
Uncommon Variants of Mature T-Cell Lymphomas (MTCLs): Imaging and Histopathologic and Clinical Features with Updates from the Fourth Edition of the World Health Organization (WHO) Classification of Lymphoid Neoplasms

Abstract
:Simple Summary
Abstract
1. Introduction
2. Changes Contained in the Updated 2016 WHO Classification of T-Cell Lymphoid Malignancies
3. Uncommon Mature T-Cell Lymphoma Variants
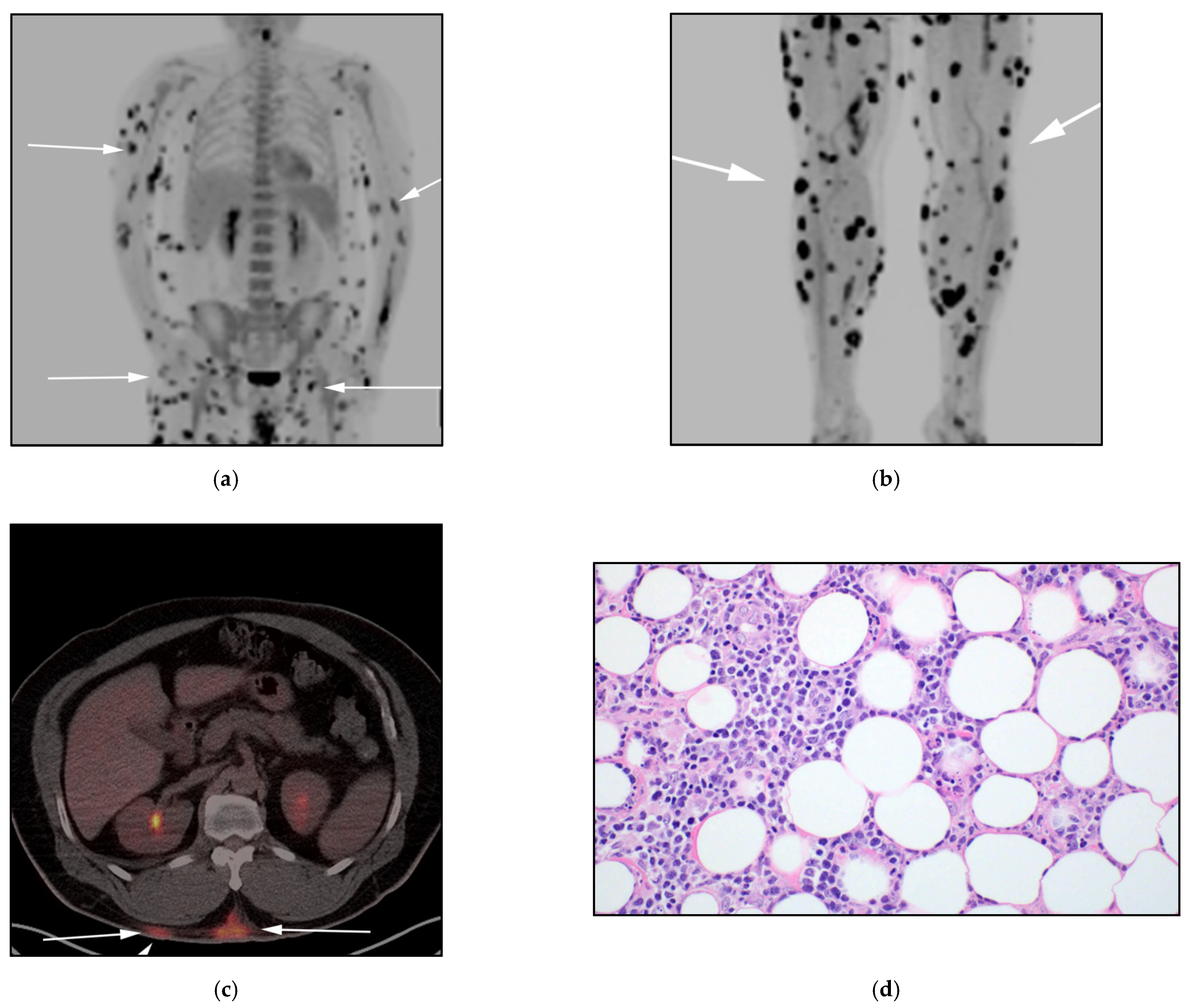
3.1. Subcutaneous Panniculitis-like T-Cell Lymphoma (SPTCL)
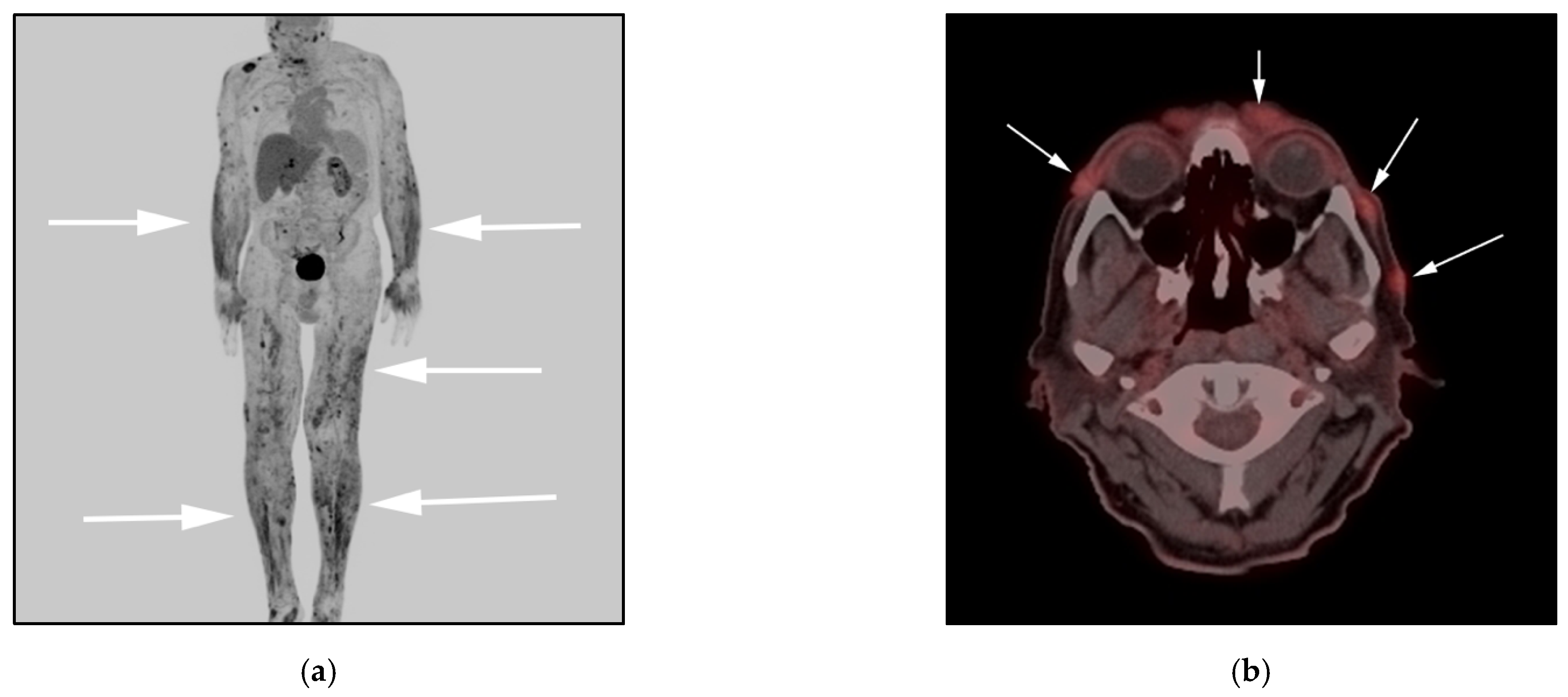
3.2. Anaplastic Large-Cell Lymphoma (ALCL)
3.3. Breast Implant-Associated Anaplastic Large-Cell Lymphoma (BIA-ALCL)
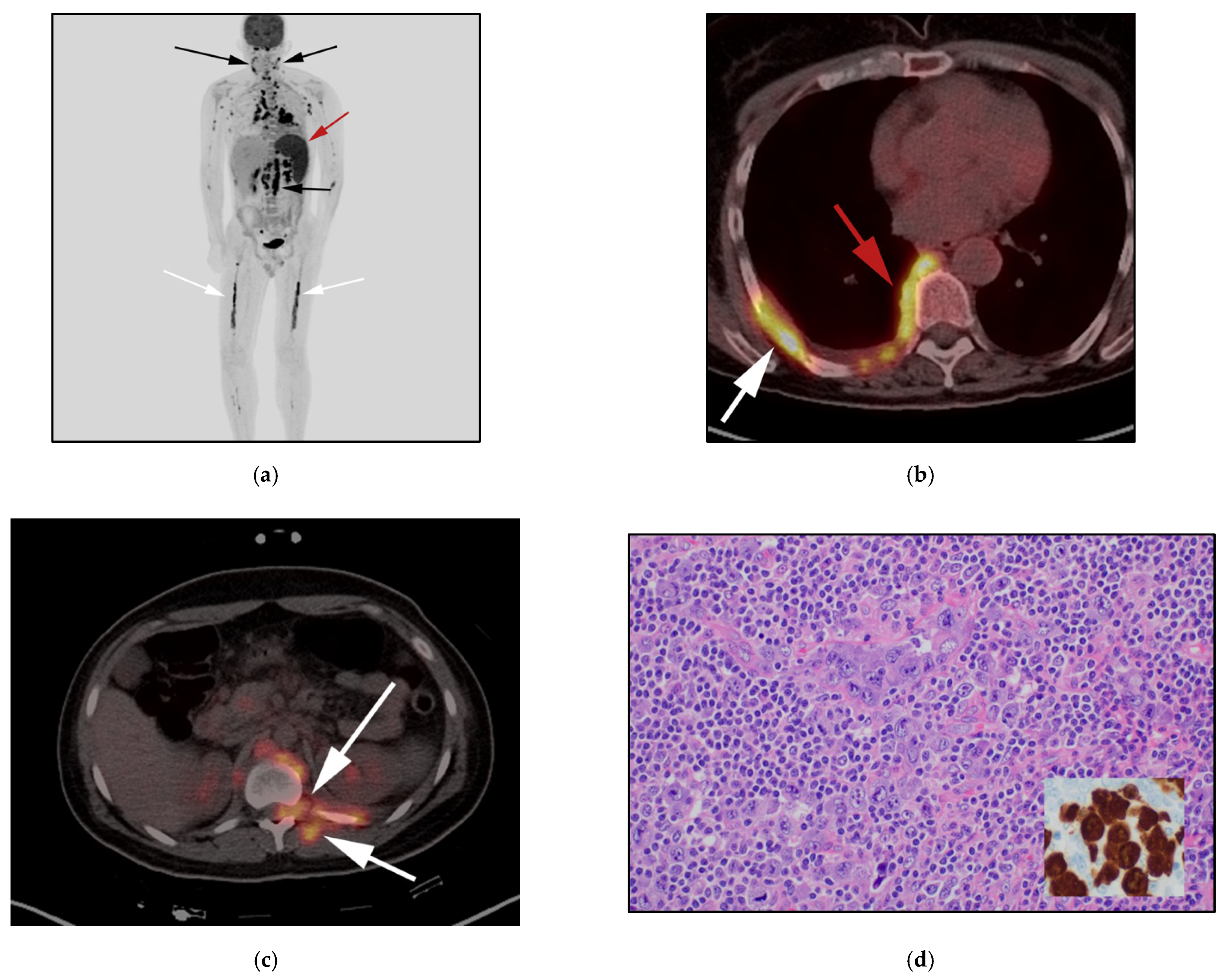
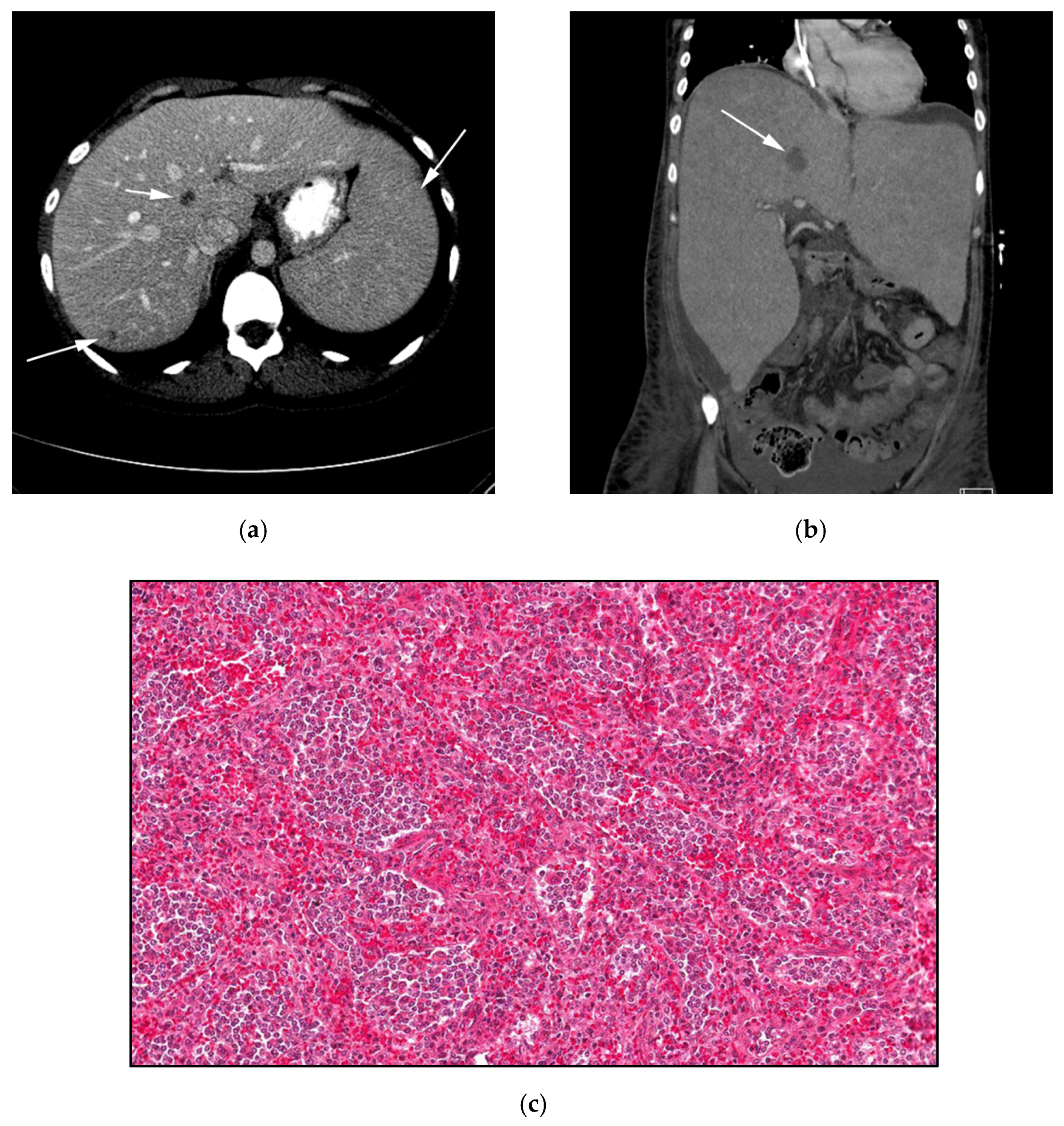
3.4. Hepatosplenic Gamma Delta T-Cell Lymphoma: (HSTL)
3.5. Peripheral T-Cell Lymphoma Not Otherwise Specified NOS: (PTCL-NOS)
3.6. Angioimmunoblastic T-Cell Lymphoma (AITL)
3.7. Enteropathy-Associated T-Cell Lymphoma of the Bowel (EATL)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Toma, P.; Granata, C.; Rossi, A.; Garaventa, A. Multimodality Imaging of Hodgkin Disease and Non-Hodgkin Lymphomas in Children. RadioGraphics 2007, 27, 1335–1354. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Lee, S.H. A poor prognostic case of peripheral T-cell lymphoma in the base of tongue with chemotherapy followed by radiation therapy. SpringerPlus 2014, 3, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, J.; Ray, S.; Sen, S.; Chandy, M. Extranodal involvement in lymphoma—A Pictorial Essay and Retrospective Analysis of 281 PET/CT studies. Asia Ocean. J. Nucl. Med. Biol. 2014, 2, 42–56. [Google Scholar] [PubMed]
- Marchi, E.; O’Connor, O.A. The rapidly changing landscape in mature T-cell lymphoma (MTCL) biology and management. CA A Cancer J. Clin. 2020, 70, 47–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, K.J. Peripheral T-cell lymphomas. Blood Rev. 2007, 21, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Cheson, B.D. Role of Functional Imaging in the Management of Lymphoma. J. Clin. Oncol. 2011, 29, 1844–1854. [Google Scholar] [CrossRef]
- Cheson, B.D.; Fisher, R.I.; Barrington, S.; Cavalli, F.; Schwartz, L.H.; Zucca, E.; Lister, T.A. Recommendations for Initial Evaluation, Staging, and Response Assessment of Hodgkin and Non-Hodgkin Lymphoma: The Lugano Classification. J. Clin. Oncol. 2014, 32, 3059–3067. [Google Scholar] [CrossRef]
- Ngeow, J.Y.Y.; Quek, R.H.H.; Ng, D.C.E.; Hee, S.W.; Tao, M.; Lim, L.C.; Tan, Y.H.; Lim, S.T. High SUV uptake on FDG–PET/CT predicts for an aggressive B-cell lymphoma in a prospective study of primary FDG–PET/CT staging in lymphoma. Ann. Oncol. 2009, 20, 1543–1547. [Google Scholar] [CrossRef]
- Valls, L.; Badve, C.; Avril, S.; Herrmann, K.; Faulhaber, P.; O’Donnell, J.; Avril, N. FDG-PET imaging in hematological malignancies. Blood Rev. 2016, 30, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Silberstein, L.; Anastasi, J. Chapter 85—T-Cell Lymphomas. In Hematology, 7th ed.; Hoffman, R., Benz, E., Heslop, H., Weitz, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1343–1380. [Google Scholar]
- Jaffe, E.S.; Barr, P.M.; Smith, S.M. Understanding the New WHO Classification of Lymphoid Malignancies: Why It’s Important and How It Will Affect Practice. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 535–546. [Google Scholar] [CrossRef]
- Phan, A.; Veldman, R.; Lechowicz, M.J. T-cell Lymphoma Epidemiology: The Known and Unknown. Curr. Hematol. Malign. Rep. 2016, 11, 492–503. [Google Scholar] [CrossRef]
- Gonzalez, C.L.; Medeiros, L.J.; Braziel, R.M.; Jaffe, E.S. T-cell lymphoma involving subcutaneous tissue. A clinicopathologic entity commonly associated with hemophagocytic syndrome. Am. J. Surg. Pathol. 1991, 15, 17–27. [Google Scholar] [CrossRef]
- Parveen, Z.; Thompson, K. Subcutaneous panniculitis-like T-cell lymphoma: Redefinition of diagnostic criteria in the recent World Health Organization-European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas. Arch. Pathol. Lab. Med. 2009, 133, 303–308. [Google Scholar] [CrossRef]
- Heyman, B.; Beaven, A. Mycophenolate Mofetil for the Treatment of Subcutaneous Panniculitis-Like T-Cell Lymphoma: Case Report and Review of the Literature. Clin. Lymphoma Myeloma Leuk. 2018, 18, e437–e440. [Google Scholar] [CrossRef]
- Fujii, K. New Therapies and Immunological Findings in Cutaneous T-Cell Lymphoma. Front. Oncol. 2018, 8, 198. [Google Scholar] [CrossRef]
- Sander, C.A.; Kind, P.; Kaudewitz, P.; Raffeld, M.; Jaffe, E.S. The Revised European-American Classification of Lymphoid Neoplasms (REAL): A new perspective for the classification of cutaneous lymphomas. J. Cutan. Pathol. 1997, 24, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Im, J.-G.; Goo, J.M.; Kim, K.W.; Choi, B.I.; Chang, K.H.; Han, J.K.; Han, M.H. Peripheral T-Cell Lymphoma: Spectrum of Imaging Findings with Clinical and Pathologic Features. RadioGraphics 2003, 23, 7–26. [Google Scholar] [CrossRef] [PubMed]
- Attygalle, A.D.; Cabeçadas, J.; Gaulard, P.; Jaffe, E.S.; De Jong, D.; Ko, Y.H.; Said, J.; Klapper, W. Peripheral T-cell and NK-cell lymphomas and their mimics; taking a step forward-report on the lymphoma workshop of the XVIth meeting of the European Association for Haematopathology and the Society for Hematopathology. Histopathology 2014, 64, 171–199. [Google Scholar] [CrossRef] [PubMed]
- Salar, A.; De Sevilla, A.F.; Romagosa, V.; Domingo-Claros, A.; González-Barca, E.; De Sanjosé, S.; Pera, J.; Servitje, O.; Grañena, A. Distribution and incidence rates of lymphoid neoplasms according to the REAL classification in a single institution. A prospective study of 940 cases. Eur. J. Haematol. 1997, 59, 231–237. [Google Scholar] [CrossRef]
- Zain, J.M. Aggressive T-cell lymphomas: 2019 updates on diagnosis, risk stratification, and management. Am. J. Hematol. 2019, 94, 929–946. [Google Scholar] [CrossRef] [Green Version]
- Keech, J.A., Jr.; Creech, B.J. Anaplastic t-cell lymphoma in proximity to a saline-filled breast implant. Plast. Reconstr. Surg. 1997, 100, 554–555. [Google Scholar] [CrossRef]
- Mehta-Shah, N.; Clemens, M.W.; Horwitz, S.M. How I treat breast implant–associated anaplastic large cell lymphoma. Blood 2018, 132, 1889–1898. [Google Scholar] [CrossRef]
- Clemens, M.W. Discussion: Breast Implant-Associated Anaplastic Large Cell Lymphoma in Australia and New Zealand: High-Surface-Area Textured Implants Are Associated with Increased Risk. Plast. Reconstr. Surg. 2017, 140, 660–662. [Google Scholar] [CrossRef]
- Clemens, M.W.; Medeiros, L.J.; Butler, C.E.; Hunt, K.K.; Fanale, M.A.; Horwitz, S.; Weisenburger, D.D.; Liu, J.; Morgan, E.; Kanagal-Shamanna, R.; et al. Complete Surgical Excision Is Essential for the Management of Patients with Breast Implant–Associated Anaplastic Large-Cell Lymphoma. J. Clin. Oncol. 2016, 34, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Di Napoli, A.; Pepe, G.; Giarnieri, E.; Cippitelli, C.; Bonifacino, A.; Mattei, M.; Martelli, M.; Falasca, C.; Cox, M.C.; Santino, I.; et al. Cytological diagnostic features of late breast implant seromas: From reactive to anaplastic large cell lymphoma. PLoS ONE 2017, 12, e0181097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastorello, R.G.; Costa, F.D.; Osório, C.A.B.T.; Makdissi, F.B.A.; Bezerra, S.M.; de Brot, M.; Campos, A.H.J.F.M.; Soares, F.A.; Vassallo, J. Breast implant-associated anaplastic large cell lymphoma in a Li-FRAUMENI patient: A case report. Diagn. Pathol. 2018, 13, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gidengil, C.A.; Predmore, Z.; Mattke, S.; van Busum, K.; Kim, B. Breast Implant-Associated Anaplastic Large Cell Lymphoma: A Systematic Review. JAMA Surg. 2017, 152, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Dashevsky, B.Z.; Gallagher, K.M.; Grabenstetter, A.; Cordeiro, P.G.; Dogan, A.; Morris, E.A.; Horwitz, S.M.; Sutton, E.J. Breast implant-associated anaplastic large cell lymphoma: Clinical and imaging findings at a large US cancer center. Breast J. 2019, 25, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otero, H.J.; Jagannathan, J.P.; Prevedello, L.; Johnston, C.J.; Ramaiya, N.H.; Abbeele, A.D.V.D.; DiPiro, P.J. CT and PET/CT Findings of T-Cell Lymphoma. Am. J. Roentgenol. 2009, 193, 349–358. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, E. Hepatosplenic T-Cell Lymphoma: A Clinicopathologic Review with an Emphasis on Diagnostic Differentiation from Other T-Cell/Natural Killer–Cell Neoplasms. Arch. Pathol. Lab. Med. 2015, 139, 1173–1180. [Google Scholar] [CrossRef]
- Yabe, M.; Miranda, R.N.; Medeiros, L.J. Hepatosplenic T-cell Lymphoma: A review of clinicopathologic features, pathogenesis, and prognostic factors. Hum. Pathol. 2018, 74, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Tripodo, C.; Iannitto, E.; Florena, A.M.; Pucillo, C.E.; Piccaluga, P.P.; Franco, V.; Pileri, S.A. Gamma-delta T-cell lymphomas. Nat. Rev. Clin. Oncol. 2009, 6, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Yabe, M.; Medeiros, L.J.; Wang, S.A.; Tang, G.; Bueso-Ramos, C.E.; Jorgensen, J.L.; Govind, B.; Weina, C.; Ken, Y.H.; Miranda, R.N.; et al. Distinguishing Between Hepatosplenic T-cell Lymphoma and gammadelta T-cell Large Granular Lymphocytic Leukemia: A Clinicopathologic, Immunophenotypic, and Molecular Analysis. Am. J. Surg. Pathol. 2017, 41, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Panwalkar, A.W.; Armitage, J.O. T-cell/NK-cell lymphomas: A review. Cancer Lett. 2007, 253, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Broccoli, A.; Zinzani, P.L. Peripheral T-cell lymphoma, not otherwise specified. Blood 2017, 129, 1103–1112. [Google Scholar] [CrossRef] [Green Version]
- Chiba, S.; Sakata-Yanagimoto, M. Advances in understanding of angioimmunoblastic T-cell lymphoma. Leukemia 2020, 34, 2592–2606. [Google Scholar] [CrossRef] [PubMed]
- Frizzera, G.; Moran, E.; Rappaport, H. Angio-Immunoblastic Lymphadenopathy with Dysproteinæmia. Lancet 1974, 303, 1070–1073. [Google Scholar] [CrossRef]
- Jain, G.; Kumar, C.; Malhotra, A.; Mallick, S.R.; Bakhshi, S.; Chopra, A. Peripheral blood involvement in angioimmunoblastic T-cell lymphoma: A case report and review of the literature. Am. J. Blood Res. 2020, 10, 257–265. [Google Scholar]
- Shao, D.; Gao, Q.; Liang, C.-H.; Wang, S.-X. Discussion of 18F-FDG PET/CT imaging characteristics and diagnostic values of angioimmunoblastic T-cell lymphoma. Leuk. Lymphoma 2017, 58, 1581–1588. [Google Scholar] [CrossRef]
- Kako, S.; Izutsu, K.; Ota, Y.; Minatani, Y.; Sugaya, M.; Momose, T.; Ohtomo, K.; Kanda, Y.; Chiba, S.; Motokura, T.; et al. FDG-PET in T-cell and NK-cell neoplasms. Ann. Oncol. 2007, 18, 1685–1690. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.S. Lymphoproliferative Disorders of the Gastrointestinal Tract: A Review and Pragmatic Guide to Diagnosis. Arch. Pathol. Lab. Med. 2011, 135, 1283–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintanilla-Martinez, L. The 2016 updated WHO classification of lymphoid neoplasias. Hematol. Oncol. 2017, 35 (Suppl. 1), 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieniawski, M.K.; Lennard, A.L. Enteropathy-Associated T-Cell Lymphoma: Epidemiology, Clinical Features, and Current Treatment Strategies. Curr. Hematol. Malign. Rep. 2011, 6, 231–240. [Google Scholar] [CrossRef]
- Sieniawski, M.; Angamuthu, N.; Boyd, K.; Chasty, R.; Davies, J.; Forsyth, P.; Jack, F.; Lyons, S.; Mounter, P.; Revell, P.; et al. Evaluation of enteropathy-associated T-cell lymphoma comparing standard therapies with a novel regimen including autologous stem cell transplantation. Blood 2010, 115, 3664–3670. [Google Scholar] [CrossRef] [Green Version]
- Mansoor, A.; Pittaluga, S.; Beck, P.L.; Wilson, W.H.; Ferry, J.A.; Jaffe, E.S. NK-cell enteropathy: A benign NK-cell lymphoproliferative disease mimicking intestinal lymphoma: Clinicopathologic features and follow-up in a unique case series. Blood 2011, 117, 1447–1452. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Lu, Z.; Yang, D.; Chen, J. Primary intestinal T-cell and NK-cell lymphomas: A clinicopathological and molecular study from China focused on type II enteropathy-associated T-cell lymphoma and primary intestinal NK-cell lymphoma. Mod. Pathol. 2011, 24, 983–992. [Google Scholar] [CrossRef] [Green Version]
- van de Water, J.M.; Cillessen, S.A.; Visser, O.J.; Verbeek, W.H.; Meijer, C.J.; Mulder, C.J. Enteropathy associated T-cell lymphoma and its precursor lesions. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Summers, T.A.; Moncur, J.T. The small cell variant of anaplastic large cell lymphoma. Arch. Pathol. Lab. Med. 2010, 134, 1706–1710. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Histologic Subtype | Molecular Change & Characteristic Immunophenotype | Clinical Characteristic |
|---|---|---|
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
| ||
|
|
|
| ||
|
|
|
| ||
|
|
|
| ||
|
|
|
|
|
|
| Entiry/Category | Updates | New Diagnostic Considerations |
|---|---|---|
| Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) |
|
|
| Hepatosplenic delta T-cell lymphoma (HSTCL) |
|
|
| Angioimmunoblastic T-cell lymphoma (AITL) |
|
|
| Enteropathy-associated T-cell lymphoma (EATL) |
|
|
| Monomorphic epitheliotropic intestinal T-cell Lymphoma (MEITL) |
|
|
| Indolent T-cell lymphoproliferative disorder of the GI tract |
|
|
| Primary cutaneous acral CD8+ T-cell lymphoma |
|
|
| Peripheral T-cell lymphoma not otherwise specified (PTCL-NOS) |
|
|
| ALK anaplastic large-cell lymphoma (ALCL) |
|
|
| Breast implant-associated anaplastic large-cell lymphoma (BIA-ALCL) |
|
|
| Extranodal | Nodal | Skin | Disseminated |
|---|---|---|---|
| Hepatosplenic T-cell lymphoma (HSTCL) |
|
| Adult T-cell Leukemia/Lymphoma (ATLL) |
| Extranodal T-cell lymphoma—nasal type (EKNTL) |
|
| T-prolymphocytic Leukemia (T-PLL) |
| Enteropathy-associated T-cell lymphoma (EATL) | Primary cutaneous ALCL (PCALCL) | T-cell large granular lymphocytes (LGL) | |
| Monomorphic, epitheliotropic, intestinal T-cell lymphoma (MEITL) | Primary cutaneous CD4+ small/medium pleomorphic T-cell lymphoma (PCSM-TCL) | Aggressive natural killer T-cell leukemia | |
| Breast implant-associated ALCL (BIA-ALCL) |
| Entity | Key Pathologic Features | Key Clinical Features | Key Imaging Features | Differential Considerations |
|---|---|---|---|---|
| Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) |
|
|
|
|
| Anaplastic large-cell lymphoma (ALCL) |
|
|
|
|
| Breast implant-associated anaplastic large-cell lymphoma (BIA-ALCL) |
|
|
|
|
| Hepatosplenic T-cell lymphoma (HSTL) |
|
|
|
|
| Peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS). |
|
|
|
|
| Angioimmunoblastic T-cell lymphoma (AITL) |
|
|
|
|
| Enteropathy-associated T-cell lymphoma of the bowel (EATL) |
|
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salem, A.E.; Zaki, Y.H.; El-Hussieny, G.; ElNoueam, K.I.; Shaaban, A.M.; Koppula, B.R.; Yang, M.; Salama, M.; Elsayes, K.M.; Covington, M.F. Uncommon Variants of Mature T-Cell Lymphomas (MTCLs): Imaging and Histopathologic and Clinical Features with Updates from the Fourth Edition of the World Health Organization (WHO) Classification of Lymphoid Neoplasms. Cancers 2021, 13, 5217. https://doi.org/10.3390/cancers13205217
Salem AE, Zaki YH, El-Hussieny G, ElNoueam KI, Shaaban AM, Koppula BR, Yang M, Salama M, Elsayes KM, Covington MF. Uncommon Variants of Mature T-Cell Lymphomas (MTCLs): Imaging and Histopathologic and Clinical Features with Updates from the Fourth Edition of the World Health Organization (WHO) Classification of Lymphoid Neoplasms. Cancers. 2021; 13(20):5217. https://doi.org/10.3390/cancers13205217
Chicago/Turabian StyleSalem, Ahmed Ebada, Yehia H. Zaki, Gamal El-Hussieny, Khaled I. ElNoueam, Akram M. Shaaban, Bhasker Rao Koppula, Ming Yang, Mohamed Salama, Khaled M. Elsayes, and Matthew F. Covington. 2021. "Uncommon Variants of Mature T-Cell Lymphomas (MTCLs): Imaging and Histopathologic and Clinical Features with Updates from the Fourth Edition of the World Health Organization (WHO) Classification of Lymphoid Neoplasms" Cancers 13, no. 20: 5217. https://doi.org/10.3390/cancers13205217
APA StyleSalem, A. E., Zaki, Y. H., El-Hussieny, G., ElNoueam, K. I., Shaaban, A. M., Koppula, B. R., Yang, M., Salama, M., Elsayes, K. M., & Covington, M. F. (2021). Uncommon Variants of Mature T-Cell Lymphomas (MTCLs): Imaging and Histopathologic and Clinical Features with Updates from the Fourth Edition of the World Health Organization (WHO) Classification of Lymphoid Neoplasms. Cancers, 13(20), 5217. https://doi.org/10.3390/cancers13205217