
Antisense Oligonucleotide-Mediated Splice Switching: Potential Therapeutic Approach for Cancer Mitigation

 ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
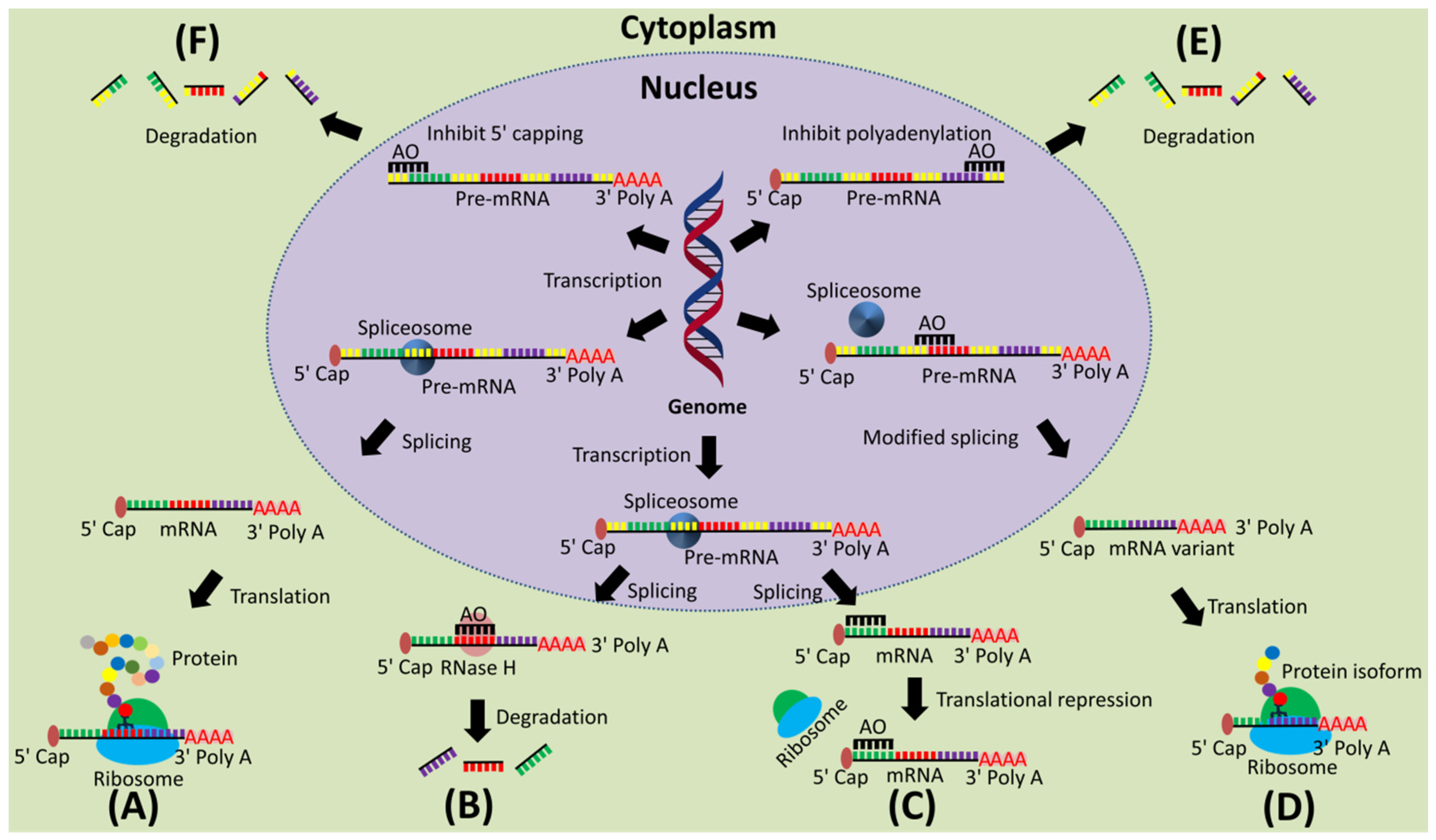
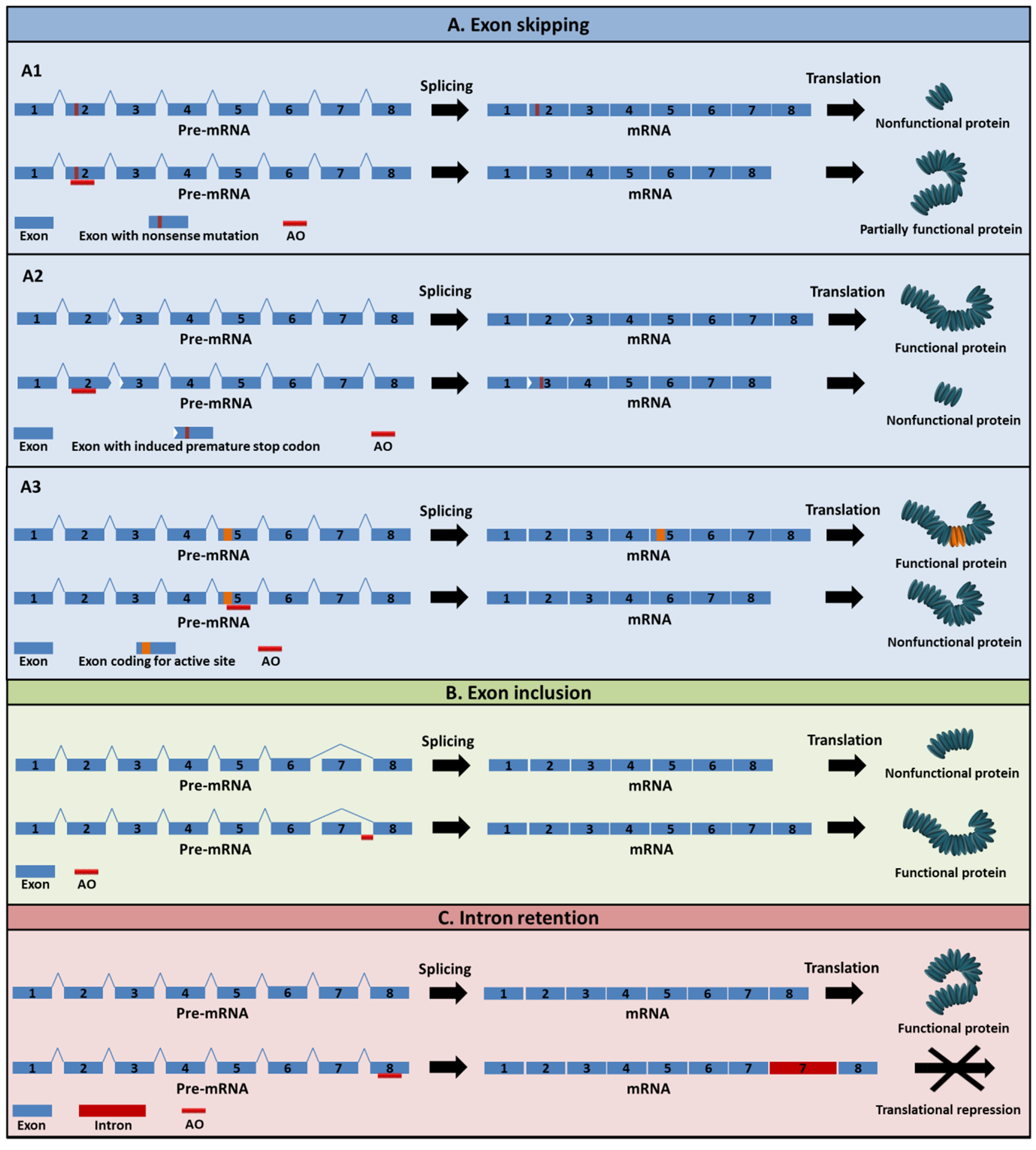
2. Mechanism of Action of AOs
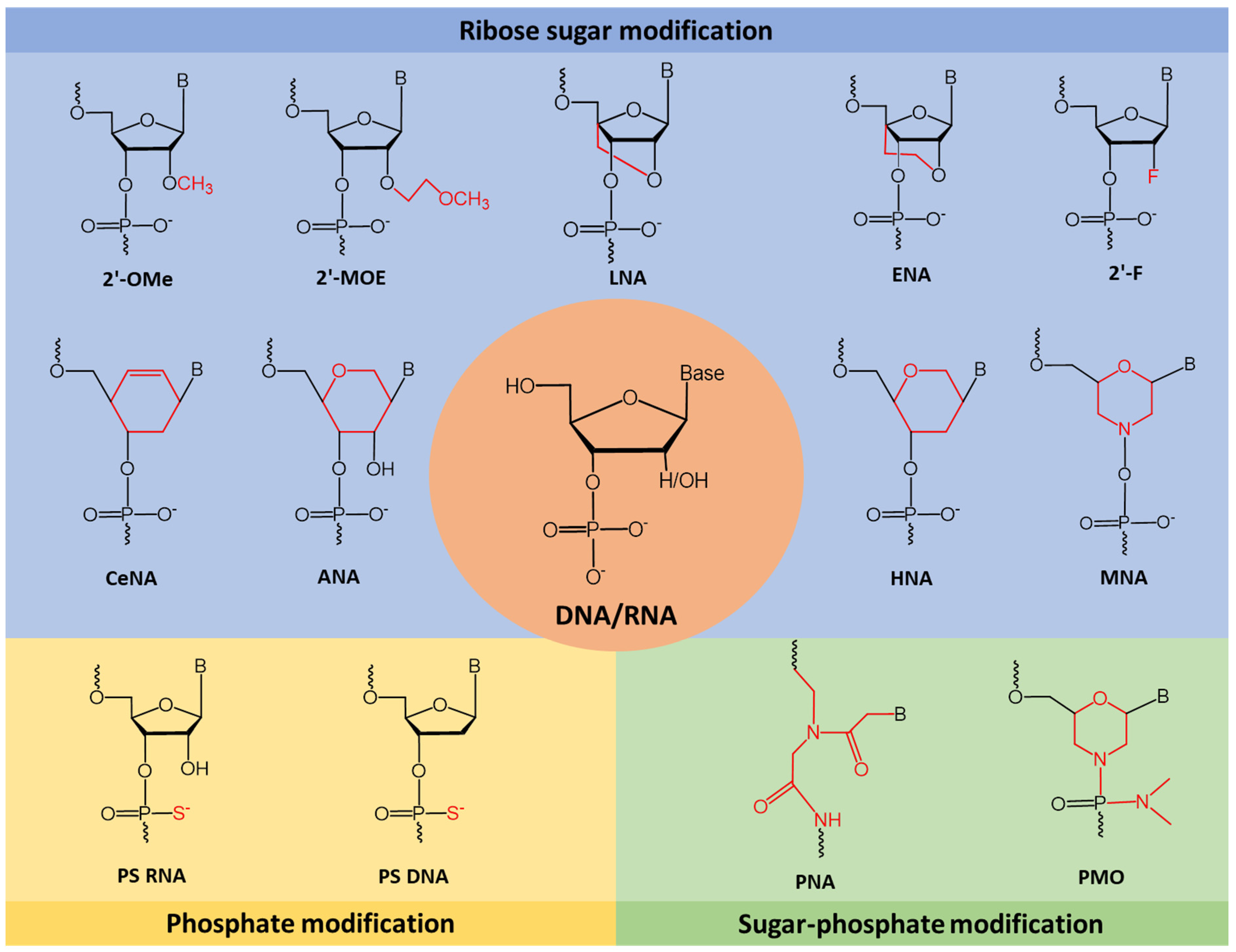
3. Chemical Modifications
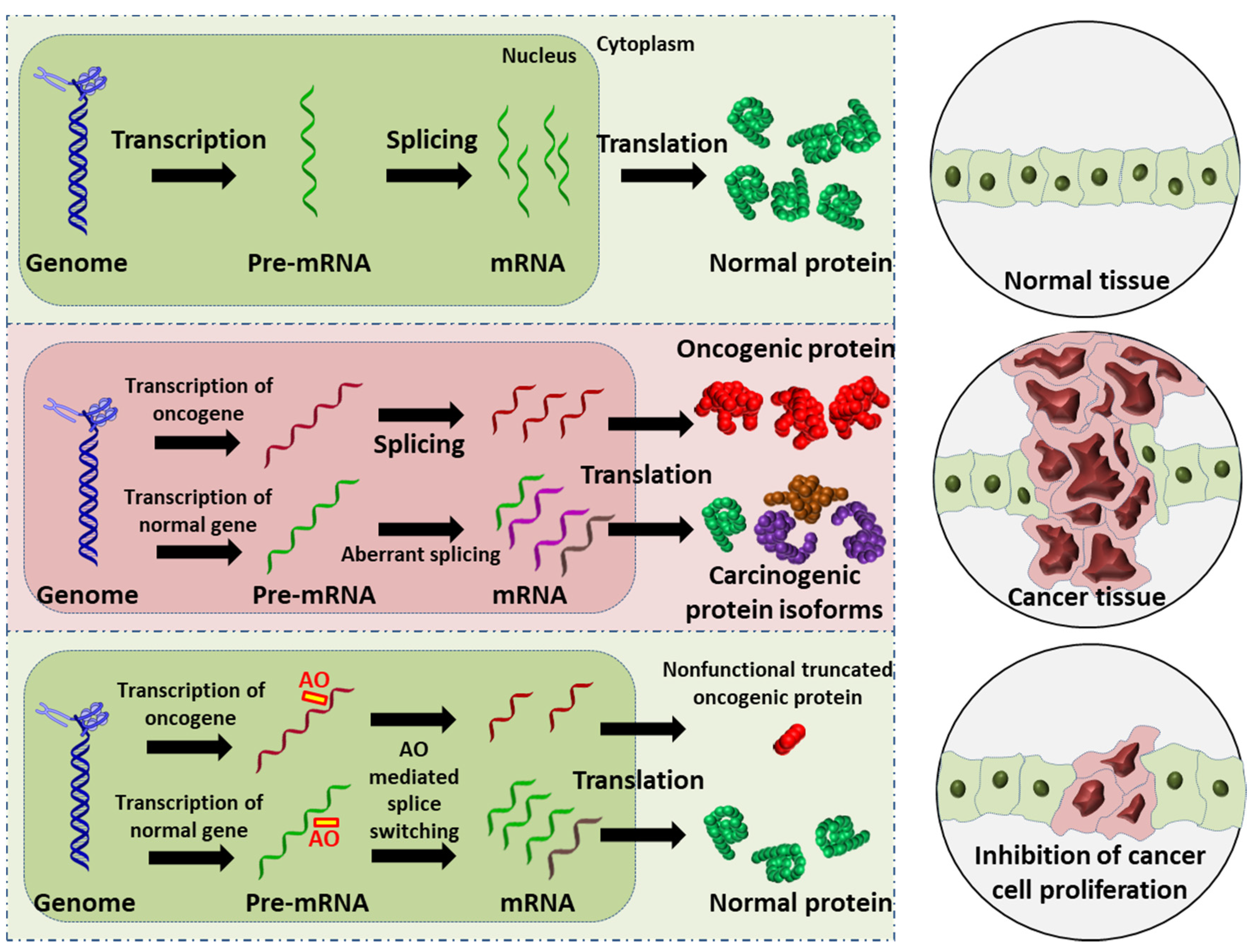
4. Alternative Splicing in Cancer
5. Exon-Skipping AOs in Cancer
5.1. Breast Cancer
5.2. Leukemia
5.3. Melanoma
6. Antisense Oligos in Clinical Trials
6.1. Apatorsen/OGX-427
6.2. AZD4785
6.3. AZD5312/ARRx
6.4. AZD9150/ISIS 481464/ISIS-STAT3Rx/Danvatirsen
6.5. BP1001
6.6. C-Myb AS ODN/G4460/LR3001
6.7. EZN-2968/RO7070179/SPC2968
6.8. G3139/Oblimersen/Genasense
6.9. GTI-2040
6.10. ISIS 3521
6.11. ISIS 5132
6.12. LErafAON
6.13. OGX-011
7. Modes of AO Delivery
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fackenthal, J.D.; Godley, L.A. Aberrant RNA splicing and its functional consequences in cancer cells. Dis. Models Mech. 2008, 1, 37–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.B.; Kawasawa, Y.I.; Mason, C.E.; Krsnik, Ž.; Coppola, G.; Bogdanović, D.; Geschwind, D.H.; Mane, S.M.; State, M.W.; Šestan, N. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 2009, 62, 494–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siva, K.; Covello, G.; Denti, M.A. Exon-skipping antisense oligonucleotides to correct missplicing in neurogenetic diseases. Nucleic Acid Ther. 2014, 24, 69–86. [Google Scholar] [PubMed] [Green Version]
- Johnson, J.M.; Castle, J.; Garrett-Engele, P.; Kan, Z.; Loerch, P.M.; Armour, C.D.; Santos, R.; Schadt, E.E.; Stoughton, R.; Shoemaker, D.D. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 2003, 302, 2141–2144. [Google Scholar]
- Clark, T.A.; Schweitzer, A.C.; Chen, T.X.; Staples, M.K.; Lu, G.; Wang, H.; Williams, A.; Blume, J.E. Discovery of tissue-specific exons using comprehensive human exon microarrays. Genome Biol. 2007, 8, R64. [Google Scholar] [CrossRef] [Green Version]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhir, A.; Buratti, E. Alternative splicing: Role of pseudoexons in human disease and potential therapeutic strategies. FEBS J. 2010, 277, 841–855. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.; Zhao, X.; Meyerson, M. Somatic alterations in the human cancer genome. Cancer Cell 2004, 6, 433–438. [Google Scholar] [CrossRef] [Green Version]
- Mendelsohn, J.; Howley, P.; Israel, M.; Gray, J.; Thompson, C. The Molecular Basis of Cancer; Saunders Elsevier: Philadelphia, PA, USA, 2008. [Google Scholar]
- Wang, B.-D.; Lee, N.H. Aberrant RNA splicing in cancer and drug resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleave, M.E.; Monia, B.P. Antisense therapy for cancer. Nat. Rev. Cancer 2005, 5, 468–479. [Google Scholar]
- Arechavala-Gomeza, V.; Khoo, B.; Aartsma-Rus, A. Splicing Modulation Therapy in the Treatment of Genetic Diseases. Appl. Clin. Genet. 2014, 7, 245–252. [Google Scholar]
- Chen, S.; Sbuh, N.; Veedu, R.N. Antisense oligonucleotides as potential therapeutics for Type 2 Diabetes. Nucleic Acid Ther. 2021, 31, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Liang, X.H.; Baker, B.F.; Crooke, R.M. Antisense technology: A review. J. Biol. Chem. 2021, 296, 100416. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F. Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Le, B.T.; Raguraman, P.; Kosbar, T.R.; Fletcher, S.; Wilton, S.D.; Veedu, R.N. Antisense oligonucleotides targeting angiogenic factors as potential cancer therapeutics. Mol. Ther.-Nucleic Acids 2019, 14, 142–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushner, D.M.; Silverman, R.H. Antisense cancer therapy: The state of the science. Curr. Oncol. Rep. 2000, 2, 23–30. [Google Scholar]
- Chan, J.H.; Lim, S.; Wong, W.F. Antisense oligonucleotides: From design to therapeutic application. Clin. Exp. Pharmacol. Physiol. 2006, 33, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Turczynski, S.; Titeux, M.; Pironon, N.; Hovnanian, A. Antisense-mediated exon skipping to reframe transcripts. In Exon Skipping; Springer: Clifton, NJ, USA, 2012; pp. 221–238. [Google Scholar]
- Aartsma-Rus, A. Overview on DMD exon skipping. Exon Skipp. 2012, 867, 97–116. [Google Scholar]
- Wilton, S.D.; Fall, A.M.; Harding, P.L.; McClorey, G.; Coleman, C.; Fletcher, S. Antisense oligonucleotide-induced exon skipping across the human dystrophin gene transcript. Mol. Ther. 2007, 15, 1288–1296. [Google Scholar] [CrossRef]
- Syed, Y.Y. Eteplirsen: First global approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.-A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef]
- Disterer, P.; Khoo, B. Antisense-mediated exon-skipping to induce gene knockdown. In Exon Skipping; Springer: Clifton, NJ, USA, 2012; pp. 289–305. [Google Scholar]
- Du, L.; Gatti, R.A. Progress toward therapy with antisense-mediated splicing modulation. Curr. Opin. Mol. Ther. 2009, 11, 116–123. [Google Scholar]
- Douglas, A.G.; Wood, M.J. Splicing therapy for neuromuscular disease. Mol. Cell. Neurosci. 2013, 56, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Prakash, V. Spinraza—A rare disease success story. Gene Ther. 2017, 24, 497. [Google Scholar]
- El Marabti, E.; Younis, I. The cancer spliceome: Reprograming of alternative splicing in cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef]
- Kurreck, J. Antisense technologies: Improvement through novel chemical modifications. Eur. J. Biochem. 2003, 270, 1628–1644. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.B.; Seth, P.P. The medicinal chemistry of therapeutic oligonucleotides. J. Med. Chem. 2016, 59, 9645–9667. [Google Scholar] [CrossRef] [PubMed]
- Sierakowska, H.; Sambade, M.J.; Agrawal, S.; Kole, R. Repair of thalassemic human β-globin mRNA in mammalian cells by antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 1996, 93, 12840–12844. [Google Scholar] [CrossRef] [Green Version]
- Dunckley, M.G.; Manoharan, M.; Villiet, P.; Eperon, I.C.; Dickson, G. Modification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotides. Hum. Mol. Genet. 1998, 7, 1083–1090. [Google Scholar] [CrossRef]
- Lu, Q.L.; Mann, C.J.; Lou, F.; Bou-Gharios, G.; Morris, G.E.; Xue, S.-A.; Fletcher, S.; Partridge, T.A.; Wilton, S.D. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat. Med. 2003, 9, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Rabinowitz, A.; Chen, Y.C.; Yokota, T.; Yin, H.; Alter, J.; Jadoon, A.; Bou-Gharios, G.; Partridge, T. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Natl. Acad. Sci. USA 2005, 102, 198–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goemans, N.M.; Tulinius, M.; Van den Hauwe, M.; Kroksmark, A.-K.; Buyse, G.; Wilson, R.J.; van Deutekom, J.C.; de Kimpe, S.J.; Lourbakos, A.; Campion, G. Long-term efficacy, safety, and pharmacokinetics of drisapersen in Duchenne muscular dystrophy: Results from an open-label extension study. PLoS ONE 2016, 11, e0161955. [Google Scholar]
- Relizani, K.; Griffith, G.; Echevarría, L.; Zarrouki, F.; Facchinetti, P.; Vaillend, C.; Leumann, C.; Garcia, L.; Goyenvalle, A. Efficacy and safety profile of tricyclo-DNA antisense oligonucleotides in Duchenne muscular dystrophy mouse model. Mol. Ther.-Nucleic Acids 2017, 8, 144–157. [Google Scholar] [PubMed] [Green Version]
- Goyenvalle, A.; Griffith, G.; Babbs, A.; El Andaloussi, S.; Ezzat, K.; Avril, A.; Dugovic, B.; Chaussenot, R.; Ferry, A.; Voit, T. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat. Med. 2015, 21, 270–275. [Google Scholar] [PubMed]
- Robin, V.; Griffith, G.; Carter, J.-P.L.; Leumann, C.J.; Garcia, L.; Goyenvalle, A. Efficient SMN rescue following subcutaneous Tricyclo-DNA antisense oligonucleotide treatment. Mol. Ther.-Nucleic Acids 2017, 7, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renneberg, D.; Leumann, C.J. Watson− crick base-pairing properties of tricyclo-DNA. J. Am. Chem. Soc. 2002, 124, 5993–6002. [Google Scholar] [CrossRef] [PubMed]
- Ittig, D.; Gerber, A.-B.; Leumann, C.J. Position-dependent effects on stability in tricyclo-DNA modified oligonucleotide duplexes. Nucleic Acids Res. 2011, 39, 373–380. [Google Scholar] [CrossRef]
- Aupy, P.; Echevarria, L.; Relizani, K.; Goyenvalle, A. The Use of Tricyclo-DNA Oligomers for the Treatment of Genetic Disorders. Biomedicines 2017, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Koshkin, A.A.; Singh, S.K.; Nielsen, P.; Rajwanshi, V.K.; Kumar, R.; Meldgaard, M.; Olsen, C.E.; Wengel, J. LNA (Locked Nucleic Acids): Synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron 1998, 54, 3607–3630. [Google Scholar]
- Veedu, R.N.; Wengel, J. Locked nucleic acid as a novel class of therapeutic agents. RNA Biol. 2009, 6, 321–323. [Google Scholar] [CrossRef] [Green Version]
- Aartsma-Rus, A.; Kaman, W.; Bremmer-Bout, M.; Janson, A.; Den Dunnen, J.; van Ommen, G.B.; Van Deutekom, J. Comparative analysis of antisense oligonucleotide analogs for targeted DMD exon 46 skipping in muscle cells. Gene Ther. 2004, 11, 1391–1398. [Google Scholar] [CrossRef] [Green Version]
- Egholm, M.; Buchardt, O.; Christensen, L.; Behrens, C.; Freier, S.M.; Driver, D.A.; Berg, R.H.; Kim, S.K.; Norden, B.; Nielsen, P.E. PNA hybridizes to complementary oligonucleotides obeying the Watson–Crick hydrogen-bonding rules. Nature 1993, 365, 566–568. [Google Scholar]
- Yin, H.; Lu, Q.; Wood, M. Effective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx mice. Mol. Ther. 2008, 16, 38–45. [Google Scholar] [CrossRef]
- Yin, H.; Betts, C.; Saleh, A.F.; Ivanova, G.D.; Lee, H.; Seow, Y.; Kim, D.; Gait, M.J.; Wood, M.J. Optimization of peptide nucleic acid antisense oligonucleotides for local and systemic dystrophin splice correction in the mdx mouse. Mol. Ther. 2010, 18, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Schmajuk, G.; Sierakowska, H.; Kole, R. Antisense oligonucleotides with different backbones: Modification of splicing pathways and efficacy of uptake. J. Biol. Chem. 1999, 274, 21783–21789. [Google Scholar]
- Fletcher, S.; Honeyman, K.; Fall, A.M.; Harding, P.L.; Johnsen, R.D.; Wilton, S.D. Dystrophin expression in the mdx mouse after localised and systemic administration of a morpholino antisense oligonucleotide. J. Gene Med. 2006, 8, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, H.A.; de Winter, C.L.; de Kimpe, S.J.; van Kuik-Romeijn, P.; Heuvelmans, N.; Platenburg, G.J.; van Ommen, G.J.B.; van Deutekom, J.C.; Aartsma-Rus, A. In vivo comparison of 2′-O-methyl phosphorothioate and morpholino antisense oligonucleotides for Duchenne muscular dystrophy exon skipping. J. Gene Med. 2009, 11, 257–266. [Google Scholar] [CrossRef]
- Betts, C.; Saleh, A.F.; Arzumanov, A.A.; Hammond, S.M.; Godfrey, C.; Coursindel, T.; Gait, M.J.; Wood, M.J. Pip6-PMO, a new generation of peptide-oligonucleotide conjugates with improved cardiac exon skipping activity for DMD treatment. Mol. Ther.-Nucleic Acids 2012, 1, e38. [Google Scholar] [CrossRef]
- Young, C.S.; Pyle, A.D. Exon Skipping Therapy. Cell 2016, 167, 1144. [Google Scholar] [CrossRef] [PubMed]
- Le, B.T.; Agarwal, S.; Veedu, R.N. Evaluation of DNA segments in 2′-modified RNA sequences in designing efficient splice switching antisense oligonucleotides. RSC Adv. 2021, 11, 14029–14035. [Google Scholar]
- Le, B.T.; Hornum, M.; Sharma, P.K.; Nielsen, P.; Veedu, R.N. Nucleobase-modified antisense oligonucleotides containing 5-(phenyltriazol)-2′-deoxyuridine nucleotides induce exon-skipping in vitro. RSC Adv. 2017, 7, 54542–54545. [Google Scholar]
- Le, B.T.; Chen, S.; Abramov, M.; Herdewijn, P.; Veedu, R.N. Evaluation of anhydrohexitol nucleic acid, cyclohexenyl nucleic acid and d-altritol nucleic acid-modified 2′-O-methyl RNA mixmer antisense oligonucleotides for exon skipping in vitro. Chem. Commun. 2016, 52, 13467–13470. [Google Scholar] [CrossRef] [PubMed]
- Le, B.T.; Adams, A.M.; Fletcher, S.; Wilton, S.D.; Veedu, R.N. Rational design of short locked nucleic acid-modified 2′-O-methyl antisense oligonucleotides for efficient exon-skipping in vitro. Mol. Ther.-Nucleic Acids 2017, 9, 155–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, B.T.; Filichev, V.V.; Veedu, R.N. Investigation of twisted intercalating nucleic acid (TINA)-modified antisense oligonucleotides for splice modulation by induced exon-skipping in vitro. RSC Adv. 2016, 6, 95169–95172. [Google Scholar] [CrossRef]
- Le, B.T.; Murayama, K.; Shabanpoor, F.; Asanuma, H.; Veedu, R.N. Antisense oligonucleotide modified with serinol nucleic acid (SNA) induces exon skipping in mdx myotubes. RSC Adv. 2017, 7, 34049–34052. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Le, B.T.; Chakravarthy, M.; Kosbar, T.R.; Veedu, R.N. Systematic evaluation of 2′-Fluoro modified chimeric antisense oligonucleotide-mediated exon skipping in vitro. Sci. Rep. 2019, 9, 6078. [Google Scholar]
- Le, B.T.; Kosbar, T.R.; Veedu, R.N. Novel disulfide-bridged bioresponsive antisense oligonucleotide induces efficient splice modulation in muscle Myotubes in Vitro. ACS Omega 2020, 5, 18035–18039. [Google Scholar] [CrossRef]
- Chen, S.; Le, B.T.; Rahimizadeh, K.; Shaikh, K.; Mohal, N.; Veedu, R.N. Synthesis of a morpholino nucleic acid (MNA)-uridine phosphoramidite, and exon skipping using MNA/2′-O-methyl mixmer antisense oligonucleotide. Molecules 2016, 21, 1582. [Google Scholar] [CrossRef] [Green Version]
- Raguraman, P.; Wang, T.; Ma, L.; Jørgensen, P.T.; Wengel, J.; Veedu, R.N. Alpha-l-Locked nucleic acid-modified antisense oligonucleotides induce efficient splice modulation in vitro. Int. J. Mol. Sci. 2020, 21, 2434. [Google Scholar] [CrossRef] [Green Version]
- Jansen, B.; Zangemeister-Wittke, U. Antisense therapy for cancer—The time of truth. Lancet Oncol. 2002, 3, 672–683. [Google Scholar] [CrossRef]
- National Cancer Institute. What Is Cancer? Available online: https://www.cancer.gov/about-cancer/understanding/what-is-cancer (accessed on 7 August 2019).
- Zhu, S.; Mott, R.T.; Fry, E.A.; Taneja, P.; Kulik, G.; Sui, G.; Inoue, K. Cooperation between Dmp1 loss and cyclin D1 overexpression in breast cancer. Am. J. Pathol. 2013, 183, 1339–1350. [Google Scholar] [PubMed] [Green Version]
- Maglic, D.; Stovall, D.B.; Cline, J.M.; Fry, E.A.; Mallakin, A.; Taneja, P.; Caudell, D.L.; Willingham, M.C.; Sui, G.; Inoue, K. DMP1β, a splice isoform of the tumour suppressor DMP1 locus, induces proliferation and progression of breast cancer. J. Pathol. 2015, 236, 90–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, K.; Fry, E.A. Aberrant splicing of the DMP1-ARF-MDM2-p53 pathway in cancer. Int. J. Cancer 2016, 139, 33–41. [Google Scholar] [CrossRef] [Green Version]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [Green Version]
- Urbanski, L.M.; Leclair, N.; Anczukow, O. Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip. Rev. RNA 2018, 9, e1476. [Google Scholar] [CrossRef]
- Bonnal, S.C.; Lopez-Oreja, I.; Valcarcel, J. Roles and mechanisms of alternative splicing in cancer—Implications for care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef]
- Escobar-Hoyos, L.; Knorr, K.; Abdel-Wahab, O. Aberrant RNA Splicing in Cancer. Annu. Rev. Cancer. Biol. 2019, 3, 167–185. [Google Scholar] [CrossRef]
- Zhang, S.; Roeder, R.G. The Long and the Short of BRD4: Two Tales in Breast Cancer. Mol. Cell 2020, 78, 993–995. [Google Scholar] [CrossRef]
- Dean, N.M.; Bennett, C.F. Antisense oligonucleotide-based therapeutics for cancer. Oncogene 2003, 22, 9087–9096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercatante, D.R.; Mohler, J.L.; Kole, R. Cellular response to an antisense-mediated shift of Bcl-x pre-mRNA splicing and antineoplastic agents. J. Biol. Chem. 2002, 277, 49374–49382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, R.G.; Hoover, R.N.; Pike, M.C.; Hildesheim, A.; Nomura, A.M.; West, D.W.; Wu-Williams, A.H.; Kolonel, L.N.; Horn-Ross, P.L.; Rosenthal, J.F. Migration patterns and breast cancer risk in Asian-American women. JNCI J. Natl. Cancer Inst. 1993, 85, 1819–1827. [Google Scholar] [CrossRef]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swain, S.M.; Baselga, J.; Kim, S.-B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.-M.; Schneeweiss, A.; Heeson, S. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moasser, M.M. Targeting the function of the HER2 oncogene in human cancer therapeutics. Oncogene 2007, 26, 6577–6592. [Google Scholar] [PubMed] [Green Version]
- Wan, J.; Sazani, P.; Kole, R. Modification of HER2 pre-mRNA alternative splicing and its effects on breast cancer cells. Int. J. Cancer 2009, 124, 772–777. [Google Scholar]
- Pankratova, S.; Nielsen, B.N.; Shiraishi, T.; Nielsen, P.E. PNA-mediated modulation and redirection of Her-2 pre-mRNA splicing: Specific skipping of erbB-2 exon 19 coding for the ATP catalytic domain. Int. J. Oncol. 2010, 36, 29–38. [Google Scholar]
- Menard, S.; Casalini, P.; Campiglio, M.; Pupa, S.; Agresti, R.; Tagliabue, E. HER2 overexpression in various tumor types, focussing on its relationship to the development of invasive breast cancer. Ann. Oncol. 2001, 12, S15–S19. [Google Scholar] [CrossRef]
- Dias, N.; Stein, C. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar]
- Nielsen, T.O.; Sorensen, S.; Dagnaes-Hansen, F.; Kjems, J.; Sorensen, B. Directing HER4 mRNA expression towards the CYT2 isoform by antisense oligonucleotide decreases growth of breast cancer cells in vitro and in vivo. Br. J. Cancer 2013, 108, 2291–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar]
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, X.; Xin, Y.; Mi, R.; Ding, J.; Wang, X.; Hu, J.; Fan, R.; Wei, X.; Song, Y.; Zhao, R.Y. Overexpression of Wilms tumor 1 gene as a negative prognostic indicator in acute myeloid leukemia. PLoS ONE 2014, 9, e92470. [Google Scholar]
- Menssen, H.; Renkl, H.; Rodeck, U.; Maurer, J.; Notter, M.; Schwartz, S.; Reinhardt, R.; Thiel, E. Presence of Wilms’ tumor gene (wt1) transcripts and the WT1 nuclear protein in the majority of human acute leukemias. Leukemia 1995, 9, 1060–1067. [Google Scholar]
- Renshaw, J.; Orr, R.M.; Walton, M.I.; Te Poele, R.; Williams, R.D.; Wancewicz, E.V.; Monia, B.P.; Workman, P.; Pritchard-Jones, K. Disruption of WT1 gene expression and exon 5 splicing following cytotoxic drug treatment: Antisense down-regulation of exon 5 alters target gene expression and inhibits cell survival. Mol. Cancer Ther. 2004, 3, 1467–1484. [Google Scholar]
- Miwa, H.; Beran, M.; Saunders, G. Expression of the Wilms’ tumor gene (WT1) in human leukemias. Leukemia 1992, 6, 405–409. [Google Scholar]
- Bollum, F.J. Terminal deoxynucleotidyl transferase as a hematopoietic cell marker. Blood 1979, 54, 1203–1215. [Google Scholar] [CrossRef] [Green Version]
- Farahat, N.; Lens, D.; Morilla, R.; Matutes, E.; Catovsky, D. Differential TdT expression in acute leukemia by flow cytometry: A quantitative study. Leukemia 1995, 9, 583–587. [Google Scholar]
- Montazersaheb, S.; Kazemi, M.; Nabat, E.; Nielsen, P.E.; Hejazi, M.S. Downregulation of TdT expression through splicing modulation by antisense peptide nucleic acid (PNA). Curr. Pharm. Biotechnol. 2019, 20, 168–178. [Google Scholar] [PubMed]
- Matthews, N.H.; Li, W.Q.; Qureshi, A.A.; Weinstock, M.A.; Cho, E. Epidemiology of Melanoma. In Cutaneous Melanoma: Etiology and Therapy; Ward, W.H., Farma, J.M., Eds.; Codon Publications, Brisbane, Australia, 2017.
- Gembarska, A.; Luciani, F.; Fedele, C.; Russell, E.A.; Dewaele, M.; Villar, S.; Zwolinska, A.; Haupt, S.; de Lange, J.; Yip, D.; et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat. Med. 2012, 18, 1239–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewaele, M.; Tabaglio, T.; Willekens, K.; Bezzi, M.; Teo, S.X.; Low, D.H.; Koh, C.M.; Rambow, F.; Fiers, M.; Rogiers, A. Antisense oligonucleotide–mediated MDM4 exon 6 skipping impairs tumor growth. J. Clin. Investig. 2016, 126, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.N.; Yu, E.Y.; Jacobs, C.; Bazov, J.; Kollmannsberger, C.; Higano, C.S.; Mukherjee, S.D.; Gleave, M.E.; Stewart, P.S.; Hotte, S.J. A phase I dose-escalation study of apatorsen (OGX-427), an antisense inhibitor targeting heat shock protein 27 (Hsp27), in patients with castration-resistant prostate cancer and other advanced cancers. Ann. Oncol. 2016, 27, 1116–1122. [Google Scholar] [CrossRef]
- Bellmunt, J.; Eigl, B.J.; Senkus-Konefka, E.; Loriot, Y.; Twardowski, P.; Castellano, D.E.; Blais, N.; Sridhar, S.S.; Sternberg, C.N.; Retz, M.; et al. First-line randomized phase II study of gemcitabine/cisplatin plus apatorsen or placebo in patients with advanced bladder cancer: The International Borealis-1 trial. J. Clin. Oncol. 2015, 33, 4503. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Hahn, N.M.; Werner, L.; Regan, M.M.; Rosenberg, J.E. Borealis-2: A randomized phase II study of OGX-427 (apatorsen) plus docetaxel versus docetaxel alone in platinum-resistant metastatic urothelial cancer (mUC) (Hoosier Cancer Research Network GU12-160). J. Clin. Oncol. 2017, 35, 289. [Google Scholar] [CrossRef]
- Ko, A.H.; Murphy, P.B.; Peyton, J.D.; Shipley, D.L.; Al-Hazzouri, A.; Rodriguez, F.A.; Womack, M.S.t.; Xiong, H.Q.; Waterhouse, D.M.; Tempero, M.A.; et al. A Randomized, Double-Blinded, Phase II Trial of Gemcitabine and Nab-Paclitaxel Plus Apatorsen or Placebo in Patients with Metastatic Pancreatic Cancer: The RAINIER Trial. Oncologist 2017, 22, 1427-e129. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Blackhall, F.; Muthukumar, D.; Lester, J.; Khan, S.; Adams, J.; Illsley, M.; Macgregor, C.; Owadally, W.; Sarker, S.J.; et al. A phase II, randomised, open-label study of gemcitabine/carboplatin first-line chemotherapy in combination with or without the antisense oligonucleotide apatorsen (OGX-427) in advanced squamous cell lung cancers. Ann. Oncol. 2017, 28. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.Y.; Ellard, S.L.; Hotte, S.J.; Gingerich, J.R.; Joshua, A.M.; Gleave, M.E.; Chi, K.N. A randomized phase 2 study of a HSP27 targeting antisense, apatorsen with prednisone versus prednisone alone, in patients with metastatic castration resistant prostate cancer. Investig. New Drugs 2018, 36, 278–287. [Google Scholar] [CrossRef]
- Spigel, D.R.; Shipley, D.L.; Waterhouse, D.M.; Jones, S.F.; Ward, P.J.; Shih, K.C.; Hemphill, B.; McCleod, M.; Whorf, R.C.; Page, R.D.; et al. A Randomized, Double-Blinded, Phase II Trial of Carboplatin and Pemetrexed with or without Apatorsen (OGX-427) in Patients with Previously Untreated Stage IV Non-Squamous-Non-Small-Cell Lung Cancer: The SPRUCE Trial. Oncologist 2019, 24, e1409–e1416. [Google Scholar] [CrossRef] [Green Version]
- Ross, S.J.; Revenko, A.S.; Hanson, L.L.; Ellston, R.; Staniszewska, A.; Whalley, N.; Pandey, S.K.; Revill, M.; Rooney, C.; Buckett, L.K.; et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci. Transl. Med. 2017, 9, eaal5253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phase I Dose-Escalation Study of AZD4785 in Patients with Advanced Solid Tumours. Available online: https://astrazenecagrouptrials.pharmacm.com/ST/Submission/View?id=24623 (accessed on 7 October 2021).
- Chowdhury, S.; Burris, H.A.; Patel, M.; Infante, J.R.; Jones, S.F.; Voskoboynik, M.; Parry, K.; Elvin, P.; Coleman, T.; Gardner, H.; et al. A phase I dose escalation, safety and pharmacokinetic (PK) study of AZD5312 (IONIS-ARRx), a first-in-class Generation 2.5 antisense oligonucleotide targeting the androgen receptor (AR). Eur. J. Cancer 2016, 69, S145. [Google Scholar]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Younes, A.; Fayad, L.; Fowler, N.H.; Hagemeister, F.B.; Mistry, R.; Nemunaitis, J.J.; Borad, M.J.; Bryce, A.H.; Yamashita, M.; et al. A phase I study of ISIS 481464 (AZD9150), a first-in-human, first-in-class, antisense oligonucleotide inhibitor of STAT3, in patients with advanced cancers. J. Clin. Oncol. 2013, 31, 8523. [Google Scholar] [CrossRef]
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: Results of a phase 1b trial. J. Immunother. Cancer 2018, 6, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribrag, V.; Lee, S.T.; Rizzieri, D.; Dyer, M.J.S.; Fayad, L.; Kurzrock, R.; Andritsos, L.; Bouabdallah, R.; Hayat, A.; Bacon, L.; et al. A Phase 1b Study to Evaluate the Safety and Efficacy of Durvalumab in Combination With Tremelimumab or Danvatirsen in Patients With Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Clin. Lymphoma Myeloma Leuk. 2021, 21, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Ohanian, M.; Ashizawa, A.T.; Garcia-Manero, G.; Pemmaraju, N.; Kadia, T.; Jabbour, E.; Ravandi, F.; Borthakur, G.; Andreeff, M.; Konopleva, M.; et al. Liposomal Grb2 antisense oligodeoxynucleotide (BP1001) in patients with refractory or relapsed haematological malignancies: A single-centre, open-label, dose-escalation, phase 1/1b trial. Lancet Haematol. 2018, 5, e136–e146. [Google Scholar] [CrossRef]
- Tantravahi, R.V.; Reddy, E.P. myb. In Encyclopedia of Cancer, 2nd ed.; Bertino, J.R., Ed.; Academic Press: New York, NY, USA, 2002; pp. 287–296. [Google Scholar] [CrossRef]
- C-myb Antisense Oligonucleotide G4460. Available online: https://ncit.nci.nih.gov/ncitbrowser/ConceptReport.jsp?dictionary=NCI_Thesaurus&ns=ncit&code=C1541 (accessed on 19 October 2021).
- Luger, S.M.; O’Brien, S.G.; Ratajczak, J.; Ratajczak, M.Z.; Mick, R.; Stadtmauer, E.A.; Nowell, P.C.; Goldman, J.M.; Gewirtz, A.M. Oligodeoxynucleotide-mediated inhibition of c-myb gene expression in autografted bone marrow: A pilot study. Blood 2002, 99, 1150–1158. [Google Scholar] [CrossRef] [Green Version]
- Greenberger, L.M.; Horak, I.D.; Filpula, D.; Sapra, P.; Westergaard, M.; Frydenlund, H.F.; Albaek, C.; Schroder, H.; Orum, H. A RNA antagonist of hypoxia-inducible factor-1alpha, EZN-2968, inhibits tumor cell growth. Mol. Cancer Ther. 2008, 7, 3598–3608. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, A.; Chiorean, E.G.; Tolcher, A.; Papadopoulos, K.; Beeram, M.; Kee, D.; Waddell, M.; Gilles, E.; Buchbinder, A. EZN-2968, a novel hypoxia-inducible factor-1α (HIF-1α) messenger ribonucleic acid (mRNA) antagonist: Results of a phase I, pharmacokinetic (PK), dose-escalation study of daily administration in patients (pts) with advanced malignancies. J. Clin. Oncol. 2009, 27, 2564. [Google Scholar] [CrossRef]
- Jeong, W.; Rapisarda, A.; Park, S.R.; Kinders, R.J.; Chen, A.; Melillo, G.; Turkbey, B.; Steinberg, S.M.; Choyke, P.; Doroshow, J.H.; et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1alpha), in patients with refractory solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Contratto, M.; Shanbhogue, K.P.; Manji, G.A.; O’Neil, B.H.; Noonan, A.; Tudor, R.; Lee, R. Evaluation of a locked nucleic acid form of antisense oligo targeting HIF-1alpha in advanced hepatocellular carcinoma. World J. Clin. Oncol. 2019, 10, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Cordon-Cardo, C.; Kelly, W.K.; Slovin, S.F.; Siedlecki, K.; Regan, K.P.; DiPaola, R.S.; Rafi, M.; Rosen, N.; Scher, H.I. Safety and biologic activity of intravenous BCL-2 antisense oligonucleotide (G3139) and taxane chemotherapy in patients with advanced cancer. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Byrd, J.C.; Dai, G.; Klisovic, M.I.; Kourlas, P.J.; Young, D.C.; Cataland, S.R.; Fisher, D.B.; Lucas, D.; Chan, K.K.; et al. Phase 1 and pharmacodynamic studies of G3139, a Bcl-2 antisense oligonucleotide, in combination with chemotherapy in refractory or relapsed acute leukemia. Blood 2003, 101, 425–432. [Google Scholar] [CrossRef] [Green Version]
- Rudin, C.M.; Otterson, G.A.; Mauer, A.M.; Villalona-Calero, M.A.; Tomek, R.; Prange, B.; George, C.M.; Szeto, L.; Vokes, E.E. A pilot trial of G3139, a bcl-2 antisense oligonucleotide, and paclitaxel in patients with chemorefractory small-cell lung cancer. Ann. Oncol. 2002, 13, 539–545. [Google Scholar] [CrossRef]
- Mita, M.M.; Ochoa, L.; Rowinsky, E.K.; Kuhn, J.; Schwartz, G.; Hammond, L.A.; Patnaik, A.; Yeh, I.T.; Izbicka, E.; Berg, K.; et al. A phase I, pharmacokinetic and biologic correlative study of oblimersen sodium (Genasense, G3139) and irinotecan in patients with metastatic colorectal cancer. Ann. Oncol. 2006, 17, 313–321. [Google Scholar] [CrossRef]
- Moore, J.; Seiter, K.; Kolitz, J.; Stock, W.; Giles, F.; Kalaycio, M.; Zenk, D.; Marcucci, G. A Phase II study of Bcl-2 antisense (oblimersen sodium) combined with gemtuzumab ozogamicin in older patients with acute myeloid leukemia in first relapse. Leuk. Res. 2006, 30, 777–783. [Google Scholar] [CrossRef]
- Agarwala, S.S.; Keilholz, U.; Gilles, E.; Bedikian, A.Y.; Wu, J.; Kay, R.; Stein, C.A.; Itri, L.M.; Suciu, S.; Eggermont, A.M. LDH correlation with survival in advanced melanoma from two large, randomised trials (Oblimersen GM301 and EORTC 18951). Eur. J. Cancer 2009, 45, 1807–1814. [Google Scholar] [CrossRef]
- Chanan-Khan, A.A.; Niesvizky, R.; Hohl, R.J.; Zimmerman, T.M.; Christiansen, N.P.; Schiller, G.J.; Callander, N.; Lister, J.; Oken, M.; Jagannath, S. Phase III randomised study of dexamethasone with or without oblimersen sodium for patients with advanced multiple myeloma. Leuk. Lymphoma 2009, 50, 559–565. [Google Scholar] [CrossRef]
- Rudin, C.M.; Kozloff, M.; Hoffman, P.C.; Edelman, M.J.; Karnauskas, R.; Tomek, R.; Szeto, L.; Vokes, E.E. Phase I study of G3139, a bcl-2 antisense oligonucleotide, combined with carboplatin and etoposide in patients with small-cell lung cancer. J. Clin. Oncol. 2004, 22, 1110–1117. [Google Scholar] [CrossRef]
- O’Brien, S.M.; Cunningham, C.C.; Golenkov, A.K.; Turkina, A.G.; Novick, S.C.; Rai, K.R. Phase I to II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in patients with advanced chronic lymphocytic leukemia. J. Clin. Oncol. 2005, 23, 7697–7702. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.; Moore, J.O.; Boyd, T.E.; Larratt, L.M.; Skotnicki, A.B.; Koziner, B.; Chanan-Khan, A.A.; Seymour, J.F.; Gribben, J.; Itri, L.M.; et al. 5-year survival in patients with relapsed or refractory chronic lymphocytic leukemia in a randomized, phase III trial of fludarabine plus cyclophosphamide with or without oblimersen. J. Clin. Oncol. 2009, 27, 5208–5212. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.R.; Marcucci, G.; Yin, J.; Blum, W.; Stock, W.; Kohlschmidt, J.; Mrozek, K.; Carroll, A.J.; Eisfeld, A.K.; Wang, E.S.; et al. Phase 3 randomized trial of chemotherapy with or without oblimersen in older AML patients: CALGB 10201 (Alliance). Blood Adv. 2021, 5, 2775–2787. [Google Scholar] [CrossRef] [PubMed]
- Bedikian, A.Y.; Garbe, C.; Conry, R.; Lebbe, C.; Grob, J.J.; Genasense Melanoma Study, G. Dacarbazine with or without oblimersen (a Bcl-2 antisense oligonucleotide) in chemotherapy-naive patients with advanced melanoma and low-normal serum lactate dehydrogenase: ‘The AGENDA trial’. Melanoma Res. 2014, 24, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.A.; Schilsky, R.L.; Young, A.; Janisch, L.; Stadler, W.M.; Vogelzang, N.J.; Cadden, S.; Wright, J.A.; Ratain, M.J. A phase I study of antisense oligonucleotide GTI-2040 given by continuous intravenous infusion in patients with advanced solid tumors. Ann. Oncol. 2005, 16, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Stadler, W.M.; Desai, A.A.; Quinn, D.I.; Bukowski, R.; Poiesz, B.; Kardinal, C.G.; Lewis, N.; Makalinao, A.; Murray, P.; Torti, F.M. A Phase I/II study of GTI-2040 and capecitabine in patients with renal cell carcinoma. Cancer Chemother. Pharmacol. 2008, 61, 689–694. [Google Scholar] [CrossRef]
- Juhasz, A.; Vassilakos, A.; Chew, H.K.; Gandara, D.; Yen, Y. Analysis of ribonucleotide reductase M2 mRNA levels in patient samples after GTI-2040 antisense drug treatment. Oncol. Rep. 2006, 15, 1299–1304. [Google Scholar] [CrossRef]
- Klisovic, R.B.; Blum, W.; Wei, X.; Liu, S.; Liu, Z.; Xie, Z.; Vukosavljevic, T.; Kefauver, C.; Huynh, L.; Pang, J.; et al. Phase I study of GTI-2040, an antisense to ribonucleotide reductase, in combination with high-dose cytarabine in patients with acute myeloid leukemia. Clin. Cancer Res. 2008, 14, 3889–3895. [Google Scholar] [CrossRef] [Green Version]
- Leighl, N.B.; Laurie, S.A.; Chen, X.E.; Ellis, P.; Shepherd, F.A.; Knox, J.J.; Goss, G.; Burkes, R.L.; Pond, G.R.; Dick, C.; et al. A phase I/II study of GTI-2040 plus docetaxel as second-line treatment in advanced non-small cell lung cancer: A study of the PMH phase II consortium. J. Thorac. Oncol. 2009, 4, 1163–1169. [Google Scholar] [CrossRef] [Green Version]
- Shibata, S.I.; Doroshow, J.H.; Frankel, P.; Synold, T.W.; Yen, Y.; Gandara, D.R.; Lenz, H.J.; Chow, W.A.; Leong, L.A.; Lim, D.; et al. Phase I trial of GTI-2040, oxaliplatin, and capecitabine in the treatment of advanced metastatic solid tumors: A California Cancer Consortium Study. Cancer Chemother. Pharmacol. 2009, 64, 1149–1155. [Google Scholar] [CrossRef] [Green Version]
- Sridhar, S.S.; Canil, C.M.; Chi, K.N.; Hotte, S.J.; Ernst, S.; Wang, L.; Chen, E.X.; Juhasz, A.; Yen, Y.; Murray, P.; et al. A phase II study of the antisense oligonucleotide GTI-2040 plus docetaxel and prednisone as first-line treatment in castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2011, 67, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Mannargudi, M.B.; Deb, S. Clinical pharmacology and clinical trials of ribonucleotide reductase inhibitors: Is it a viable cancer therapy? J. Cancer Res. Clin. Oncol. 2017, 143, 1499–1529. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Holmlund, J.T.; Kraynak, M.; Richards, D.; Bruce, J.; Ognoskie, N.; Kwoh, T.J.; Geary, R.; Dorr, A.; Von Hoff, D.; et al. Phase I Evaluation of ISIS 3521, an Antisense Oligodeoxynucleotide to Protein Kinase C-Alpha, in Patients With Advanced Cancer. J. Clin. Oncol. 1999, 17, 3586–3595. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Douillard, J.Y.; Koralewski, P.; Manegold, C.; Smit, E.F.; Reyes, J.M.; Chang, G.C.; John, W.J.; Peterson, P.M.; Obasaju, C.K.; et al. Phase III study of gemcitabine and cisplatin with or without aprinocarsen, a protein kinase C-alpha antisense oligonucleotide, in patients with advanced-stage non-small-cell lung cancer. J. Clin. Oncol. 2006, 24, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.C.; Holmlund, J.T.; Schiller, J.H.; Geary, R.S.; Kwoh, T.J.; Dorr, A.; Nemunaitis, J. A Phase I Trial of c-Raf Kinase Antisense Oligonucleotide ISIS 5132 Administered as a Continuous Intravenous Infusion in Patients with Advanced Cancer. Clin. Cancer Res. 2000, 6, 1626–1631. [Google Scholar] [PubMed]
- Oza, A.M.; Elit, L.; Swenerton, K.; Faught, W.; Ghatage, P.; Carey, M.; McIntosh, L.; Dorr, A.; Holmlund, J.T.; Eisenhauer, E. Phase II study of CGP 69846A (ISIS 5132) in recurrent epithelial ovarian cancer: An NCIC clinical trials group study (NCIC IND.116)☆. Gynecol. Oncol. 2003, 89, 129–133. [Google Scholar] [CrossRef]
- Cripps, M.C.; Figueredo, A.T.; Oza, A.M.; Taylor, M.J.; Fields, A.L.; Holmlund, J.T.; McIntosh, L.W.; Geary, R.S.; Eisenhauer, E.A. Phase II randomized study of ISIS 3521 and ISIS 5132 in patients with locally advanced or metastatic colorectal cancer: A National Cancer Institute of Canada clinical trials group study. Clin. Cancer Res. 2002, 8, 2188–2192. [Google Scholar]
- Rudin, C.M.; Marshall, J.L.; Huang, C.H.; Kindler, H.L.; Zhang, C.; Kumar, D.; Gokhale, P.C.; Steinberg, J.; Wanaski, S.; Kasid, U.N.; et al. Delivery of a liposomal c-raf-1 antisense oligonucleotide by weekly bolus dosing in patients with advanced solid tumors: A phase I study. Clin. Cancer Res. 2004, 10, 7244–7251. [Google Scholar] [CrossRef] [Green Version]
- Dritschilo, A.; Huang, C.H.; Rudin, C.M.; Marshall, J.; Collins, B.; Dul, J.L.; Zhang, C.; Kumar, D.; Gokhale, P.C.; Ahmad, A.; et al. Phase I study of liposome-encapsulated c-raf antisense oligodeoxyribonucleotide infusion in combination with radiation therapy in patients with advanced malignancies. Clin. Cancer Res. 2006, 12, 1251–1259. [Google Scholar] [CrossRef] [Green Version]
- Chi, K.N.; Eisenhauer, E.; Fazli, L.; Jones, E.C.; Goldenberg, S.L.; Powers, J.; Tu, D.; Gleave, M.E. A phase I pharmacokinetic and pharmacodynamic study of OGX-011, a 2′-methoxyethyl antisense oligonucleotide to clusterin, in patients with localized prostate cancer. J. Natl. Cancer Inst. 2005, 97, 1287–1296. [Google Scholar] [CrossRef]
- Chi, K.N.; Siu, L.L.; Hirte, H.; Hotte, S.J.; Knox, J.; Kollmansberger, C.; Gleave, M.; Guns, E.; Powers, J.; Walsh, W.; et al. A phase I study of OGX-011, a 2′-methoxyethyl phosphorothioate antisense to clusterin, in combination with docetaxel in patients with advanced cancer. Clin. Cancer Res. 2008, 14, 833–839. [Google Scholar] [CrossRef] [Green Version]
- Chia, S.; Dent, S.; Ellard, S.; Ellis, P.M.; Vandenberg, T.; Gelmon, K.; Powers, J.; Walsh, W.; Seymour, L.; Eisenhauer, E.A. Phase II trial of OGX-011 in combination with docetaxel in metastatic breast cancer. Clin. Cancer Res. 2009, 15, 708–713. [Google Scholar] [CrossRef] [Green Version]
- Chi, K.N.; Hotte, S.J.; Yu, E.Y.; Tu, D.; Eigl, B.J.; Tannock, I.; Saad, F.; North, S.; Powers, J.; Gleave, M.E.; et al. Randomized phase II study of docetaxel and prednisone with or without OGX-011 in patients with metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2010, 28, 4247–4254. [Google Scholar] [CrossRef] [PubMed]
- Saad, F.; Hotte, S.; North, S.; Eigl, B.; Chi, K.; Czaykowski, P.; Wood, L.; Pollak, M.; Berry, S.; Lattouf, J.B.; et al. Randomized phase II trial of Custirsen (OGX-011) in combination with docetaxel or mitoxantrone as second-line therapy in patients with metastatic castrate-resistant prostate cancer progressing after first-line docetaxel: CUOG trial P-06c. Clin. Cancer Res. 2011, 17, 5765–5773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beer, T.M.; Hotte, S.J.; Saad, F.; Alekseev, B.; Matveev, V.; Fléchon, A.; Gravis, G.; Joly, F.; Chi, K.N.; Malik, Z.; et al. Custirsen (OGX-011) combined with cabazitaxel and prednisone versus cabazitaxel and prednisone alone in patients with metastatic castration-resistant prostate cancer previously treated with docetaxel (AFFINITY): A randomised, open-label, international, phase 3 trial. Lancet Oncol. 2017, 18, 1532–1542. [Google Scholar] [CrossRef]
- Chi, K.N.; Sweeney, C.; Jacobs, C.; Stewart, P.S.; Hahn, N.M. The Pacific trial: A randomized phase II study of OGX-427 in men with metastatic castration-resistant prostate cancer (mCRPC) and PSA progression while receiving abiraterone acetate (AA). J. Clin. Oncol. 2013, 31, TPS5101. [Google Scholar] [CrossRef]
- Roschewski, M.; Izumi, R.; Hamdy, A.; Patel, M.R.; Arkenau, H.-T.; de Vos, S.; Reagan, P.M.; Zinzani, P.L.; Davies, A.; Pagel, J.M.; et al. PRISM: A Platform Protocol for the Treatment of Relapsed/Refractory Aggressive Non-Hodgkin Lymphoma. Blood 2019, 134, 2869. [Google Scholar] [CrossRef]
- A Phase I/Ib Study of AZD9150 (ISIS-STAT3Rx) in Patients with Advanced/Metastatic Hepatocellular Carcinoma. Available online: https://clinicaltrials.gov/ct2/show/NCT01839604 (accessed on 7 October 2021).
- Study of AZD9150 and MEDI4736 (Durvalumab) in Japanese Adult Patients With Advanced Solid Malignancies. Available online: https://astrazenecagrouptrials.pharmacm.com/ST/Submission/View?id=25189 (accessed on 8 October 2021).
- Bardelli, V.; Arniani, S.; Pierini, V.; Pierini, T.; Di Giacomo, D.; Gorello, P.; Moretti, M.; Pellanera, F.; Elia, L.; Vitale, A.; et al. MYB rearrangements and over-expression in T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer 2021, 60, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.; Panda, S.; Samal, S.K.; Shriwas, O.; Rath, R.; Pellecchia, M.; Emdad, L.; Das, S.K.; Fisher, P.B.; Dash, R. Bcl-2 Antiapoptotic Family Proteins and Chemoresistance in Cancer. Adv. Cancer Res. 2018, 137, 37–75. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.; Nelson, K.; Patel, V. Emerging therapies for rare cutaneous cancers: A systematic review. Cancer Treat. Rev. 2021, 100, 102266. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, K.; Gogtay, N.J. Therapeutic nucleic acids: Current clinical status. Br. J. Clin. Pharmacol. 2016, 82, 659–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rheingold, S.R.; Hogarty, M.D.; Blaney, S.M.; Zwiebel, J.A.; Sauk-Schubert, C.; Chandula, R.; Krailo, M.D.; Adamson, P.C. Phase I Trial of G3139, a bcl-2 antisense oligonucleotide, combined with doxorubicin and cyclophosphamide in children with relapsed solid tumors: A Children’s Oncology Group Study. J. Clin. Oncol. 2007, 25, 1512–1518. [Google Scholar] [CrossRef]
- Marcucci, G.; Stock, W.; Dai, G.; Klisovic, R.B.; Liu, S.; Klisovic, M.I.; Blum, W.; Kefauver, C.; Sher, D.A.; Green, M.; et al. Phase I study of oblimersen sodium, an antisense to Bcl-2, in untreated older patients with acute myeloid leukemia: Pharmacokinetics, pharmacodynamics, and clinical activity. J. Clin. Oncol. 2005, 23, 3404–3411. [Google Scholar] [CrossRef]
- Mays, T.A.; Mita, A.C.; Takimoto, C.; Petro, D.; Egorin, M.J.; Patnaik, A.; Papodopolous, K.; Rowinsky, E.; Goldston, M.; Tolcher, A. Bcl-2 biomodulation with oblimersen sodium in combination with FOLFOX4 chemotherapy: A phase I study in metastatic colon carcinoma. J. Clin. Oncol. 2005, 23, 3158. [Google Scholar] [CrossRef]
- Sternberg, C.N.; Dumez, H.; Van Poppel, H.; Skoneczna, I.; Sella, A.; Daugaard, G.; Gil, T.; Graham, J.; Carpentier, P.; Calabro, F.; et al. Docetaxel plus oblimersen sodium (Bcl-2 antisense oligonucleotide): An EORTC multicenter, randomized phase II study in patients with castration-resistant prostate cancer. Ann. Oncol. 2009, 20, 1264–1269. [Google Scholar] [CrossRef]
- Knox, J.J.; Chen, X.E.; Feld, R.; Nematollahi, M.; Cheiken, R.; Pond, G.; Zwiebel, J.A.; Gill, S.; Moore, M. A phase I-II study of oblimersen sodium (G3139, Genasense) in combination with doxorubicin in advanced hepatocellular carcinoma (NCI # 5798). Investig. New Drugs 2008, 26, 193–194. [Google Scholar] [CrossRef]
- Giudice, V.; Mensitieri, F.; Izzo, V.; Filippelli, A.; Selleri, C. Aptamers and Antisense Oligonucleotides for Diagnosis and Treatment of Hematological Diseases. Int. J. Mol. Sci. 2020, 21, 3252. [Google Scholar] [CrossRef]
- Pro, B.; Leber, B.; Smith, M.; Fayad, L.; Romaguera, J.; Hagemeister, F.; Rodriguez, A.; McLaughlin, P.; Samaniego, F.; Zwiebel, J.; et al. Phase II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in combination with rituximab in patients with recurrent B-cell non-Hodgkin lymphoma. Br. J. Haematol. 2008, 143, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Suvarna, V.; Singh, V.; Murahari, M. Current overview on the clinical update of Bcl-2 anti-apoptotic inhibitors for cancer therapy. Eur. J. Pharmacol. 2019, 862, 172655. [Google Scholar] [CrossRef] [PubMed]
- Faleiro, I.; Leão, R.; Binnie, A.; de Mello, R.A.; Maia, A.-T.; Castelo-Branco, P. Epigenetic therapy in urologic cancers: An update on clinical trials. Oncotarget 2017, 8, 12484–12500. [Google Scholar] [CrossRef] [Green Version]
- Raab, R.; Sparano, J.A.; Ocean, A.J.; Christos, P.; Ramirez, M.; Vinciguerra, V.; Kaubisch, A. A phase I trial of oblimersen sodium in combination with cisplatin and 5-fluorouracil in patients with advanced esophageal, gastroesophageal junction, and gastric carcinoma. Am. J. Clin. Oncol. 2010, 33, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Bedikian, A.Y.; Millward, M.; Pehamberger, H.; Conry, R.; Gore, M.; Trefzer, U.; Pavlick, A.C.; DeConti, R.; Hersh, E.M.; Hersey, P.; et al. Bcl-2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: The Oblimersen Melanoma Study Group. J. Clin. Oncol. 2006, 24, 4738–4745. [Google Scholar] [CrossRef] [PubMed]
- Tentori, L.; Lacal, P.M.; Graziani, G. Challenging resistance mechanisms to therapies for metastatic melanoma. Trends Pharmacol. Sci. 2013, 34, 656–666. [Google Scholar] [CrossRef]
- Badros, A.Z.; Goloubeva, O.; Rapoport, A.P.; Ratterree, B.; Gahres, N.; Meisenberg, B.; Takebe, N.; Heyman, M.; Zwiebel, J.; Streicher, H.; et al. Phase II study of G3139, a Bcl-2 antisense oligonucleotide, in combination with dexamethasone and thalidomide in relapsed multiple myeloma patients. J. Clin. Oncol. 2005, 23, 4089–4099. [Google Scholar] [CrossRef]
- Klisovic, R.B.; Blum, W.; Liu, Z.; Xie, Z.; Kefauver, C.; Huynh, L.; Zwiebel, J.A.; Devine, S.M.; Byrd, J.C.; Grever, M.R.; et al. Phase I study of GTI-2040, a ribonucleotide reductase antisense, with high dose cytarabine in patients with relapsed/refractory acute myeloid leukemia. Leuk. Lymphoma 2014, 55, 1332–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschbaum, M.H.; Frankel, P.; Synold, T.W.; Xie, Z.; Yen, Y.; Popplewell, L.; Chen, R.; Aljitawi, O.; Tuscano, J.M.; Chan, K.K.; et al. A phase I pharmacodynamic study of GTI-2040, an antisense oligonucleotide against ribonuclotide reductase, in acute leukemias: A California Cancer Consortium study. Leuk. Lymphoma 2016, 57, 2307–2314. [Google Scholar] [CrossRef] [Green Version]
- Malik, L.; Zwiebel, A.; Cooper, J. A phase I pharmacokinetic and pharmacodynamic study of GTI-2040 in combination with gemcitabine in patients with solid tumors. Cancer Chemother. Pharmacol. 2018, 82, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Ritch, P.; Rudin, C.M.; Bitran, J.D.; Edelman, M.J.; Makalinao, A.; Irwin, D.; Lilenbaum, R.; Peterson, P.; John, W.J. Phase II study of PKC-alpha antisense oligonucleotide aprinocarsen in combination with gemcitabine and carboplatin in patients with advanced non-small cell lung cancer. Lung Cancer 2006, 52, 173–180. [Google Scholar] [CrossRef]
- Gradishar, W.; O’Neill, A.; Cobleigh, M.; Goldstein, L.; Davidson, N. A phase II trial with antisense oligonucleotide ISIS 3521/Cgp 64128a in patients (Pts) with metastatic breast cancer (MBC): ECOG Trial 3197. In Proceedings of the 37th Annual Meeting of the American Society of Clinical Oncology, San Francisco, CA, USA, 12–15 May 2001. [Google Scholar]
- Steinberg, J.L.; Mendelson, D.S.; Block, H.; Green, S.B.; Shu, V.S.; Parker, K.; Cullinan, P.; Dul, J.L.; von Hoff, D.D.; Gordon, M.S. Phase I study of LErafAON-ETU, an easy-to-use formulation of liiposome entrapped c-raf antisense oligonucleotide, in advanced cancer patients. J. Clin. Oncol. 2005, 23, 3214. [Google Scholar] [CrossRef]
- Xiu, P.; Dong, X.F.; Li, X.P.; Li, J. Clusterin: Review of research progress and looking ahead to direction in hepatocellular carcinoma. World J. Gastroenterol. 2015, 21, 8262–8270. [Google Scholar] [CrossRef]
- Laskin, J.J.; Nicholas, G.; Lee, C.; Gitlitz, B.; Vincent, M.; Cormier, Y.; Stephenson, J.; Ung, Y.; Sanborn, R.; Pressnail, B.; et al. Phase I/II trial of custirsen (OGX-011), an inhibitor of clusterin, in combination with a gemcitabine and platinum regimen in patients with previously untreated advanced non-small cell lung cancer. J. Thorac. Oncol. 2012, 7, 579–586. [Google Scholar] [CrossRef] [Green Version]
- Blumenstein, B.; Saad, F.; Hotte, S.; Chi, K.N.; Eigl, B.; Gleave, M.; Jacobs, C. Reduction in serum clusterin is a potential therapeutic biomarker in patients with castration-resistant prostate cancer treated with custirsen. Cancer Med. 2013, 2, 468–477. [Google Scholar] [CrossRef]
- de Liano, A.G.; Reig, O.; Mellado, B.; Martin, C.; Rull, E.U.; Maroto, J.P. Prognostic and predictive value of plasma testosterone levels in patients receiving first-line chemotherapy for metastatic castrate-resistant prostate cancer. Br. J. Cancer 2014, 110, 2201–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, K.N.; Higano, C.S.; Blumenstein, B.; Ferrero, J.-M.; Reeves, J.; Feyerabend, S.; Gravis, G.; Merseburger, A.S.; Stenzl, A.; Bergman, A.M.; et al. Custirsen in combination with docetaxel and prednisone for patients with metastatic castration-resistant prostate cancer (SYNERGY trial): A phase 3, multicentre, open-label, randomised trial. Lancet Oncol. 2017, 18, 473–485. [Google Scholar] [CrossRef]
- Juliano, R.; Alam, M.R.; Dixit, V.; Kang, H. Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucleic Acids Res. 2008, 36, 4158–4171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Mukhopadhyay, T.; Donehower, L.A.; Georges, R.N.; Roth, J.A. Retroviral vector-mediated transduction of K-ras antisense RNA into human lung cancer cells inhibits expression of the malignant phenotype. Hum. Gene Ther. 1993, 4, 451–460. [Google Scholar] [CrossRef]
- Gokirmak, T.; Nikan, M.; Wiechmann, S.; Prakash, T.P.; Tanowitz, M.; Seth, P.P. Overcoming the challenges of tissue delivery for oligonucleotide therapeutics. Trends Pharmacol. Sci. 2021, 42, 588–604. [Google Scholar] [CrossRef]
- Raizada, M.K.; Katovich, M.J.; Wang, H.; Berecek, K.H.; Gelband, C.H. Is antisense gene therapy a step in the right direction in the control of hypertension? Am. J. Physiol.-Heart Circ. Physiol. 1999, 277, H423–H432. [Google Scholar] [CrossRef] [PubMed]
- Felgner, P.L.; Ringold, G. Cationic liposome-mediated transfection. Nature 1989, 337, 387–388. [Google Scholar]
- Capaccioli, S.; Dipasquale, G.; Mini, E.; Mazzei, T.; Quattrone, A. Cationic lipids improve antisense oligonucleotide uptake and prevent degradation in cultured cells and in human serum. Biochem. Biophys. Res. Commun. 1993, 197, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Farhood, H.; Bottega, R.; Epand, R.M.; Huang, L. Effect of cationic cholesterol derivatives on gene transfer and protein kinase C activity. Biochim. Et Biophys. Acta BBA-Biomembr. 1992, 1111, 239–246. [Google Scholar] [CrossRef]
- Gagliardi, M.; Ashizawa, A.T. The Challenges and Strategies of Antisense Oligonucleotide Drug Delivery. Biomedicines 2021, 9, 433. [Google Scholar] [CrossRef] [PubMed]
- Hunt, M.A.; Currie, M.J.; Robinson, B.A.; Dachs, G.U. Optimizing transfection of primary human umbilical vein endothelial cells using commercially available chemical transfection reagents. J. Biomol. Tech. JBT 2010, 21, 66–72. [Google Scholar] [PubMed]
- Akhtar, S.; Hughes, M.D.; Khan, A.; Bibby, M.; Hussain, M.; Nawaz, Q.; Double, J.; Sayyed, P. The delivery of antisense therapeutics. Adv. Drug Deliv. Rev. 2000, 44, 3–21. [Google Scholar] [CrossRef]
- Bielinska, A.; Kukowska-Latallo, J.F.; Johnson, J.; Tomalia, D.A.; Baker, J.R., Jr. Regulation of in vitro gene expression using antisense oligonucleotides or antisense expression plasmids transfected using starburst PAMAM dendrimers. Nucleic Acids Res. 1996, 24, 2176–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.Y.; Wu, C.H. Receptor-mediated gene delivery and expression in vivo. J. Biol. Chem. 1988, 263, 14621–14624. [Google Scholar] [CrossRef]
- Lewis, K.J.; Irwin, W.J.; Akhtar, S. Development of a sustained-release biodegradable polymer delivery system for site-specific delivery of oligonucleotides: Characterization of P (LA-GA) copolymer microspheres in vitro. J. Drug Target. 1998, 5, 291–302. [Google Scholar] [CrossRef]
- Putney, S.D.; Brown, J.; Cucco, C.; Lee, R.; Skorski, T.; Leonetti, C.; Geiser, T.; Calabretta, B.; Zupi, G.; Zon, G. Enhanced anti-tumor effects with microencapsulated c-myc antisense oligonucleotide. Antisense Nucleic Acid Drug Dev. 1999, 9, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Sommer, W.; Fuxe, K.; Akhtar, S. Site-specific administration of antisense oligonucleotides using biodegradable polymer microspheres provides sustained delivery and improved subcellular biodistribution in the neostriatum of the rat brain. J. Drug Target. 2000, 8, 319–334. [Google Scholar] [CrossRef]
- Kher, G.; Trehan, S.; Misra, A. Antisense oligonucleotides and rna interference. Chall. Deliv. Ther. Genom. Proteom. 2011, 325–386. [Google Scholar]
- Dong, L.; Xia, S.; Chen, H.; Chen, J.; Zhang, J. Spleen-specific suppression of TNF-α by cationic hydrogel-delivered antisense nucleotides for the prevention of arthritis in animal models. Biomaterials 2009, 30, 4416–4426. [Google Scholar] [CrossRef]
- Lou, X.; Garrett, K.L.; Rakoczy, P.E.; Chirila, T.V. Synthetic hydrogels as carriers in antisense therapy: Preliminary evaluation of an oligodeoxynucleotide covalent conjugate with a copolymer of 1-vinyl-2-pyrrolidinone and 2-hydroxyethyl methacrylate. J. Biomater. Appl. 2001, 15, 307–320. [Google Scholar] [CrossRef]
- Javadzadeh, Y.; Bahari, L.A. Therapeutic nanostructures for dermal and transdermal drug delivery. In Nano-and Microscale Drug Delivery Systems; Elsevier: Amsterdam, The Netherlands, 2017; pp. 131–146. [Google Scholar]
- Prochiantz, A. Messenger proteins: Homeoproteins, TAT and others. Curr. Opin. Cell Biol. 2000, 12, 400–406. [Google Scholar] [CrossRef]
- McClorey, G.; Banerjee, S. Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics. Biomedicines 2018, 6, 51. [Google Scholar] [CrossRef] [Green Version]
- Pichon, C.; Freulon, I.; Midoux, P.; Mayer, R.; Monsigny, M.; Roche, A.-C. Cytosolic and nuclear delivery of oligonucleotides mediated by an amphiphilic anionic peptide. Antisense Nucleic Acid Drug Dev. 1997, 7, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Bolhassani, A.; Jafarzade, B.S.; Mardani, G. In vitro and in vivo delivery of therapeutic proteins using cell penetrating peptides. Peptides 2017, 87, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Novak, J.S.; Jaiswal, J.K.; Partridge, T.A. The macrophage as a Trojan horse for antisense oligonucleotide delivery. Expert Opin. Ther. Targets 2018, 22, 463–466. [Google Scholar] [PubMed]
- Sun, D.; Zhuang, X.; Zhang, S.; Deng, Z.-B.; Grizzle, W.; Miller, D.; Zhang, H.-G. Exosomes are endogenous nanoparticles that can deliver biological information between cells. Adv. Drug Deliv. Rev. 2013, 65, 342–347. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar]
- Liu, R.; Liu, J.; Ji, X.; Liu, Y. Synthetic nucleic acids delivered by exosomes: A potential therapeutic for generelated metabolic brain diseases. Metab. Brain Dis. 2013, 28, 551–562. [Google Scholar]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Jeong, K.; Jeong, S.; Kim, J.A.; Rhee, W.J. Exosome-based antisense locked nucleic acid delivery for inhibition of type II collagen degradation in chondrocyte. J. Ind. Eng. Chem. 2019, 74, 126–135. [Google Scholar] [CrossRef]
- Zewert, T.E.; Pliquett, U.F.; Langer, R.; Weaver, J.C. Transdermal transport of DNA antisense oligonucleotides by electroporation. Biochem. Biophys. Res. Commun. 1995, 212, 286–292. [Google Scholar] [CrossRef]
- Regnier, V.; Préat, V. Localization of a FITC-labeled phosphorothioate oligodeoxynucleotide in the skin after topical delivery by iontophoresis and electroporation. Pharm. Res. 1998, 15, 1596–1602. [Google Scholar] [CrossRef] [PubMed]
- Bergan, R.; Connell, Y.; Fahmy, B.; Neckers, L. Electroporation enhances c-myc antisense oligodeoxynucleotide efficacy. Nucleic Acids Res. 1993, 21, 3567–3573. [Google Scholar] [CrossRef] [Green Version]
- Regnier, V.; De Morre, N.; Jadoul, A.; Préat, V. Mechanisms of a phosphorothioate oligonucleotide delivery by skin electroporation. Int. J. Pharm. 1999, 184, 147–156. [Google Scholar] [CrossRef]
- Zavaglia, D.; Normand, N.; Brewis, N.; O’hare, P.; Favrot, M.-C.; Coll, J.-l. VP22-mediated and light-activated delivery of an anti-c-raf1 antisense oligonucleotide improves its activity after intratumoral injection in nude mice. Mol. Ther. 2003, 8, 840–845. [Google Scholar] [PubMed]
- Brewis, N.; Phelan, A.; Normand, N.; Choolun, E.; O’hare, P. Particle assembly incorporating a VP22–BH3 fusion protein, facilitating intracellular delivery, regulated release, and apoptosis. Mol. Ther. 2003, 7, 262–270. [Google Scholar] [CrossRef]
- Normand, N.; Valamanesh, F.; Savoldelli, M.; Mascarelli, F.; BenEzra, D.; Courtois, Y.; Behar-Cohen, F. VP22 light controlled delivery of oligonucleotides to ocular cells in vitro and in vivo. Mol. Vis. 2005, 11, 184–191. [Google Scholar]
- Kontturi, L.-S.; Van Den Dikkenberg, J.; Urtti, A.; Hennink, W.E.; Mastrobattista, E. Light-triggered cellular delivery of oligonucleotides. Pharmaceutics 2019, 11, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haag, P.; Frauscher, F.; Gradl, J.; Seitz, A.; Schäfer, G.; Lindner, J.R.; Klibanov, A.L.; Bartsch, G.; Klocker, H.; Eder, I.E. Microbubble-enhanced ultrasound to deliver an antisense oligodeoxynucleotide targeting the human androgen receptor into prostate tumours. J. Steroid Biochem. Mol. Biol. 2006, 102, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Unger, E.C.; Hersh, E.; Vannan, M.; McCreery, T. Gene delivery using ultrasound contrast agents. Echocardiography 2001, 18, 355–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negishi, Y.; Ishii, Y.; Nirasawa, K.; Sasaki, E.; Endo-Takahashi, Y.; Suzuki, R.; Maruyama, K. PMO delivery system using bubble liposomes and ultrasound exposure for duchenne muscular dystrophy treatment. In Duchenne Muscular Dystrophy; Springer: Clifton, NJ, USA, 2018; pp. 185–192. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Research | Ref. | |
|---|---|---|---|
| 1 | AO | SSO111 is a 20mer fully modified 2′-MOE-PS AO-targeting oncogene HER2. SSO111 induced exon 15 skipping during splicing, leading to the generation of a novel mRNA transcript that excludes exon 15. | [81] |
| Mechanism |  | ||
| 2 | AO | Acr-PNA 2794 is a 15mer fully modified PNA AO conjugated with Acr targeting HER2. Acr-PNA 2794 induced exon-19 skipping, leading to the generation of a novel mRNA transcript that excludes exon-19. | [82] |
| Mechanism |  | ||
| 3 | AO | SSOe26 is a 15mer LNA-modified mixmer AO targeting HER4. SSOe26 induced exon 26 skipping, leading to the generation of a novel mRNA transcript that excludes exon 26 (CYT2 isoform). | [85] |
| Mechanism |  | ||
| 4 | AO | ASWT1exon5 is a 20mer 2′-MOE-PS gapmer AO targeting oncogene WT1. It induces RNase H-mediated degradation of exon 5-containing transcripts, thus increasing the proportion of transcripts that exclude exon 5. | [91] |
| Mechanism |  | ||
| 5 | AO | PNA 4577, 4578, 4580, and 4581 are 16mer fully modified PNA AOs conjugated with octaarginine or cholic acid-targeting oncogene TdT. These four PNAs all induced intron 7 retention, leading to the generation of a novel mRNA transcript that included intron-7. | [95] |
| Mechanism |  | ||
| 6 | AO | Morpholino MDM4 is a 25mer fully modified PMO AO targeting MDM4. Morpholino MDM4 induced exon 6 skipping, leading to nonsense-mediated decay of the mRNA transcript that excludes exon-6. | [98] |
| Mechanism |  | ||
| No | Drug Name | Target | Condition | Clinical Trial Number | Development Stage | Chemistry | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | Apatorsen (OGX-427) | Hsp27 | Prostate, ovarian, NSCLC, breast or bladder cancer | NCT00487786 | Phase I | 2′-MOE PS | [99] |
| Prostate cancer | NCT01120470 | Phase II | [104] | ||||
| NCT01681433 | Phase II | [154] | |||||
| Urologic neoplasms | NCT01454089 | Phase II | [100] | ||||
| Urothelial carcinoma | NCT01780545 | Phase II | [101] | ||||
| Non-squamous NSCLC | NCT01829113 | Phase II | [105] | ||||
| Advanced squamous cell lung cancers | NCT02423590 | [103] | |||||
| Pancreatic cancer | NCT01844817 | Phase II | [102] | ||||
| 2 | AZD4785 | KRAS | NSCLC/advanced solid tumors | NCT03101839 | Phase I | 2′-4′ cEt | [107] |
| 3 | AZD5312 (ARRx) | Androgen receptor | Androgen receptor-dependent advanced solid tumors | NCT02144051 | Phase I | PS cEt | [108] |
| Prostate cancer/solid tumors | NCT03300505 | Phase Ib/II (recruiting) | [109] | ||||
| 4 | AZD9150 | STAT3 | DLBCL/NHL | NCT01563302 | Phase I/II | cEt gapmer | [110] |
| NCT03527147 | Phase I | [155] | |||||
| HCC and metastasis | NCT01839604 | Phase I | [156] | ||||
| Advanced solid malignancies | NCT03394144 | Phase I | [157] | ||||
| DLBCL | NCT02549651 | Phase Ib | [112] | ||||
| Advanced solid tumors | NCT03421353 | Phase I | Ongoing | ||||
| Muscle-invasive bladder cancer | NCT02546661 | Phase I | Ongoing | ||||
| Metastatic NSCLC | NCT03819465 | Phase I | Ongoing | ||||
| NSCLC | NCT03334617 | Phase II | Ongoing | ||||
| Advanced solid tumors and metastatic squamous cell carcinoma of the head and neck | NCT02499328 | Phase II | Ongoing | ||||
| Advanced pancreatic, NSCLC, and mismatch repair-deficient CRC | NCT02983578 | Phase II | Ongoing | ||||
| 5 | BP1001 | Grb2 | Recurrent adult AML/ALL Myelodysplastic syndrome Ph1-positive CML | NCT01159028 | Phase I | DNAbilize® technology | [113] |
| Solid tumors | NCT04196257 | Phase I | Ongoing | ||||
| AML | NCT02781883 | Phase II | Ongoing | ||||
| 6 | c-myb AS ODN | c-myb | Hematologic malignancies | NCT00780052 | Phase I | PS | [158] |
| Leukemia | NCT00002592 | Phase II | [116] | ||||
| 7 | EZN-2968 | HIF-1 | Carcinoma/lymphoma | NCT00466583 | Phase I | LNA | [118] |
| Liver metastases/neoplasms | NCT01120288 | Phase I | [119] | ||||
| HCC | NCT02564614 | Phase I | [120] | ||||
| 8 | G3139 (Oblimersen) | Bcl-2 | Waldenström Macroglobulinemia | NCT00062244 | Phase 1 Phase 2 | PS | [159] |
| Merkel cell carcinoma | NCT00079131 | Phase 2 | [160] | ||||
| Solid tumors | NCT00003103 | Phase 1 Phase 2 | [121] | ||||
| NCT00054548 | Phase 1 | [161] | |||||
| NCT00543231 | Phase 1 | ||||||
| NCT00636545 | Phase 1 | [161] | |||||
| Relapsed or refractory solid tumors | NCT00039481 | Phase 1 | [162] | ||||
| Leukemia (AML or ALL) | NCT00004862 | Phase 1 | [122] | ||||
| AML | NCT00039117 | Phase 1 | [163] | ||||
| NCT00017589 | Phase 2 | [125] | |||||
| NCT00085124 | Phase 3 | [131] | |||||
| CRC | NCT00004870 | Phase 1 Phase 2 | [124] | ||||
| NCT00055822 | Phase 1 Phase 2 | [164] | |||||
| Prostate cancer | NCT00085228 | Phase 2 | [165] | ||||
| HCC | NCT00047229 | Phase 2 | [166] | ||||
| Recurrent SCLC | NCT00005032 | Phase 1 Phase 2 | [123] | ||||
| Extensive stage SCLC | NCT00017251 | Phase 1 | [128] | ||||
| NCT00042978 | Phase 2 | [128] | |||||
| CML | NCT00049192 | Phase 2 | [167] | ||||
| NHL | NCT00086944 | Phase 1 Phase 2 | [168] | ||||
| Recurrent B-cell NHL | NCT00054639 | Phase 2 | [168] | ||||
| CLL | NCT00078234 | Phase 1 Phase 2 | [169] | ||||
| NCT00021749 | Phase 1 Phase 2 | [129] | |||||
| NCT00024440 | Phase 3 | [130] | |||||
| Metastatic RCC | NCT00059813 | Phase 2 | [170] | ||||
| Advanced esophageal, gastro-esophageal junction and gastric cancer | NCT00064259 | Phase 1 Phase 2 | [171] | ||||
| Melanoma | NCT00409383 | Phase 1 | Unknown | ||||
| NCT00542893 | Phase 1 | [172] | |||||
| NCT00518895 | Phase 3 | [173] | |||||
| NCT00016263 | Phase 3 | [126] | |||||
| NCT00070343 | Not applicable | Ongoing | |||||
| NSCLC | NCT00030641 | Phase 2 Phase 3 | Ongoing | ||||
| Multiple myeloma and plasma cell neoplasm | NCT00049374 | Phase 2 | [174] | ||||
| NCT00017602 | Phase 3 | [127] | |||||
| DLBCL | NCT00070083 | Phase 1 | |||||
| 9 | GTI-2040 | R2 component of RNR | Breast cancer | NCT00068588 | Phase 2 | PS | [135] |
| AML | NCT00070551 | Phase 1 | [175] | ||||
| NCT00565058 | Phase 2 | [136] | |||||
| Acute leukemia, high-grade myelodysplastic syndromes, or refractory or blastic phase CML | NCT00459212 | Phase 1 | [176] | ||||
| RCC | NCT00056173 | Phase 1 Phase 2 | [134] | ||||
| NSCLC, prostate cancer, or other solid tumors | NCT00074022 | Phase 1 Phase 2 | [137] | ||||
| CRC or other solid tumors | NCT00084643 | Phase 1 | [138] | ||||
| Prostate cancer | NCT00087165 | Phase 2 | [139] | ||||
| Metastatic or unresectable solid tumors | NCT00078962 | Phase 1 | [177] | ||||
| 10 | ISIS 3521 | Pkc-Alpha | NSCLC/ melanoma (skin) | NCT00003989 | Phase II | PS | |
| NSCLC | NCT00017407 | Phase III | |||||
| NCT00042679 | Phase II | [178] | |||||
| NCT00034268 | Phase III | [142] | |||||
| 11 | ISIS 5132 | C-raf | Ovarian cancer | NCT00003892 | Phase II | PS | [144] |
| 12 | ISIS 3521 + ISIS 5132 | Pkc-Alpha C-raf | Breast cancer | NCT00003236 | Phase II | [179] | |
| 13 | LErafAON | Raf-1 | Advanced solid tumors | NCT00024661 | Phase I | Liposome encapsulated PS | [146] |
| NCT00100672 | Phase I | [180] | |||||
| NCT00024648 | Phase I | [147] | |||||
| 14 | OGX-011 | Clusterin | Cancer | NCT01497470 | Phase I | 2′-MOE PS | [181] |
| NSCLC | NCT00138658 | Phase I/II | [182] | ||||
| NCT01630733 | Phase III | Ongoing | |||||
| Prostate cancer | NCT00054106 | Phase I | [148] | ||||
| NCT00258388 | Phase II | [151] | |||||
| NCT00138918 | Phase II | [181] | |||||
| NCT00327340 | Phase II | [152,183] | |||||
| NCT01578655 | Phase III | [153] | |||||
| NCT01188187 | Phase III | [184,185] | |||||
| Breast cancer | NCT00258375 | Phase II | [150] | ||||
| Solid tumors | NCT00471432 | Phase I | [149] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raguraman, P.; Balachandran, A.A.; Chen, S.; Diermeier, S.D.; Veedu, R.N. Antisense Oligonucleotide-Mediated Splice Switching: Potential Therapeutic Approach for Cancer Mitigation. Cancers 2021, 13, 5555. https://doi.org/10.3390/cancers13215555
Raguraman P, Balachandran AA, Chen S, Diermeier SD, Veedu RN. Antisense Oligonucleotide-Mediated Splice Switching: Potential Therapeutic Approach for Cancer Mitigation. Cancers. 2021; 13(21):5555. https://doi.org/10.3390/cancers13215555
Chicago/Turabian StyleRaguraman, Prithi, Akilandeswari Ashwini Balachandran, Suxiang Chen, Sarah D. Diermeier, and Rakesh N. Veedu. 2021. "Antisense Oligonucleotide-Mediated Splice Switching: Potential Therapeutic Approach for Cancer Mitigation" Cancers 13, no. 21: 5555. https://doi.org/10.3390/cancers13215555
APA StyleRaguraman, P., Balachandran, A. A., Chen, S., Diermeier, S. D., & Veedu, R. N. (2021). Antisense Oligonucleotide-Mediated Splice Switching: Potential Therapeutic Approach for Cancer Mitigation. Cancers, 13(21), 5555. https://doi.org/10.3390/cancers13215555