, and Associated Cancers. Cancers 2021, 13, 5121. https://doi.org/10.3390/cancers13205121
Klimkowski S, Ibrahim M, Ibarra Rovira JJ, Elshikh M, Javadi S, Klekers AR, Abusaif AA, Moawad AW, Ali K, Elsayes KM. Peutz–Jeghers Syndrome and the Role of Imaging: Pathophysiology, Diagnosis, and Associated Cancers. Cancers. 2021; 13(20):5121. https://doi.org/10.3390/cancers13205121
Chicago/Turabian StyleKlimkowski, Sergio, Mohamed Ibrahim, Juan J. Ibarra Rovira, Mohamed Elshikh, Sanaz Javadi, Albert R. Klekers, Abdelraham A. Abusaif, Ahmed W. Moawad, Kamran Ali, and Khaled M. Elsayes. 2021. "Peutz–Jeghers Syndrome and the Role of Imaging: Pathophysiology, Diagnosis, and Associated Cancers" Cancers 13, no. 20: 5121. https://doi.org/10.3390/cancers13205121
APA StyleKlimkowski, S., Ibrahim, M., Ibarra Rovira, J. J., Elshikh, M., Javadi, S., Klekers, A. R., Abusaif, A. A., Moawad, A. W., Ali, K., & Elsayes, K. M. (2021). Peutz–Jeghers Syndrome and the Role of Imaging: Pathophysiology, Diagnosis, and Associated Cancers. Cancers, 13(20), 5121. https://doi.org/10.3390/cancers13205121