
Updates on Immunotherapy and Immune Landscape in Renal Clear Cell Carcinoma


 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Immunotherapeutic Updates of ccRCC
2.1. Cytokine-Based Immunotherapy
2.2. Tyrosine Kinase and mTOR Inhibitors
2.3. Immune Checkpoint Inhibitors
2.4. Ongoing Clinical Trials
3. Single-Cell Genomics to Study the Tumor Microenvironment
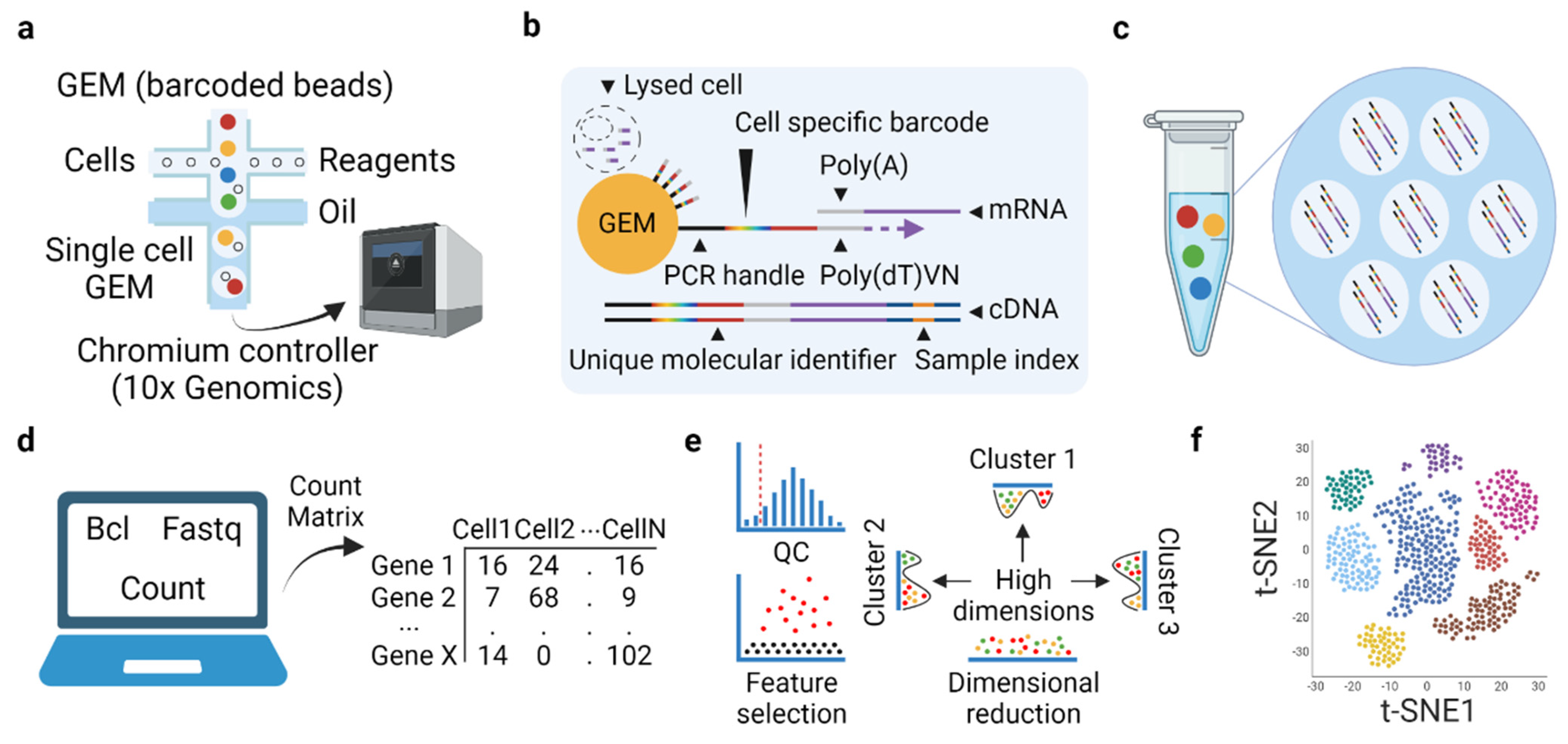
3.1. Basic Concept and Experiment-Related Workflow of Microfluidic-Based scRNAseq
3.2. ScRNAseq in ccRCC
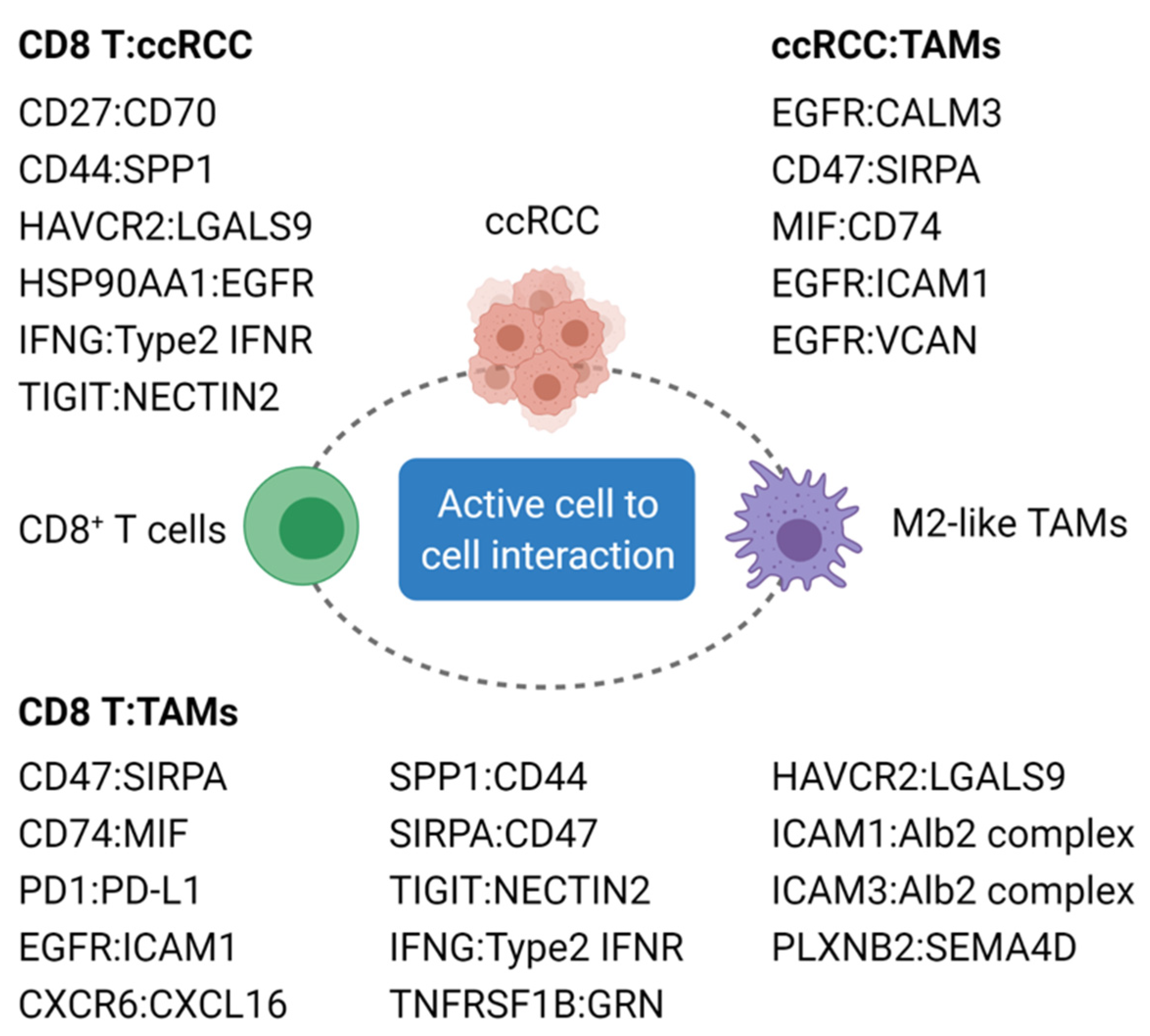
3.3. Major Immune Cell Types Associated with Poor Prognosis and Resistance to ICIs
3.4. Limitations and Challenges in scRNAseq Technology
4. Perspectives and Clinical Implications
4.1. Consensus in Nomenclature
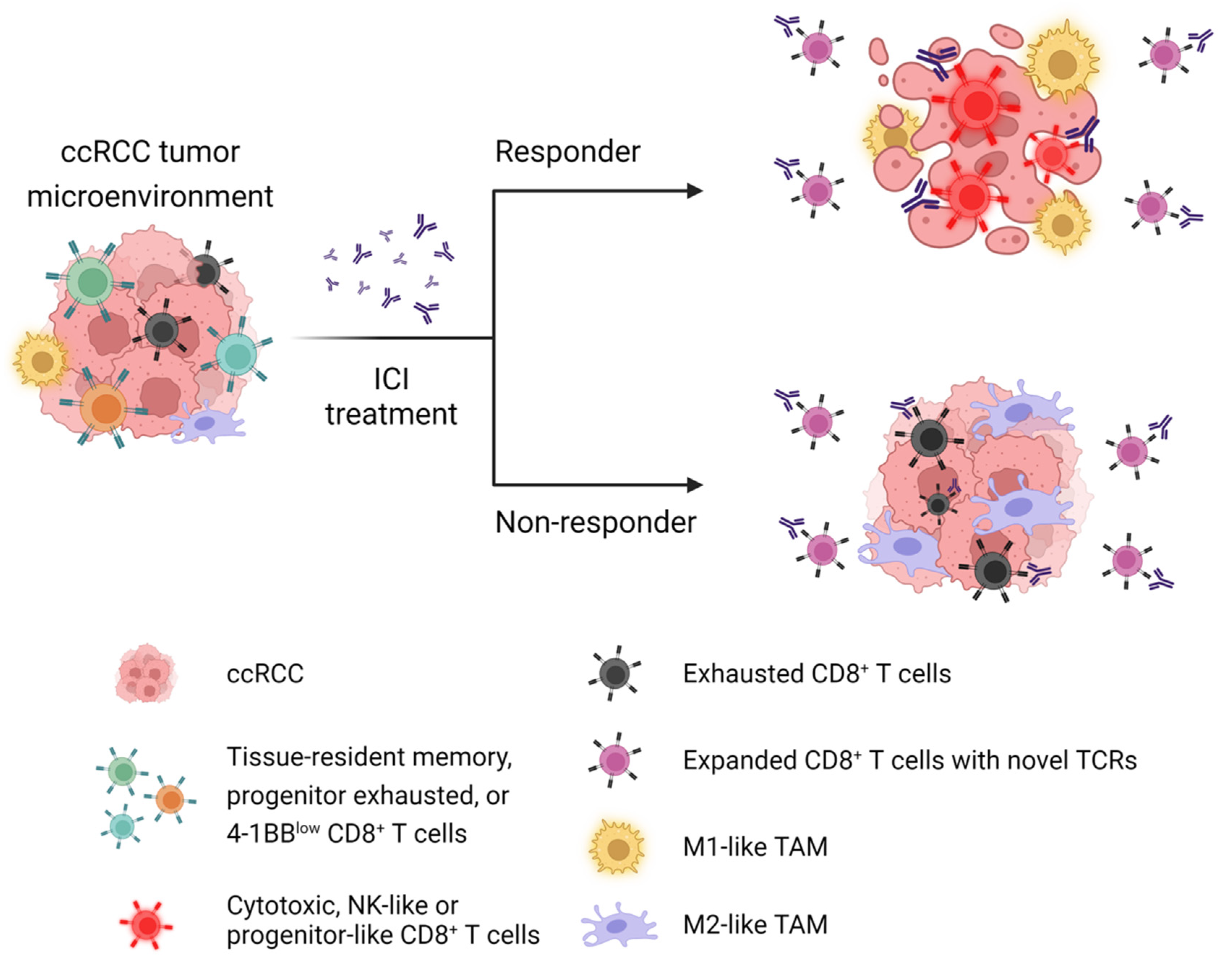
4.2. ScRNAseq Reveals Mechanisms of Immune Activation
4.3. Conclusions Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rini, B.I.; Campbell, S.C.; Escudier, B. Renal cell carcinoma. Lancet 2009, 373, 1119–1132. [Google Scholar] [CrossRef]
- Chow, W.-H.; Dong, L.M.; Devesa, S.S. Epidemiology and risk factors for kidney cancer. Nat. Rev. Urol. 2010, 7, 245–257. [Google Scholar] [CrossRef]
- Gong, J.; Maia, M.C.; Dizman, N.; Govindarajan, A.; Pal, S.K. Metastasis in renal cell carcinoma: Biology and implications for therapy. Asian J. Urol. 2016, 3, 286–292. [Google Scholar] [CrossRef] [Green Version]
- Janzen, N.K.; Kim, H.L.; Figlin, R.A.; Belldegrun, A.S. Surveillance after radical or partial nephrectomy for localized renal cell carcinoma and management of recurrent disease. Urol. Clin. 2003, 30, 843–852. [Google Scholar] [CrossRef]
- Nickerson, M.L.; Jaeger, E.; Shi, Y.; Durocher, J.A.; Mahurkar, S.; Zaridze, D.; Matveev, V.; Janout, V.; Kollarova, H.; Bencko, V. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin. Cancer Res. 2008, 14, 4726–4734. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. Von hippel-lindau disease. Annu. Rev. Pathol. Mech. Dis. 2007, 2, 145–173. [Google Scholar] [CrossRef]
- Vuong, L.; Kotecha, R.R.; Voss, M.H.; Hakimi, A.A. Tumor microenvironment dynamics in clear-cell renal cell carcinoma. Cancer Discov. 2019, 9, 1349–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albiges, L.; Oudard, S.; Negrier, S.; Caty, A.; Gravis, G.; Joly, F.; Duclos, B.; Geoffrois, L.; Rolland, F.; Guillot, A. Complete remission with tyrosine kinase inhibitors in renal cell carcinoma. J. Clin. Oncol. 2012, 30, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Buczek, M.; Escudier, B.; Bartnik, E.; Szczylik, C.; Czarnecka, A. Resistance to tyrosine kinase inhibitors in clear cell renal cell carcinoma: From the patient’s bed to molecular mechanisms. Biochim. Biophys. Acta-Rev. Cancer 2014, 1845, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Challis, G.; Stam, H. The spontaneous regression of cancer: A review of cases from 1900 to 1987. Acta Oncol. 1990, 29, 545–550. [Google Scholar] [CrossRef]
- Everson, T.C.; Cole, W.H. Spontaneous regression of cancer: Preliminary report. Ann. Surg. 1956, 144, 366. [Google Scholar] [CrossRef]
- Snow, R.M.; Schellhammer, P.F. Spontaneous regression of metastatic renal cell carcinoma. Urology 1982, 20, 177–181. [Google Scholar] [CrossRef]
- Janiszewska, A.D.; Poletajew, S.; Wasiutyński, A. Spontaneous regression of renal cell carcinoma. Contemp. Oncol. 2013, 17, 123. [Google Scholar] [CrossRef]
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Frontera, O.A.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; Oyervides Juárez, V.M.; Hsieh, J.J.; Basso, U.; Shah, A.Y. Nivolumab plus cabozantinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2021, 384, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): A multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019, 393, 2404–2415. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Motzer, R.; Alekseev, B.; Rha, S.-Y.; Porta, C.; Eto, M.; Powles, T.; Grünwald, V.; Hutson, T.E.; Kopyltsov, E.; Méndez-Vidal, M.J. Lenvatinib plus pembrolizumab or everolimus for advanced renal cell carcinoma. N. Engl. J. Med. 2021, 384, 1289–1300. [Google Scholar] [CrossRef]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Frontera, O.A.; Hammers, H.J.; Carducci, M.A.; Salman, P.; Escudier, B.; Beuselinck, B.; Amin, A. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: Extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1370–1385. [Google Scholar] [CrossRef]
- Massari, F.; Mollica, V.; Rizzo, A.; Cosmai, L.; Rizzo, M.; Porta, C. Safety evaluation of immune-based combinations in patients with advanced renal cell carcinoma: A systematic review and meta-analysis. Expert. Opin. Drug. Saf. 2020, 19, 1329–1338. [Google Scholar] [CrossRef]
- Cella, D.; Grünwald, V.; Escudier, B.; Hammers, H.J.; George, S.; Nathan, P.; Grimm, M.O.; Rini, B.I.; Doan, J.; Ivanescu, C.; et al. Patient-reported outcomes of patients with advanced renal cell carcinoma treated with nivolumab plus ipilimumab versus sunitinib (CheckMate 214): A randomised, phase 3 trial. Lancet Oncol. 2019, 20, 297–310. [Google Scholar] [CrossRef]
- Atkins, M.B.; Rini, B.I.; Motzer, R.J.; Powles, T.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Gurney, H. Patient-reported outcomes from the phase III Randomized IMmotion151 Trial: Atezolizumab + Bevacizumab versus sunitinib in treatment-naive metastatic renal cell carcinoma. Clin. Cancer Res. 2020, 26, 2506–2514. [Google Scholar]
- Motzer, R.J.; Banchereau, R.; Hamidi, H.; Powles, T.; McDermott, D.; Atkins, M.B.; Escudier, B.; Liu, L.-F.; Leng, N.; Abbas, A.R. Molecular subsets in renal cancer determine outcome to checkpoint and angiogenesis blockade. Cancer Cell 2020, 38, 803–817.e804. [Google Scholar] [CrossRef] [PubMed]
- Abou Alaiwi, S.; Nassar, A.H.; Xie, W.; Bakouny, Z.; Berchuck, J.E.; Braun, D.A.; Baca, S.C.; Nuzzo, P.V.; Flippot, R.; Mouhieddine, T.H. Mammalian SWI/SNF complex genomic alterations and immune checkpoint blockade in solid tumors. Cancer Immunol. Res. 2020, 8, 1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motzer, R.J.; Robbins, P.B.; Powles, T.; Albiges, L.; Haanen, J.B.; Larkin, J.; Mu, X.J.; Ching, K.A.; Uemura, M.; Pal, S.K. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: Biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat. Med. 2020, 26, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Li, W.; Martini, D.J.; Norton, C.; Bossé, D.; Wankowicz, S.M.; Cullen, D. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.A.; Hou, Y.; Bakouny, Z.; Ficial, M.; Sant’Angelo, M.; Forman, J.; Ross-Macdonald, P.; Berger, A.C.; Jegede, O.A.; Elagina, L. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 2020, 26, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Ishii, Y.; Walsh, A.M.; Van Allen, E.M.; Wu, C.J.; Shukla, S.A.; Choueiri, T.K. Clinical validation of PBRM1 alterations as a marker of immune checkpoint inhibitor response in renal cell carcinoma. JAMA Oncol. 2019, 5, 1631–1633. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef]
- Au, L.; Hatipoglu, E.; Robert de Massy, M.; Litchfield, K.; Beattie, G.; Rowan, A.; Schnidrig, D.; Thompson, R.; Byrne, F.; Horswell, S.; et al. Determinants of anti-PD-1 response and resistance in clear cell renal cell carcinoma. Cancer Cell 2021, 39, 1497–1518.e1411. [Google Scholar] [CrossRef]
- Erlmeier, F.; Weichert, W.; Schrader, A.J.; Autenrieth, M.; Hartmann, A.; Steffens, S.; Ivanyi, P. Prognostic impact of PD-1 and its ligands in renal cell carcinoma. Med. Oncol. 2017, 34, 99. [Google Scholar] [CrossRef]
- Labriola, M.K.; Zhu, J.; Gupta, R.; McCall, S.; Jackson, J.; Kong, E.F.; White, J.R.; Cerqueira, G.; Gerding, K.; Simmons, J.K. Characterization of tumor mutation burden, PD-L1 and DNA repair genes to assess relationship to immune checkpoint inhibitors response in metastatic renal cell carcinoma. J. Immunother. Cancer 2020, 8, e000319. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Zitvogel, L.; Sautès–Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Nakano, O.; Sato, M.; Naito, Y.; Suzuki, K.; Orikasa, S.; Aizawa, M.; Suzuki, Y.; Shintaku, I.; Nagura, H.; Ohtani, H. Proliferative activity of intratumoral CD8+ T-lymphocytes as a prognostic factor in human renal cell carcinoma: Clinicopathologic demonstration of antitumor immunity. Cancer Res. 2001, 61, 5132–5136. [Google Scholar] [PubMed]
- Murakami, T.; Tanaka, N.; Takamatsu, K.; Hakozaki, K.; Fukumoto, K.; Masuda, T.; Mikami, S.; Shinojima, T.; Kakimi, K.; Tsunoda, T. Multiplexed single-cell pathology reveals the association of CD8 T-cell heterogeneity with prognostic outcomes in renal cell carcinoma. Cancer Immunol. Immunother. 2021, 70, 3001–3013. [Google Scholar] [CrossRef] [PubMed]
- Jansen, C.S.; Prokhnevska, N.; Master, V.A.; Sanda, M.G.; Carlisle, J.W.; Bilen, M.A.; Cardenas, M.; Wilkinson, S.; Lake, R.; Sowalsky, A.G. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 2019, 576, 465–470. [Google Scholar] [CrossRef]
- Şenbabaoğlu, Y.; Gejman, R.S.; Winer, A.G.; Liu, M.; Van Allen, E.M.; de Velasco, G.; Miao, D.; Ostrovnaya, I.; Drill, E.; Luna, A. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol. 2016, 17, 231. [Google Scholar] [CrossRef] [Green Version]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H. An immune atlas of clear cell renal cell carcinoma. Cell 2017, 169, 736–749.e718. [Google Scholar] [CrossRef] [Green Version]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.D.; Puligandla, M.; Shuch, B.M.; Leibovich, B.C.; Kapoor, A.; Master, V.A.; Drake, C.G.; Heng, D.Y.; Lara, P.N.; Choueiri, T.K. The future of perioperative therapy in advanced renal cell carcinoma: How can we PROSPER? Future Oncol. 2019, 15, 1683–1695. [Google Scholar] [CrossRef]
- Baine, M.K.; Turcu, G.; Zito, C.R.; Adeniran, A.J.; Camp, R.L.; Chen, L.; Kluger, H.M.; Jilaveanu, L.B. Characterization of tumor infiltrating lymphocytes in paired primary and metastatic renal cell carcinoma specimens. Oncotarget 2015, 6, 24990. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.-S.; Shea, L.K. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef]
- Sun, G.; Li, Z.; Rong, D.; Zhang, H.; Shi, X.; Yang, W.; Zheng, W.; Sun, G.; Wu, F.; Cao, H. Single-cell RNA sequencing in cancer: Applications, advances, and emerging challenges. Mol. Ther. Oncolytics 2021, 21, 183–206. [Google Scholar] [CrossRef] [PubMed]
- Papalexi, E.; Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2018, 18, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, G.; Stegle, O.; Reik, W. Single-cell epigenomics: Recording the past and predicting the future. Science 2017, 358, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Lu, W.; Su, C.; Lv, Y.; Ye, Y.; Guo, B.; Liu, D.; Yan, H.; Mi, H.; Li, T. Single-cell RNA-seq identification of the cellular molecular characteristics of sporadic bilateral clear cell renal cell carcinoma. Front. Oncol. 2021, 11, 1825. [Google Scholar] [CrossRef] [PubMed]
- Krishna, C.; DiNatale, R.G.; Kuo, F.; Srivastava, R.M.; Vuong, L.; Chowell, D.; Gupta, S.; Vanderbilt, C.; Purohit, T.A.; Liu, M. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell 2021, 39, 662–677.e666. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Narayanan, S.P.; Mannan, R.; Raskind, G.; Wang, X.; Vats, P.; Su, F.; Hosseini, N.; Cao, X.; Kumar-Sinha, C. Single-cell analyses of renal cell cancers reveal insights into tumor microenvironment, cell of origin, and therapy response. Proc. Natl. Acad. Sci. USA 2021, 118, e2103240118. [Google Scholar] [CrossRef]
- Young, M.D.; Mitchell, T.J.; Braga, F.A.V.; Tran, M.G.; Stewart, B.J.; Ferdinand, J.R.; Collord, G.; Botting, R.A.; Popescu, D.-M.; Loudon, K.W. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 2018, 361, 594–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borcherding, N.; Vishwakarma, A.; Voigt, A.P.; Bellizzi, A.; Kaplan, J.; Nepple, K.; Salem, A.K.; Jenkins, R.W.; Zakharia, Y.; Zhang, W. Mapping the immune environment in clear cell renal carcinoma by single-cell genomics. Commun. Biol. 2021, 4, 1–11. [Google Scholar] [CrossRef]
- Bi, K.; He, M.X.; Bakouny, Z.; Kanodia, A.; Napolitano, S.; Wu, J.; Grimaldi, G.; Braun, D.A.; Cuoco, M.S.; Mayorga, A. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell 2021, 39, 649–661.e645. [Google Scholar] [CrossRef]
- Braun, D.A.; Street, K.; Burke, K.P.; Cookmeyer, D.L.; Denize, T.; Pedersen, C.B.; Gohil, S.H.; Schindler, N.; Pomerance, L.; Hirsch, L. Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell 2021, 39, 632–648.e638. [Google Scholar] [CrossRef]
- Obradovic, A.; Chowdhury, N.; Haake, S.M.; Ager, C.; Wang, V.; Vlahos, L.; Guo, X.V.; Aggen, D.H.; Rathmell, W.K.; Jonasch, E. Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell 2021, 184, 2988–3005.e2916. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Madireddi, S.; de Almeida, P.E.; Banchereau, R.; Chen, Y.-J.J.; Chitre, A.S.; Chiang, E.Y.; Iftikhar, H.; O’Gorman, W.E.; Au-Yeung, A. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 2020, 579, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Malek, T.R. The biology of interleukin-2. Annu. Rev. Immunol. 2008, 26, 453–479. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.I.; Rosenberg, S.A.; Fyfe, G. Long-term survival update for high-dose recombinant interleukin-2 in patients with renal cell carcinoma. Cancer J. Sci. Am. 2000, 6, S55–S57. [Google Scholar]
- Fyfe, G.; Fisher, R.I.; Rosenberg, S.A.; Sznol, M.; Parkinson, D.R.; Louie, A.C. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J. Clin. Oncol. 1995, 13, 688–696. [Google Scholar] [CrossRef]
- Klapper, J.A.; Downey, S.G.; Smith, F.O.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Sherry, R.M.; Royal, R.E.; Steinberg, S.M.; Rosenberg, S. High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma: A retrospective analysis of response and survival in patients treated in the surgery branch at the National Cancer Institute between 1986 and 2006. Cancer 2008, 113, 293–301. [Google Scholar] [CrossRef] [Green Version]
- McDermott, D.F.; Regan, M.M.; Clark, J.I.; Flaherty, L.E.; Weiss, G.R.; Logan, T.F.; Kirkwood, J.M.; Gordon, M.S.; Sosman, J.A.; Ernstoff, M.S. Randomized phase III trial of high-dose interleukin-2 versus subcutaneous interleukin-2 and interferon in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2005, 23, 133–141. [Google Scholar] [CrossRef] [Green Version]
- McDermott, D.F.; Cheng, S.-C.; Signoretti, S.; Margolin, K.A.; Clark, J.I.; Sosman, J.A.; Dutcher, J.P.; Logan, T.F.; Curti, B.D.; Ernstoff, M.S. The high-dose aldesleukin “select” trial: A trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin. Cancer Res. 2015, 21, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Minasian, L.M.; Motzer, R.J.; Gluck, L.; Mazumdar, M.; Vlamis, V.; Krown, S.E. Interferon alfa-2a in advanced renal cell carcinoma: Treatment results and survival in 159 patients with long-term follow-up. J. Clin. Oncol. 1993, 11, 1368–1375. [Google Scholar] [CrossRef]
- Negrier, S.; Escudier, B.; Lasset, C.; Douillard, J.-Y.; Savary, J.; Chevreau, C.; Ravaud, A.; Mercatello, A.; Peny, J.; Mousseau, M. Recombinant human interleukin-2, recombinant human interferon alfa-2a, or both in metastatic renal-cell carcinoma. N. Engl. J. Med. 1998, 338, 1272–1278. [Google Scholar] [CrossRef]
- Motzer, R.J.; Murphy, B.A.; Bacik, J.; Schwartz, L.H.; Nanus, D.M.; Mariani, T.; Loehrer, P.; Wilding, G.; Fairclough, D.L.; Cella, D. Phase III trial of interferon alfa-2a with or without 13-cis-retinoic acid for patients with advanced renal cell carcinoma. J. Clin. Oncol. 2000, 18, 2972–2980. [Google Scholar] [CrossRef]
- Dutcher, J. Current status of interleukin-2 therapy for metastatic renal cell carcinoma and metastatic melanoma. Oncology 2002, 16, 4–10. [Google Scholar] [PubMed]
- Motzer, R.J.; Bacik, J.; Murphy, B.A.; Russo, P.; Mazumdar, M. Interferon-alfa as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. J. Clin. Oncol. 2002, 20, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Hessel, C.; Halabi, S.; Sanford, B.; Michaelson, M.D.; Hahn, O.; Walsh, M.; Olencki, T.; Picus, J.; Small, E.J. Cabozantinib versus sunitinib as initial therapy for metastatic renal cell carcinoma of intermediate or poor risk (Alliance A031203 CABOSUN randomised trial): Progression-free survival by independent review and overall survival update. Eur. J. Cancer 2018, 94, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef]
- Gore, M.E.; Szczylik, C.; Porta, C.; Bracarda, S.; Bjarnason, G.A.; Oudard, S.; Hariharan, S.; Lee, S.-H.; Haanen, J.; Castellano, D. Safety and efficacy of sunitinib for metastatic renal-cell carcinoma: An expanded-access trial. Lancet Oncol. 2009, 10, 757–763. [Google Scholar] [CrossRef]
- Hutson, T.E.; Lesovoy, V.; Al-Shukri, S.; Stus, V.P.; Lipatov, O.N.; Bair, A.H.; Rosbrook, B.; Chen, C.; Kim, S.; Vogelzang, N.J. Axitinib versus sorafenib as first-line therapy in patients with metastatic renal-cell carcinoma: A randomised open-label phase 3 trial. Lancet Oncol. 2013, 14, 1287–1294. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Cella, D.; Reeves, J.; Hawkins, R.; Guo, J.; Nathan, P.; Staehler, M.; de Souza, P.; Merchan, J.R. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N. Engl. J. Med. 2013, 369, 722–731. [Google Scholar] [CrossRef] [Green Version]
- Choueiri, T.K.; Halabi, S.; Sanford, B.L.; Hahn, O.; Michaelson, M.D.; Walsh, M.K.; Feldman, D.R.; Olencki, T.; Picus, J.; Small, E.J. Cabozantinib versus sunitinib as initial targeted therapy for patients with metastatic renal cell carcinoma of poor or intermediate risk: The alliance A031203 CABOSUN trial. J. Clin. Oncol. 2017, 35, 591. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef] [Green Version]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1α expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toschi, A.; Edelstein, J.; Rockwell, P.; Ohh, M.; Foster, D. HIFα expression in VHL-deficient renal cancer cells is dependent on phospholipase D. Oncogene 2008, 27, 2746–2753. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G.V.; Tran, C.; Mellinghoff, I.K.; Welsbie, D.S.; Chan, E.; Fueger, B.; Czernin, J.; Sawyers, C.L. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat. Med. 2006, 12, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet 2008, 372, 449–456. [Google Scholar] [CrossRef]
- Mollica, V.; Di Nunno, V.; Gatto, L.; Santoni, M.; Scarpelli, M.; Cimadamore, A.; Lopez-Beltran, A.; Cheng, L.; Battelli, N.; Montironi, R. Resistance to systemic agents in renal cell carcinoma predict and overcome genomic strategies adopted by tumor. Cancers 2019, 11, 830. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.C.; Hughes, M.; Kammula, U.; Royal, R.; Sherry, R.M.; Topalian, S.L.; Suri, K.B.; Levy, C.; Allen, T.; Mavroukakis, S. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J. Immunother. 2007, 30, 825. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Redman, B.G.; Kuzel, T.M.; Harrison, M.R.; Vaishampayan, U.N.; Drabkin, H.A.; George, S.; Logan, T.F. Nivolumab for metastatic renal cell carcinoma: Results of a randomized phase II trial. J. Clin. Oncol. 2015, 33, 1430. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Plimack, E.R.; Procopio, G.; McDermott, D.F. Nivolumab versus everolimus in patients with advanced renal cell carcinoma: Updated results with long-term follow-up of the randomized, open-label, phase 3 CheckMate 025 trial. Cancer 2020, 126, 4156–4167. [Google Scholar] [CrossRef]
- Amin, A.; Plimack, E.R.; Ernstoff, M.S.; Lewis, L.D.; Bauer, T.M.; McDermott, D.F.; Carducci, M.; Kollmannsberger, C.; Rini, B.I.; Heng, D.Y. Safety and efficacy of nivolumab in combination with sunitinib or pazopanib in advanced or metastatic renal cell carcinoma: The CheckMate 016 study. J. Immunother. Cancer 2018, 6, 109. [Google Scholar] [CrossRef]
- Hammers, H.J.; Plimack, E.R.; Infante, J.R.; Rini, B.I.; McDermott, D.F.; Lewis, L.D.; Voss, M.H.; Sharma, P.; Pal, S.K.; Razak, A.R.A. Safety and efficacy of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma: The CheckMate 016 study. J. Clin. Oncol. 2017, 35, 3851. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; Frontera, O.A.; Melichar, B.; Powles, T.; Donskov, F.; Plimack, E.R.; Barthélémy, P.; Hammers, H.J. Survival outcomes and independent response assessment with nivolumab plus ipilimumab versus sunitinib in patients with advanced renal cell carcinoma: 42-month follow-up of a randomized phase 3 clinical trial. J. Immunother. Cancer 2020, 8, e000891. [Google Scholar] [CrossRef]
- Griffioen, A.W. Anti-angiogenesis: Making the tumor vulnerable to the immune system. Cancer Immunol. Immunother. 2008, 57, 1553–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusmartsev, S.; Eruslanov, E.; Kübler, H.; Tseng, T.; Sakai, Y.; Su, Z.; Kaliberov, S.; Heiser, A.; Rosser, C.; Dahm, P. Oxidative stress regulates expression of VEGFR1 in myeloid cells: Link to tumor-induced immune suppression in renal cell carcinoma. J. Immunol. 2008, 181, 346–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adotevi, O.; Pere, H.; Ravel, P.; Haicheur, N.; Badoual, C.; Merillon, N.; Medioni, J.; Peyrard, S.; Roncelin, S.; Verkarre, V. A decrease of regulatory T cells correlates with overall survival after sunitinib-based antiangiogenic therapy in metastatic renal cancer patients. J. Immunother. 2010, 33, 991–998. [Google Scholar] [CrossRef]
- Hirsch, L.; Flippot, R.; Escudier, B.; Albiges, L. Immunomodulatory roles of VEGF pathway inhibitors in renal cell carcinoma. Drugs 2020, 80, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Plimack, E.R.; Soulières, D.; Waddell, T.; Stus, V.; Gafanov, R.; Nosov, D.; Pouliot, F.; Melichar, B.; Vynnychenko, I. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): Extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol. 2020, 21, 1563–1573. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Waddell, T.; Gafanov, R.; Pouliot, F.; Nosov, D.; Melichar, B.; Soulieres, D.; Borchiellini, D. Pembrolizumab (pembro) plus axitinib (axi) versus sunitinib as first-line therapy for advanced clear cell renal cell carcinoma (ccRCC): Results from 42-month follow-up of KEYNOTE-426. J. Clin. Oncol. 2021, 39, 4500. [Google Scholar] [CrossRef]
- Motzer, R.J.; Choueiri, T.K.; Powles, T.; Burotto, M.; Bourlon, M.T.; Hsieh, J.J.; Maruzzo, M.; Shah, A.Y.; Suarez, C.; Barrios, C.H. Nivolumab+ cabozantinib (NIVO+ CABO) versus sunitinib (SUN) for advanced renal cell carcinoma (aRCC): Outcomes by sarcomatoid histology and updated trial results with extended follow-up of CheckMate 9ER. J. Clin. Oncol. 2021, 39, 308. [Google Scholar] [CrossRef]
- Haanen, J.B.; Larkin, J.; Choueiri, T.K.; Albiges, L.; Rini, B.I.; Atkins, M.B.; Schmidinger, M.; Penkov, K.; Thomaidou, D.; Wang, J. Efficacy of avelumab+ axitinib (A+ Ax) versus sunitinib (S) by IMDC risk group in advanced renal cell carcinoma (aRCC): Extended follow-up results from JAVELIN Renal 101. J. Clin. Oncol. 2021, 39, 4574. [Google Scholar] [CrossRef]
- Choueiri, T.; Motzer, R.; Rini, B.; Haanen, J.; Campbell, M.; Venugopal, B.; Kollmannsberger, C.; Gravis-Mescam, G.; Uemura, M.; Lee, J. Updated efficacy results from the JAVELIN Renal 101 trial: First-line avelumab plus axitinib versus sunitinib in patients with advanced renal cell carcinoma. Ann. Oncol. 2020, 31, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Albiges, L.; Tannir, N.M.; Burotto, M.; McDermott, D.; Plimack, E.R.; Barthélémy, P.; Porta, C.; Powles, T.; Donskov, F.; George, S. Nivolumab plus ipilimumab versus sunitinib for first-line treatment of advanced renal cell carcinoma: Extended 4-year follow-up of the phase III CheckMate 214 trial. ESMO Open 2020, 5, e001079. [Google Scholar] [CrossRef]
- Tomita, Y.; Motzer, R.J.; Choueiri, T.K.; Rini, B.I.; Miyake, H.; Oya, M.; Albiges, L.; Fujii, Y.; Umeyama, Y.; Wang, J. Efficacy of avelumab plus axitinib (A+ Ax) versus sunitinib (S) by number of IMDC risk factors and tumor sites at baseline in advanced renal cell carcinoma (aRCC): Extended follow-up results from JAVELIN Renal 101. J. Clin. Oncol. 2021, 39, 302. [Google Scholar] [CrossRef]
- Bex, A.; Russo, P.; Tomita, Y.; Grünwald, V.; Ramirez, L.-M.; McHenry, B.M.; Motzer, R.J. A phase III, randomized, placebo-controlled trial of nivolumab or nivolumab plus ipilimumab in patients with localized renal cell carcinoma at high-risk of relapse after radical or partial nephrectomy (CheckMate 914). J. Clin. Oncol. 2020, 38, TPS5099. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Quinn, D.I.; Zhang, T.; Gurney, H.; Doshi, G.K.; Cobb, P.W.; Parnis, F.; Lee, J.-L.; Park, S.H.; Semenov, A. KEYNOTE-564: A phase 3, randomized, double blind, trial of pembrolizumab in the adjuvant treatment of renal cell carcinoma. J. Clin. Oncol. 2018, 36, TPS4599. [Google Scholar] [CrossRef]
- Uzzo, R.; Bex, A.; Rini, B.I.; Albiges, L.; Suarez, C.; Donaldson, F.; Asakawa, T.; Schiff, C.; Pal, S.K. A phase III study of atezolizumab (atezo) vs. placebo as adjuvant therapy in renal cell carcinoma (RCC) patients (pts) at high risk of recurrence following resection (IMmotion010). J. Clin. Oncol. 2017, 35, TPS4598. [Google Scholar] [CrossRef]
- Pal, S.K.; Albiges, L.; Suarez Rodriguez, C.; Liu, B.; Doss, J.; Khurana, S.; Scheffold, C.; Voss, M.H.; Choueiri, T.K. CONTACT-03: Randomized, open-label phase III study of atezolizumab plus cabozantinib versus cabozantinib monotherapy following progression on/after immune checkpoint inhibitor (ICI) treatment in patients with advanced/metastatic renal cell carcinoma. J. Clin. Oncol. 2021, 39, TPS370. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Albiges, L.; Powles, T.; Scheffold, C.; Wang, F.; Motzer, R.J. A phase III study (COSMIC-313) of cabozantinib (C) in combination with nivolumab (N) and ipilimumab (I) in patients (pts) with previously untreated advanced renal cell carcinoma (aRCC) of intermediate or poor risk. J. Clin. Oncol. 2020, 38, TPS767. [Google Scholar] [CrossRef]
- Tannir, N.M.; Agarwal, N.; Pal, S.K.; Cho, D.C.; Formiga, M.; Guo, J.; George, D.J.; Tagliaferri, M.A.; Singel, S.M.; O’Keeffe, B.A. PIVOT-09: A phase III randomized open-label study of bempegaldesleukin (NKTR-214) plus nivolumab versus sunitinib or cabozantinib (investigator’s choice) in patients with previously untreated advanced renal cell carcinoma (RCC). J. Clin. Oncol. 2020, 38, TPS763. [Google Scholar] [CrossRef]
- Emamekhoo, H.; Olsen, M.; Carthon, B.C.; Drakaki, A.; Percent, I.J.; Molina, A.M.; Cho, D.C.; Bendell, J.C.; Gordan, L.N.; Rezazadeh Kalebasty, A. Safety and efficacy of nivolumab plus ipilimumab (NIVO+ IPI) in patients with advanced renal cell carcinoma (aRCC) with brain metastases: Interim analysis of CheckMate 920. J. Clin. Oncol. 2019, 37, 4517. [Google Scholar] [CrossRef]
- Zhang, T.; Ballman, K.V.; Choudhury, A.D.; Chen, R.C.; Watt, C.; Wen, Y.; Zemla, T.; Emamekhoo, H.; Gupta, S.; Morris, M.J. PDIGREE: An adaptive phase 3 trial of PD-inhibitor nivolumab and ipilimumab (IPI-NIVO) with VEGF TKI cabozantinib (CABO) in metastatic untreated renal cell cancer (Alliance A031704). J. Clin. Oncol. 2019, 39, TPS366. [Google Scholar] [CrossRef]
- Harshman, L.C.; Puligandla, M.; Haas, N.B.; Allaf, M.; Drake, C.G.; McDermott, D.F.; Signoretti, S.; Cella, D.; Gupta, R.T.; Shuch, B.M. PROSPER: A phase III randomized study comparing perioperative nivolumab (nivo) versus observation in patients with localized renal cell carcinoma (RCC) undergoing nephrectomy (ECOG-ACRIN 8143). J. Clin. Oncol. 2019, 37, TPS684. [Google Scholar] [CrossRef]
- Tykodi, S.S.; Gordan, L.N.; Alter, R.S.; Arrowsmith, E.; Harrison, M.R.; Percent, I.J.; Singal, R.; Van Veldhuizen, P.J.; George, D.J.; Hutson, T.E. Nivolumab plus ipilimumab in patients with advanced non-clear cell renal cell carcinoma (nccRCC): Safety and efficacy from CheckMate 920. J. Clin. Oncol. 2021, 39, 309. [Google Scholar] [CrossRef]
- Oza, B.; Frangou, E.; Smith, B.; Bryant, H.; Kaplan, R.; Choodari-Oskooei, B.; Powles, T.; Stewart, G.D.; Albiges, L.; Bex, A.; et al. RAMPART: A phase III multi-arm multi-stage trial of adjuvant checkpoint inhibitors in patients with resected primary renal cell carcinoma (RCC) at high or intermediate risk of relapse. Contemp. Clin. Trials 2021, 108, 106482. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Kaelin, W.G. Targeting the HIF2–VEGF axis in renal cell carcinoma. Nat. Med. 2020, 26, 1519–1530. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The technology and biology of single-cell RNA sequencing. Mol. Cell 2015, 58, 610–620. [Google Scholar] [CrossRef] [Green Version]
- Davis-Marcisak, E.F.; Deshpande, A.; Stein-O’Brien, G.L.; Ho, W.J.; Laheru, D.; Jaffee, E.M.; Fertig, E.J.; Kagohara, L.T. From bench to bedside: Single-cell analysis for cancer immunotherapy. Cancer Cell 2021, 39, 1062–1080. [Google Scholar] [CrossRef]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [Green Version]
- Ziegenhain, C.; Vieth, B.; Parekh, S.; Reinius, B.; Guillaumet-Adkins, A.; Smets, M.; Leonhardt, H.; Heyn, H.; Hellmann, I.; Enard, W. Comparative analysis of single-cell RNA sequencing methods. Mol. Cell 2017, 65, 631–643.e634. [Google Scholar] [CrossRef] [Green Version]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 319. [Google Scholar] [CrossRef] [Green Version]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Amezquita, R.A.; Lun, A.T.; Becht, E.; Carey, V.J.; Carpp, L.N.; Geistlinger, L.; Marini, F.; Rue-Albrecht, K.; Risso, D.; Soneson, C. Orchestrating single-cell analysis with Bioconductor. Nat. Methods 2020, 17, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Beksac, A.T.; Paulucci, D.J.; Blum, K.A.; Yadav, S.S.; Sfakianos, J.P.; Badani, K.K. Heterogeneity in renal cell carcinoma. Oncol. Semin. Orig. Investig. 2017, 35, 507–515. [Google Scholar] [CrossRef] [PubMed]
- van den Heuvel, C.N.; van Ewijk, A.; Zeelen, C.; de Bitter, T.; Huynen, M.; Mulders, P.; Oosterwijk, E.; Leenders, W.P. Molecular profiling of druggable targets in clear cell renal cell carcinoma through targeted RNA sequencing. Front. Oncol. 2019, 9, 117. [Google Scholar] [CrossRef] [Green Version]
- Network, C.G.A.R. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43. [Google Scholar]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 2018, 173, 400–416.e411. [Google Scholar] [CrossRef] [Green Version]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R. The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018, 23, 313–326.e315. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef]
- Liao, J.; Yu, Z.; Chen, Y.; Bao, M.; Zou, C.; Zhang, H.; Liu, D.; Li, T.; Zhang, Q.; Li, J. Single-cell RNA sequencing of human kidney. Sci. Data 2020, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.-T.; Lee, H.W.; Lee, H.-O.; Song, H.J.; Shin, S.; Kim, H.; Shin, Y.; Nam, D.-H.; Jeong, B.C.; Kirsch, D.G. Application of single-cell RNA sequencing in optimizing a combinatorial therapeutic strategy in metastatic renal cell carcinoma. Genome Biol. 2016, 17, 80. [Google Scholar] [CrossRef] [Green Version]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell 2017, 170, 142–157.e119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Xia, Y.; Lin, Z.; Qu, Y.; Qi, Y.; Chen, Y.; Zhou, Q.; Zeng, H.; Wang, J.; Chang, Y. Tumor-infiltrating CD39+ CD8+ T cells determine poor prognosis and immune evasion in clear cell renal cell carcinoma patients. Cancer Immunol. Immunother. 2020, 69, 1565–1576. [Google Scholar] [CrossRef]
- Aggen, D.H.; Ager, C.R.; Obradovic, A.Z.; Chowdhury, N.; Ghasemzadeh, A.; Mao, W.; Chaimowitz, M.G.; Lopez-Bujanda, Z.A.; Spina, C.S.; Hawley, J.E. Blocking IL1 beta promotes tumor regression and remodeling of the myeloid compartment in a renal cell carcinoma model: Multidimensional analyses. Clin. Cancer Res. 2021, 27, 608–621. [Google Scholar] [CrossRef]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef]
- Lizio, M.; Abugessaisa, I.; Noguchi, S.; Kondo, A.; Hasegawa, A.; Hon, C.C.; De Hoon, M.; Severin, J.; Oki, S.; Hayashizaki, Y. Update of the FANTOM web resource: Expansion to provide additional transcriptome atlases. Nucleic Acids Res. 2019, 47, D752–D758. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.C.; Borcherding, N.; Ahmed, K.K.; Voigt, A.P.; Vishwakarma, A.; Kolb, R.; Kluz, P.N.; Pandey, G.; De, U.; Drashansky, T.; et al. CD177 modulates the function and homeostasis of tumor-infiltrating regulatory T cells. Nat Commun. 2021, 12, 5764. [Google Scholar] [CrossRef] [PubMed]
- Jensen, H.K.; Donskov, F.; Nordsmark, M.; Marcussen, N.; von der Maase, H. Increased intratumoral FOXP3-positive regulatory immune cells during interleukin-2 treatment in metastatic renal cell carcinoma. Clin. Cancer Res. 2009, 15, 1052–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauch, D.A.; Conlon, K.C.; Janakiram, M.; Brammer, J.E.; Harding, J.C.; Ye, B.H.; Zang, X.; Ren, X.; Olson, S.; Cheng, X. Rapid progression of adult T-cell leukemia/lymphoma as tumor-infiltrating Tregs after PD-1 blockade. Blood J. Am. Soc. Hematol. 2019, 134, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Kolb, R.; De, U.; Khan, S.; Luo, Y.; Kim, M.-C.; Yu, H.; Wu, C.; Mo, J.; Zhang, X.; Zhang, P. Proteolysis-targeting chimera against BCL-X L destroys tumor-infiltrating regulatory T cells. Nat. Commun. 2021, 12, 1–9. [Google Scholar]
- Stoeckius, M.; Zheng, S.; Houck-Loomis, B.; Hao, S.; Yeung, B.Z.; Mauck, W.M., 3rd; Smibert, P.; Satija, R. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 2018, 19, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, A.; Malik, L.; Smith, T.; Sudbery, I.; Patro, R. Alevin efficiently estimates accurate gene abundances from dscRNA-seq data. Genome Biol. 2019, 20, 65. [Google Scholar] [CrossRef] [Green Version]
- Melsted, P.; Booeshaghi, A.S.; Liu, L.; Gao, F.; Lu, L.; Min, K.H.J.; da Veiga Beltrame, E.; Hjörleifsson, K.E.; Gehring, J.; Pachter, L. Modular, efficient and constant-memory single-cell RNA-seq preprocessing. Nat. Biotechnol. 2021, 39, 813–818. [Google Scholar] [CrossRef]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef]
- Qiu, P. Embracing the dropouts in single-cell RNA-seq analysis. Nat. Commun. 2020, 11, 1169. [Google Scholar] [CrossRef] [Green Version]
- Kharchenko, P.V. The triumphs and limitations of computational methods for scRNA-seq. Nat. Methods 2021, 18, 723–732. [Google Scholar] [CrossRef]
- Hughes, T.K.; Wadsworth, M.H., 2nd; Gierahn, T.M.; Do, T.; Weiss, D.; Andrade, P.R.; Ma, F.; de Andrade Silva, B.J.; Shao, S.; Tsoi, L.C.; et al. Second-Strand Synthesis-Based Massively Parallel scRNA-Seq Reveals Cellular States and Molecular Features of Human Inflammatory Skin Pathologies. Immunity 2020, 53, 878–894.e877. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Ji, Z.; Ji, H.; Hicks, S.C. A systematic evaluation of single-cell RNA-sequencing imputation methods. Genome Biol. 2020, 21, 218. [Google Scholar] [CrossRef] [PubMed]
- van den Brink, S.C.; Sage, F.; Vértesy, Á.; Spanjaard, B.; Peterson-Maduro, J.; Baron, C.S.; Robin, C.; Van Oudenaarden, A. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 2017, 14, 935–936. [Google Scholar] [CrossRef] [PubMed]
- Cang, Z.; Nie, Q. Inferring spatial and signaling relationships between cells from single cell transcriptomic data. Nat. Commun. 2020, 11, 2084. [Google Scholar] [CrossRef]
- Lareau, C.A.; Ludwig, L.S.; Muus, C.; Gohil, S.H.; Zhao, T.; Chiang, Z.; Pelka, K.; Verboon, J.M.; Luo, W.; Christian, E.; et al. Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling. Nat. Biotechnol. 2021, 39, 451–461. [Google Scholar] [CrossRef]
- VanHorn, S.; Morris, S.A. Next-Generation Lineage Tracing and Fate Mapping to Interrogate Development. Dev. Cell. 2021, 56, 7–21. [Google Scholar] [CrossRef]
- Singh, M.; Al-Eryani, G.; Carswell, S.; Ferguson, J.M.; Blackburn, J.; Barton, K.; Roden, D.; Luciani, F.; Giang Phan, T.; Junankar, S.; et al. High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes. Nat. Commun. 2019, 10, 3120. [Google Scholar] [CrossRef] [Green Version]
- van der Leun, A.M.; Thommen, D.S.; Schumacher, T.N. CD8(+) T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer 2020, 20, 218–232. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 2018, 175, 998–1013.e1020. [Google Scholar] [CrossRef] [Green Version]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, N.A.; Becht, E.; Vano, Y.; Petitprez, F.; Lacroix, L.; Validire, P.; Sanchez-Salas, R.; Ingels, A.; Oudard, S.; Moatti, A. Tumor-infiltrating and peripheral blood T-cell immunophenotypes predict early relapse in localized clear cell renal cell carcinoma. Clin. Cancer Res. 2017, 23, 4416–4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noessner, E.; Brech, D.; Mendler, A.N.; Masouris, I.; Schlenker, R.; Prinz, P.U. Intratumoral alterations of dendritic-cell differentiation and CD8+ T-cell anergy are immune escape mechanisms of clear cell renal cell carcinoma. Oncoimmunology 2012, 1, 1451–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkins, D.; Ferrone, S.; Schmahl, G.E.; Störkel, S.; Seliger, B. Down-regulation of HLA class I antigen processing molecules: An immune escape mechanism of renal cell carcinoma? J. Urol. 2004, 171, 885–889. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Study Name | Identifier | Agent | Target | Total | ORR | TRAE 3+ | Citations |
|---|---|---|---|---|---|---|---|
| CheckMate 025 * | NCT01668784 | Nivolumab | PD-1 | 821 | 23% | 19% | [15,89] |
| CheckMate 214 | NCT02231749 | Nivolumab | PD-1 | 1096 | 39.1% | 47.9% | [21,27,102] |
| Ipilimumab | CTLA-4 | ||||||
| IMmotion 151 | NCT02420821 | Atezolizumab | PD-L1 | 915 | 37% | 40% | [24,30] |
| Bevacizumab | VEGF | ||||||
| JAVELIN Renal 101 | NCT02684006 | Avelumab | PD-L1 | 886 | 52.5% | 71.2% | [23,101,103] |
| Axitinib | RTK | ||||||
| CLEAR | NCT02811861 | Pembrolizumab | PD-1 | 1069 | 71% | 82.4% | [26] |
| Lenvatinib | RTK | ||||||
| Keynote 426 | NCT02853331 | Pembrolizumab | PD-1 | 861 | 60.4% | 66.4% | [25,97,98] |
| Axitinib | RTK | ||||||
| CheckMate 9ER | NCT03141177 | Nivolumab | PD-1 | 651 | 56.6% | 75.3% | [22] |
| Cabozantinib | RTK |
| Study Name | Identifier | Agent | Target | Control |
|---|---|---|---|---|
| COSMIC-313 | NCT03937219 [108] | Nivolumab | PD-1 | Nivolumab and Ipilimumab |
| Ipilimumab | CTLA-4 | |||
| Cabozantinib | RTK | |||
| na | NCT03729245 [109] | Bempegaldesleukin | IL-2 agonist | Sunitinib |
| Nivolumab | PD-1 | Cabozantinib | ||
| Keynote 564 | NCT03142334 [105] | Pembrolizumab | PD-1 | Placebo |
| Contact 03 | NCT04338269 [107] | Atezolizumab | PD-L1 | Cabozantinib |
| Cabozantinib | RTK | |||
| IMmotion 010 | NCT03024996 [106] | Atezolizumab | PD-L1 | Placebo following nephrectomy |
| PDIGREE | NCT03793166 [111] | Nivolumab | PD-1 | Nivolumab following Nivolumab and Ipilimumab |
| Cabozantinib | RTK | |||
| CheckMate 914 | NCT03138512 [104] | Nivolumab | PD-1 | Placebo following nephrectomy |
| Ipilimumab | CTLA-4 | |||
| PROSPER | NCT03055013 [112] | Nivolumab | PD-1 | Monitoring after nephrectomy |
| CheckMate 920 | NCT02982954 [113] | Nivolumab | PD-1 | This clinical trial examines the safety of ICI in RCC patients with either brain metastasis or Karnofsky Performance Status 50–60% |
| Ipilimumab | CTLA-4 | |||
| na | NCT04736706 | Pembrolizumab | PD-1 | Pembrolizumab and lenvatinib |
| Quavonlimab | CTLA-4 | |||
| Lenvatinib | RTK | |||
| Belzutifan | HIF2 | |||
| na | NCT04523272 | TQB2450 | PD-L1 | Sunitinib |
| Anlotinib | RTK | |||
| na | NCT04394975 | Toripalimab | PD-1 | Sunitinib |
| Axitinib | RTK | |||
| na | NCT03873402 | Nivolumab | PD-1 | Nivolumab |
| Ipilimumab | CTLA-4 | |||
| RAMPART | NCT03288532 [114] | Durvalumab | PD-1 | Monitoring after nephrectomy |
| Tremelimumab | CTLA-4 | |||
| CheckMate 67T | NCT04810078 | Nivolumab | PD-1 | This clinical trial examines the safety and efficacy of subcutaneous Nivolumab injection |
| PROBE | NCT04510597 | Nivolumab | PD-1 | This clinical trial examines the efficacy of cytoreductive nephrectomy in combination with ICI |
| Pembrolizumab | PD-1 | |||
| Axitinib | RTK | |||
| Avelumab | PD-L1 | |||
| na | NCT04157985 | Nivolumab | PD-1 | This clinical trial examines the length of treatment with ICI. |
| Pembrolizumab | PD-1 | |||
| Ipilimumab | CTLA-4 | |||
| Atezolizumab | PD-L1 |
| Patient Number | Control Group | Experimental Group | Cell Number | Platform | Citation |
|---|---|---|---|---|---|
| 3 | PB | ccRCC | 25,688 | 10× Genomics droplet-based | [58] |
| 11 | ANT | ccRCC | 163,905 | 10× Genomics droplet-based | [61] |
| 9 | ANT | ccRCC | 29,131 | 10× Genomics droplet-based | [56] |
| 13 | ANT | Advance stages of ccRCC | 164,722 | 10× Genomics droplet-based | [60] |
| 8 | Primary and metastatic ccRCC (LN), ICI-untreated | Primary and metastatic ccRCC (LN, lung, abdomen), ICI-treated | 34,326 | 10× Genomics droplet-based | [59] |
| 6 | ANT and primary ccRCC, ICI-untreated | PB, ANT, and multi-regions of primary and metastatic ccRCC (LN), ICI-treated | 167,283 | 10× Genomics droplet-based | [55] |
| 2 | PB and multi-regions of primary ccRCC, ICI-untreated | PB and multi-regions of primary and metastatic ccRCC (adrenal gland, bone, nephrectomy bed), ICI-treated | 26,456 | 10× Genomics droplet-based | [38] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.-C.; Jin, Z.; Kolb, R.; Borcherding, N.; Chatzkel, J.A.; Falzarano, S.M.; Zhang, W. Updates on Immunotherapy and Immune Landscape in Renal Clear Cell Carcinoma. Cancers 2021, 13, 5856. https://doi.org/10.3390/cancers13225856
Kim M-C, Jin Z, Kolb R, Borcherding N, Chatzkel JA, Falzarano SM, Zhang W. Updates on Immunotherapy and Immune Landscape in Renal Clear Cell Carcinoma. Cancers. 2021; 13(22):5856. https://doi.org/10.3390/cancers13225856
Chicago/Turabian StyleKim, Myung-Chul, Zeng Jin, Ryan Kolb, Nicholas Borcherding, Jonathan Alexander Chatzkel, Sara Moscovita Falzarano, and Weizhou Zhang. 2021. "Updates on Immunotherapy and Immune Landscape in Renal Clear Cell Carcinoma" Cancers 13, no. 22: 5856. https://doi.org/10.3390/cancers13225856
APA StyleKim, M. -C., Jin, Z., Kolb, R., Borcherding, N., Chatzkel, J. A., Falzarano, S. M., & Zhang, W. (2021). Updates on Immunotherapy and Immune Landscape in Renal Clear Cell Carcinoma. Cancers, 13(22), 5856. https://doi.org/10.3390/cancers13225856