
Molecular Mechanisms of Resistance to Immunotherapy and Antiangiogenic Treatments in Clear Cell Renal Cell Carcinoma

 , , , and
, , , and 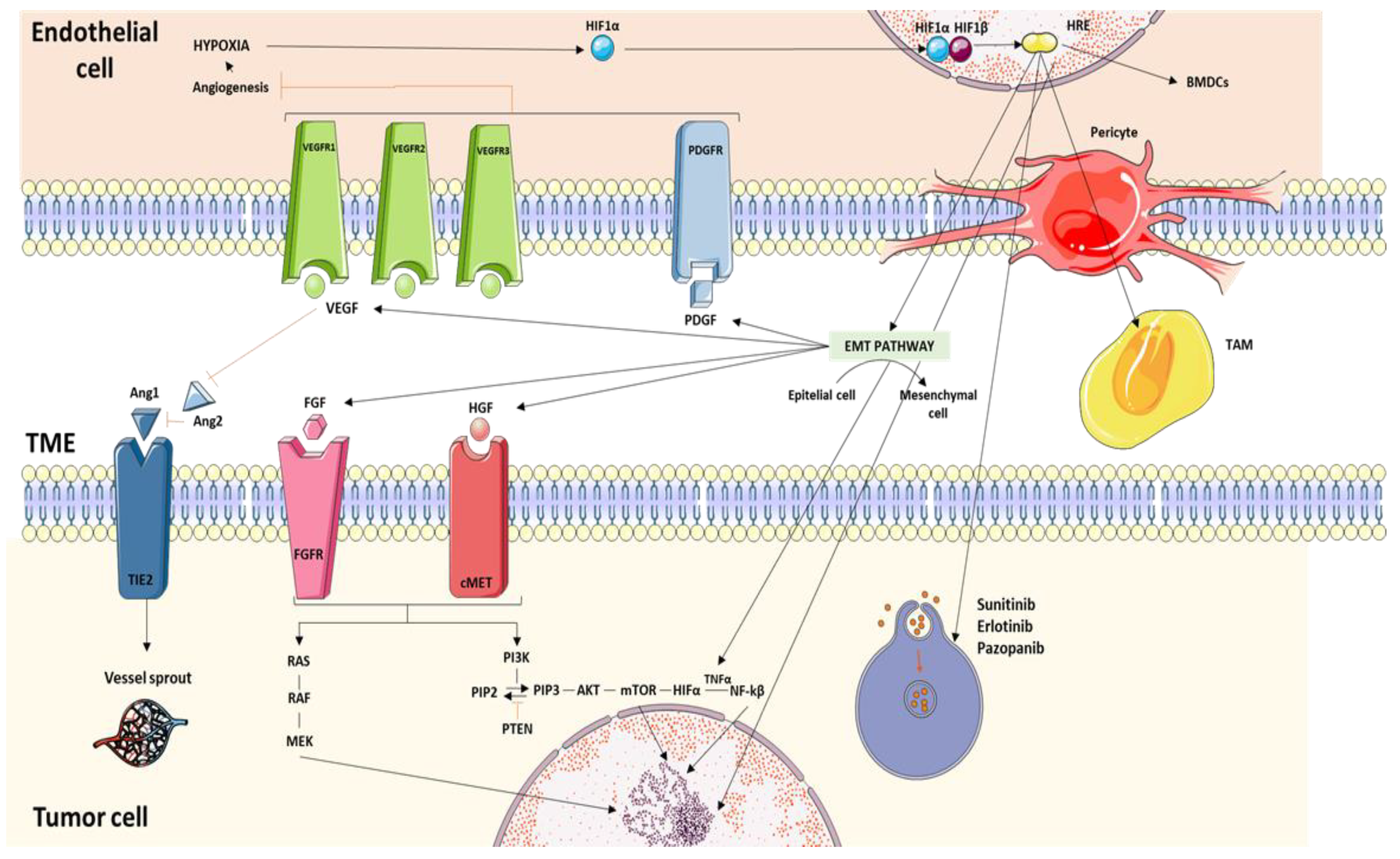{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Molecular Pathways Associated with Resistance to Treatment with Tyrosine-Kinase Inhibitors
2.1. Hypoxia as a Resistance Inductor
2.2. Angiogenic Switch
2.3. Epithelial–Mesenchymal Transition (EMT)
2.4. Activating Bypass Pathways
2.4.1. VEGF
2.4.2. PTEN
2.4.3. FGF
2.4.4. Axl and c-MET
2.4.5. TNF-α
2.4.6. Angiopoietin/Tie Pathway
2.4.7. Enhancer of Zeste Homologue 2 (EZH2)
2.5. Lysosomal Sequestration of TKIs
2.6. Noncoding RNAs (ncRNA) and Single Nucleotide Polymorphisms
2.7. Tumor Microenvironment Factors Related to Resistance to TKIs
2.7.1. Tumor Endothelial Cells (TECs)
2.7.2. Bone Marrow-Derived Proangiogenic Inflammatory Cell Recruitment
2.7.3. Pericyte Coverage
2.7.4. Tumor-Associated Fibroblasts (TAFs)
2.7.5. Tumor-Associated Macrophages
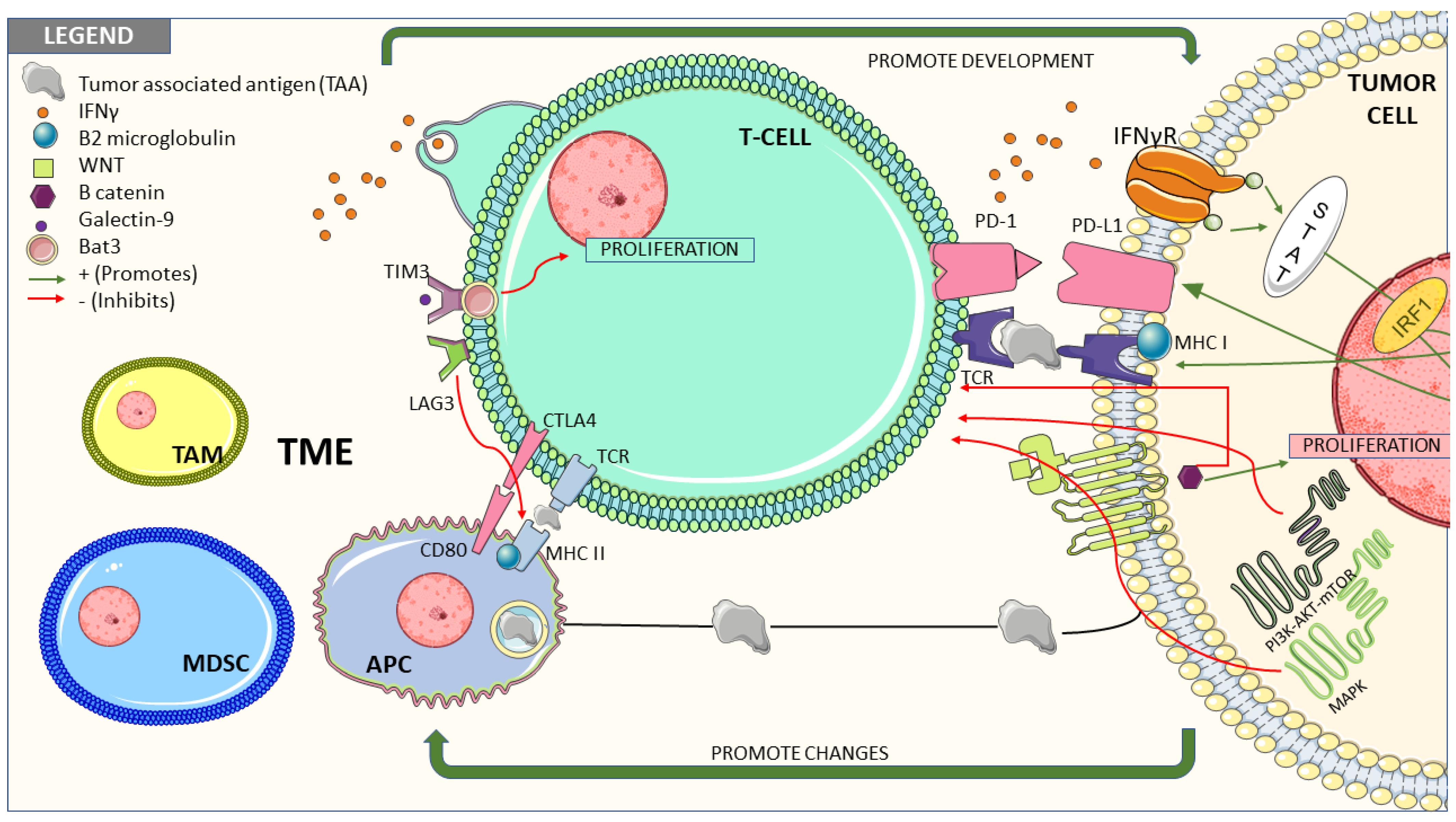
3. Molecular Pathways Associated with Resistance to Treatment with Immune Checkpoint Inhibitors
3.1. Tumor Cells-Intrinsic Factors
3.1.1. Interferon Gamma Signaling Pathway
3.1.2. Wnt/β-catenin Pathway
3.1.3. Mitogen-Activated Protein Kinases (MAPK) Pathway
3.1.4. PI3K/AKT/m-TOR Pathway
3.1.5. Cell Cycle Checkpoint Pathway
3.1.6. Loss of MHC
3.2. Tumor Microenvironment Related Factors and their Role in Resistance to Immune Response
3.2.1. T Cells
3.2.2. Innate Immune System
3.2.3. B Cells and Tertiary Lymphoid Structures
3.2.4. Proinflammatory Cytokines
3.2.5. Hypoxia
3.2.6. Protein Polybromo-1(PBRM-1) Expression
3.2.7. Immune Escape Related to Other Immune Checkpoints
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356. [Google Scholar] [CrossRef] [PubMed]
- Capitanio, U.; Bemsalah, K.; Bex, A.; Boorjian, S.A.; Bray, F.; Coleman, J.; Gore, J.L.; Sun, M.; Wood, C.; Russo, P. Epidemiology of Renal Cell Carcinoma. Eur. Urol. 2019, 75, 74. [Google Scholar] [CrossRef] [PubMed]
- Dabestani, S.; Thorstenson, A.; Lindblad, P.; Harmenberg, U.; Ljungberg, B.; Lundstam, S. Renal cell carcinoma recurrences and metastases in primary non-metastatic patients: A population-based study. World J. Urol. 2016, 34, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Levi, F.; Ferlay, J.; Galeone, C.; Lucchini, F.; Negri, E.; Boyle, P.; La Vecchia, C. The changing pattern of kidney cancer incidence and mortality in Europe. BJU Int. 2008, 101, 949. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Motzer, R.J. Systemic therapy for metastatic renal cell carcinoma. N. Engl. J. Med. 2017, 376, 354–366. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnarra, J.R.; Tory, K.; Weng, Y.; Schmidt, L.; Wei, M.H.; Li, H.; Latif, F.; Liu, S.; Chen, F.; Duh, F.M.; et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat. Genet. 1994, 7, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. The von Hippel-Lindau tumour suppressor protein and clear cell renal carcinoma. Clin. Cancer Res. 2007, 13, 680s–684s. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Invest. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Nosov, D.; Eisen, T.; Bondarenko, I.; Lesovoy, V.; Lipatov, O.; Tomczak, P.; Lyulko, O.; Alyasova, A.; Harza, M.; et al. Tivozanib versus sorafenib as initial targeted therapy for patients with metastatic renal cell carcinoma: Results from a phase III trial. J. Clin. Oncol. 2013, 31, 3791–3799. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.I.; Wong, M.K.K.; Kaufman, H.L.; Daniels, G.; Morse, M.A.; McDermott, D.F.; Agarwala, S.S.; Lewis, L.D.; Stewart, J.H. Vaishampayan, U.; et al. Impact of sequencing targeted therapies with high-dose interleukin-2 immunotherapy: An analysis of outcome and survival of patients with metastatic renal cell carcinoma from an on-going observational Il-2 clinical trial: PROCLAIMSM. Clin. Genitourin. Cancer. 2017, 15, 31–41.e4. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senbabaoglu, Y.; Gejman, R.S.; Winer, A.G.; Liu, M.; van Allen, E.M.; de Velasco, G.; Miao, D.; Ostrovnaya, I.; Drill, E.; Luna, A.; et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol. 2016, 17, 231. [Google Scholar] [CrossRef] [Green Version]
- Dietz, S.; Sultmann, H.; Du, Y.; Reisinger, E.; Riediger, A.L.; Volckmar, A.L.; Stenzinger, A.; Schlesner, M.; Jäger, D.; Hohenfellner, M.; et al. Patient-specific molecular alterations are associated with metastatic clear cell renal cell cancer progressing under tyrosine kinase inhibitor therapy. Oncotarget 2017, 8, 74049–74057. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Escalona, R.; Leung, D.; Chan, E.; Kannourakis, G. Tumour microenvironment and metabolic plasticity in cancer and cancer stem cells: Perspectives on metabolic and immune regulatory signatures in chemoresistant ovarian cancer stem cells. Semin. Cancer Biol. 2018, 53, 265–281. [Google Scholar] [CrossRef]
- Donnem, T.; Reynolds, A.R.; Kuczynski, E.A.; Gatter, K.; Vermeulen, P.B.; Kerbel, R.S.; Harris, A.L.; Pezzella, F. Non-angiogenic tumours and their influence on cancer biology. Nat. Rev. Cancer 2018, 18, 323–336. [Google Scholar] [CrossRef]
- Pezzella, F.; Ribatti, D. Vascular co-option and vasculogenic mimicry mediate resistance to antiangiogenic strategies. Cancer Rep. 2020, e1318. [Google Scholar] [CrossRef] [PubMed]
- Seftor, R.E.; Seftor, E.A.; Koshikawa, N.; Meltzer, P.S.; Gardner, L.M.; Bilban, M.; Stetler-Stevenson, W.G.; Quaranta, V.; Hendrix, M.J. Cooperative interactions of laminin 5 gamma2 chain, matrix metalloproteinase-2, and membrane type-1-matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma. Cancer Res. 2001, 61, 6322–6327. [Google Scholar]
- Saravanan, S.; Vimalraj, S.; Pavani, K.; Nikarika, R.; Sumantran, V.N. Intussusceptive angiogenesis as a key therapeutic target for cancer therapy. Life Sci. 2020, 252, 117670. [Google Scholar] [CrossRef]
- Mentzer, S.J.; Mentzer, M.A. Konerding Intussusceptive angiogenesis: Expansion and remodeling of microvascular networks. Angiogenesis 2014, 17, 499–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Magi-Galluzzi, C. Epithelial-to-mesenchymal transition in renal neoplasms. Adv. Anat. Pathol. 2014, 21, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Krishnamachary, B.; Zagzag, D.; Nagasawa, H.; Rainey, K.; Okuyama, H.; Baek, J.H.; Semenza, G.L. Hypoxia-inducible Factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor–null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006, 66, 2725–2731. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Yang, D.; Zong, H.; Zhu, L.; Wang, L.; Wang, X.; Zhu, X.; Song, X.; Wang, J. Growth-induced stress enhances epithelial-mesenchymal transition induced by IL-6 in clear cell renal cell carcinoma via the Akt/GSK-3β/β-catenin signaling pathway. Oncogenesis 2017, 6, e375. [Google Scholar] [CrossRef]
- Boström, A.K.; Möller, C.; Nilsson, E.; Elfving, P.; Axelson, H.; Johansson, M.E. Sarcomatoid conversion of clear cell renal cell carcinoma in relation to epithelial-to-mesenchymal transition. Hum. Pathol. 2012, 43, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Tammela, T.; Zarkada, G.; Wallgard, E.; Murtomäki, A.; Suchting, S.; Wirzenius, M.; Waltari, M.; Hellström, M.; Schomber, T.; Peltonen, R.; et al. Blocking vegfr-3 suppresses angiogenic sprouting and vascular network formation. Nature 2008, 454, 656–660. [Google Scholar] [CrossRef]
- van der Mijn, J.C.; Mier, J.W.; Broxterman, H.J.; Verheul, H.M. Predictive biomarkers in renal cell cancer: Insights in drug resistance mechanisms. Drug Resist. Updates 2014, 17, 77–88. [Google Scholar] [CrossRef]
- Welti, J.C.; Gourlaouen, M.; Powles, T.; Kudahetti, S.C.; Wilson, P.; Berney, D.M.; Reynolds, A.R. Fibroblast growth factor 2 regulates endothelial cell sensitivity to sunitinib. Oncogene 2011, 30, 1183–1193. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Liu, X.D.; Sun, M.; Falcón, B.; Hashizume, H.; Yao, L.C.; Aftab, D.T.; McDonald, D.M. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene 2016, 35, 2687–2697. [Google Scholar] [CrossRef]
- Gustafsson, A.; Martuszewska, D.; Johansson, M.; Ekman, C.; Hafizi, S.; Ljungberg, B.; Dahlbäck, B. Differential expression of Axl and Gas6 in renal cell carcinoma reflecting tumor advancement and survival. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 4742–4749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibney, G.T.; Aziz, S.A.; Camp, R.L.; Conrad, P.; Schwartz, B.E.; Chen, C.R.; Kelly, W.K.; Kluger, H.M. c-Met is a prognostic marker and potential therapeutic target in clear cell renal cell carcinoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol./ESMO. 2013, 24, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer. 2009, 9, 361–371. [Google Scholar] [CrossRef]
- Ho, M.Y.; Tang, S.J.; Chuang, M.J.; Cha, T.L.; Li, J.Y.; Sun, G.H.; Sun, K.H. TNF-α induces epithelial-mesenchymal transition of renal cell carcinoma cells via a GSK3β-dependent mechanism. Mol. Cancer Res. 2012, 10, 1109–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, K.; Guo, G.; Gerber, D.E.; Gao, B.; Peyton, M.; Huang, C.; Minna, J.D.; Hatanpaa, K.J.; Kernstine, K.; Cai, L. TNF-driven adaptive response mediates resistance to EGFR inhibition in lung cancer. J. Clin. Invest. 2018, 128, 2500–2518. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lin, G.; Yan, Y.; Li, X.; Hu, Y.; Wang, J.; Yin, B.; Wu, Y.; Li, Z.; Yang, X.P. Transmembrane TNF-alpha promotes chemoresistance in breast cancer cells. Oncogene 2018, 37, 3456–3470. [Google Scholar] [CrossRef]
- Hwang, H.S.; Park, Y.Y.; Shin, S.J.; Go, H.; Park, J.M.; Yoon, S.Y.; Lee, J.L.; Cho, Y.M. Involvement of the TNF-α Pathway in TKI Resistance and Suggestion of TNFR1 as a Predictive Biomarker for TKI Responsiveness in Clear Cell Renal Cell Carcinoma. J. Korean Med. Sci. 2020, 35, e31. [Google Scholar] [CrossRef]
- Rigamonti, N.; Kadioglu, E.; Keklikoglou, I.; Rmili, C.W.; Leow, C.C.; de Palma, M. Role of angiopoietin-2 in adaptive tumor resistance to VEGF signaling blockade. Cell Rep. 2014, 8, 696–706. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Bullock, A.J.; Zhang, L.; Wei, L.; Yu, D.; Mahagaokar, K.; Alsop, D.C.; Mier, J.W.; Atkins, M.B.; Coxon, A.; et al. The role of angiopoietins as potential therapeutic targets in renal cell carcinoma. Transl. Oncol. 2014, 7, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Xu, Z.; Zhong, L.; Wang, H.; Jiang, S.; Long, Q.; Xu, J.; Guo, J. Enhancer of zeste homolog 2 (EZH2) promotes tumour cell migration and invasion via epigenetic repression of E-cadherin in renal cell carcinoma. BJU Int. 2016, 117, 351–362. [Google Scholar] [CrossRef]
- Adelaiye-Ogala, R.; Budka, J.; Damayanti, N.P.; Arrington, J.; Ferris, M.; Hsu, C.C.; Chintala, S.; Orillion, A.; Miles, K.M.; Shen, L.; et al. EZH2 Modifies Sunitinib Resistance in Renal Cell Carcinoma by Kinome Reprogramming. Cancer Res. 2017, 77, 6651–6666. [Google Scholar] [CrossRef] [Green Version]
- Adelaiye, R.; Ciamporcero, E.; Miles, K.M.; Sotomayor, P.; Bard, J.; Tsompana, M.; Conroy, D.; Shen, L.; Ramakrishnan, S.; Ku, S.Y.; et al. Sunitinib dose escalation overcomes transient resistance in clear cell renal cell carcinoma and is associated with epigenetic modifications. Mol. Cancer 2015, 14, 513–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotink, K.J.; Broxterman, H.J.; Labots, M.; de Haas, R.R.; Dekker, H.; Honeywell, R.J.; Rudek, M.A.; Beerepoot, L.V.; Musters, R.J.; Jansen, G.; et al. Lysosomal sequestration of sunitinib: A novel mechanism of drug resistance. Clin. Cancer Res. 2011, 17, 7337–7346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotink, K.J.; Rovithi, M.; de Haas, R.R.; Honeywell, R.J.; Dekker, H.; Poel, D.; Azijli, K.; Peters, G.J.; Broxterman, H.J.; Verheul, H.M.W. Cross-resistance to clinically used tyrosine kinase inhibitors sunitinib, sorafenib and pazopanib. Cell Oncol. 2015, 38, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azijli, K.; Gotink, K.J.; Verheul, H.M.W. The Potential Role of Lysosomal Sequestration in Sunitinib Resistance of Renal Cell Cancer. J. Kidney Cancer VHL. 2015, 2, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Giuliano, S.; Cormerais, Y.; Dufies, M.; Grépin, R.; Colosetti, P.; Belaid, A.; Parola, J.; Martin, A.; Lacas-Gervais, S.; Mazure, N.M.; et al. Resistance to sunitinib in renal clear cell carcinoma results from sequestration in lysosomes and inhibition of the autophagic flux. Autophagy 2015, 11, 1891–1904. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Siddig, S.; Uzu, M.; Suzuki, S.; Nomura, Y.; Kashiba, T.; Gushimiyagi, K.; Sekine, Y.; Uehara, T.; Arano, Y.; et al. Elacridar enhances the cytotoxic effects of sunitinib and prevents multidrug resistance in renal carcinoma cells. Eur. J. Pharm. 2015, 746, 258–266. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget 2015, 6, 1143–1156. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Zhao, A.; Li, G.; Péoc’h, M.; Genin, C.; Gigante, M. Serum miR-210 as a novel biomarker for molecular diagnosis of clear cell renal cell carcinoma. Exp. Mol. Pathol. 2013, 94, 115–120. [Google Scholar] [CrossRef]
- Dias, F.; Teixeira, A.L.; Ferreira, M.; Adem, B.; Bastos, N.; Vieira, J.; Fernandes, M.; Sequeira, M.I.; Maurício, J.; Lobo, F.; et al. Plasmatic miR-210, miR-221 and miR1233 profile: Potential liquid biopsies candidates for renal cell carcinoma. Oncotarget 2017, 8, 103315–103326. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wu, C.; Yang, Q.; Ding, M.; Zhong, J.; Zhang, C.; Ge, J.; Wang, J.; Zhang, C. miR-28-5p acts as a tumor suppressor in renal cell carcinoma for multiple antitumor effects by targeting RAP1B. Oncotarget 2016, 7, 73888–73902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Li, Y.; Wen, H.; Feng, C. Overexpression of miR-15b promotes resistance to Sunitinib in renal cell carcinoma. J. Cancer 2019, 10, 3389–3396. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Osaki, M.; Onuma, K.; Yumioka, T.; Iwamoto, H.; Sejima, T.; Kugoh, H.; Takenaka, A.; Okada, F. Identification of MicroRNAs involved in resistance to Sunitinib in renal cell carcinoma cells. Anticancer Res. 2017, 37, 2985–2992. [Google Scholar]
- Yoshino, H.; Enokida, H.; Itesako, T.; Tatarano, S.; Kinoshita, T.; Fuse, M.; Kojima, S.; Nakagawa, M.; Seki, N. Epithelial–mesenchymal transition-related microRNA-200s regulate molecular targets and pathways in renal cell carcinoma. J. Hum. Genet. 2013, 58, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Kourembanas, S. Exosomes: Vehicles of intercellular signaling, biomarkers, and vectors of cell therapy. Annu. Rev. Physiol. 2015, 77, 13–27. [Google Scholar] [CrossRef] [Green Version]
- Qu, L.; Ding, J.; Chen, C.; Wu, Z.J.; Liu, B.; Gao, Y. Exosome-Transmitted lncARSR Promotes Sunitinib Resistance in Renal Cancer by Acting as a Competing Endogenous RNA. Cancer Cell 2016, 29, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Beuselinck, B.; Karadimou, A.; Lambrechts, D.; Claes, B.; Wolter, P.; Couchy, G.; Berkers, J.; Paridaens, R.; Schöffski, P.; Méjean, A.; et al. Single-nucleotide polymorphisms associated with outcome in metastatic renal cell carcinoma treated with sunitinib. Br. J. Cancer 2013, 108, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Diekstra, M.H.; Swen, J.J.; Boven, E.; Castellano, D.; Gelderblom, H.; Mathijssen, R.H.J.; Rodríguez-Antona, C.; García-Donas, J.; Rini, B.I.; Guchelaar, H.-J. Cyp3a5 and abcb1 polymorphisms as predictors for sunitinib outcome in metastatic renal cell carcinoma. Eur. Urol. 2015, 68, 621–629. [Google Scholar] [CrossRef]
- Makhov, P.; Joshi, S.; Ghatalia, P.; Kutikov, A.; Uzzo, R.G.; Kolenko, V.M. Resistance to Systemic Therapies in Clear Cell Renal Cell Carcinoma: Mechanisms and Management Strategies. Mol. Cancer Ther. 2018, 17, 1355–1364. [Google Scholar] [CrossRef] [Green Version]
- van der Veldt, A.A.; Vroling, L.; de Haas, R.R.; Koolwijk, P.; van den Eertwegh, A.J.; Haanen, J.B.A.G.; van Hinsbergh, V.W.M.; Broxterman, H.J.; Boven, E. Sunitinib-induced changes in circulating endothelial cell-related proteins in patients with metastatic renal cell cancer. Int. J. Cancer 2012, 131, E484–E493. [Google Scholar] [CrossRef]
- Miles, K.M.; Seshadri, M.; Ciamporcero, E.; Adelaiye, R.; Gillard, B.; Sotomayor, P.; Attwood, K.; Shen, L.; Conroy, D.; Kuhnert, F.; et al. Dll4 blockade potentiates the anti-tumor effects of VEGF inhibition in renal cell carcinoma patient-derived xenografts. PLoS ONE 2014, 9, e112371. [Google Scholar]
- Xiao, W.; Gao, Z.; Duan, Y.; Yuan, W.; Ke, Y. Notch signaling plays a crucial role in cancer stem-like cells maintaining stemness and mediating chemotaxis in renal cell carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 41. [Google Scholar] [CrossRef] [Green Version]
- Shojaei, F.; Wu, X.; Qu, X.; Kowanetz, M.; Yu, L.; Tan, M.; Meng, Y.G.; Ferrara, N. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc. Natl. Acad. Sci. USA 2009, 106, 6742–6747. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.S.; Rayman, P.; Ireland, J.; Swaidani, S.; Li, G.; Bunting, K.D.; Rini, B.; Finke, J.H.; Cohen, P.A. Direct and differential suppression of myeloid-derived suppressor cell subsets by sunitinib is compartmentally constrained. Cancer Res. 2010, 70, 3526–3536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geevarghese, A.; Herman, I.M. Pericyte-endothelial crosstalk: Implications and opportunities for advanced cellular therapies. Transl. Res. 2014, 163, 296–306. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Mestas, J.; Burdick, M.D.; Phillips, R.J.; Thomas, G.V.; Reckamp, K. Stromal derived factor-1 (SDF-1/CXCL12) and CXCR4 in renal cell carcinoma metastasis. Mol. Cancer 2006, 5, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagzag, D.; Krishnamachary, B.; Yee, H.; Okuyama, H.; Chiriboga, L.; Ali, M.A. Stromal cell-derived factor-1α and CXCR4 expression in hemangioblastoma and clear cell-renal cell carcinoma: Von Hippel-Lindau loss-of-function induces expression of a ligand and its receptor. Cancer Res. 2005, 65, 6178–6188. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Lu, Y.; Song, J.; Dong, B.; Kong, W.; Xue, W. Cancer-associated fibroblasts promote renal cell carcinoma progression. Tumor Biol. 2015, 36, 3483–3488. [Google Scholar] [CrossRef]
- Kakarla, S.; Song, X.T.; Gottschalk, S. Cancer-associated fibroblasts as targets for immunotherapy. Immunotherapy 2012, 4, 1129–1138. [Google Scholar] [CrossRef] [Green Version]
- Errarte, P.; Guarch, R.; Pulido, R.; Blanco, L.; Nunes-Xavier, C.E.; Beitia, M.; Gil, J.; Angulo, J.C.; Lopez, J.L.; Larrinaga, G. The expression of fibroblast activation protein in clear cell renal cell carcinomas is associated with synchronous lymph node metastases. PLoS ONE 2016, 11, e0169105. [Google Scholar] [CrossRef]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Krug, S.; Abbassi, R.; Griesmann, H.; Sipos, B.; Wiese, D.; Rexin, P.; Blank, A.; Perren, A.; Haybaeck, J.; Hüttelmaier, S. Therapeutic targeting of tumor-associated macrophages in pancreatic neuroendocrine tumors. Int. J. Cancer. 2018, 143, 1806–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Esquivel, F.; Kawakami, Y.; Yewdell, J.W.; Mulé, J.J.; Rosenberg, S.A.; Bennink, J.R. Identification of human cancers deficient in antigen processing. J. Exp. Med. 1993, 177, 265–272. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Sweis, R.F.; Spranger, S.; Bao, R.; Paner, G.P.; Stadler, W.M.; Steinberg, G.; Gajewski, T.F. Molecular drivers of the non-T-cell-inflamed tumor microenvironment in urothelial bladder cancer. Cancer Immunol. Res. 2016, 4, 563–568. [Google Scholar] [CrossRef] [Green Version]
- Seiwert, T.Y.; Zuo, Z.; Keck, M.K.; Khattri, A.; Pedamallu, C.S.; Stricker, T.; Brown, C.; Pugh, T.J.; Stojanov, P.; Cho, J.; et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin. Cancer Res. 2015, 21, 632–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Sánchez, A.; Memon, D.; Pourpe, S.; Veeraraghavan, H.; Li, Y.; Vargas, H.A.; Gill, M.B.; Park, K.J.; Zivanovic, O.; Konner, J.; et al. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell 2017, 170, 927–938.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Xiao, C.; Evans, K.S.; Theivanthiran, T.; DeVito, N.; Holtzhausen, A.; Liu, J.; Liu, X.; Boczkowski, D.; Nair, S.; et al. Paracrine Wnt5a-β-catenin signaling triggers a metabolic program that drives dendritic cell tolerization. Immunity 2018, 48, 147–160.e7. [Google Scholar] [CrossRef] [Green Version]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.-N.J.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef] [Green Version]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018, 23, 313–326.e5. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Uberllacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.-R.; et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 2018, 175, 984–997.e24. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist 2019, 2, 141–160. [Google Scholar] [CrossRef] [Green Version]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar]
- Giraldo, N.A.; Becht, E.; Pagès, F.; Skliris, G.; Verkarre, V.; Vano, Y.; Mejean, A.; Saint-Aubert, N.; Lacroix, L.; Natario, I.; et al. Orchestration and prognostic significance of immune checkpoints in the microenvironment of primary and metastatic renal cell cancer. Clin. Cancer Res. 2015, 21, 3031–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Tannir, N.M.; Mainwaring, P.N.; Rini, B.I.; Hammers, H.J.; Donskov, F.; Roth, B.J.; Peltola, K.; et al. METEOR investigators. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): Final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2016, 17, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Giraldo, N.A.; Becht, E.; Vano, Y.; Petitprez, F.; Lacroix, L.; Validire, P.; Sanchez-Salas, R.; Ingels, A.; Oudard, S.; Moatti, A.; et al. Tumor-infiltrating and peripheral blood T-cell immunophenotypes predict early relapse in localized clear cell renal cell carcinoma. Clin. Cancer Res. 2017, 23, 4416–4428. [Google Scholar]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 6, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Banchereau, R.; Hamidi, H.; Powles, T.; McDermott, D.; Atkins, M.B.; Escudier, B.; Liu, L.-F.; Leng, N.; Abbas, A.R.; et al. Molecular Subsets in Renal Cancer Determine Outcome to Checkpoint and Angiogenesis Blockade. Cancer Cell 2020, 38, 803–817.e4. [Google Scholar]
- Seeber, A.; Klinglmair, G.; Fritz, J.; Steinkohl, F.; Zimmer, K.-C.; Aigner, F.; Horninger, W.; Gastl, G.; Zelger, B.; Brunner, A.; et al. High IDO-1 expression in tumor endothelial cells is associated with response to immunotherapy in metastatic renal cell carcinoma. Cancer Sci. 2018, 109, 1583–1591. [Google Scholar] [CrossRef] [Green Version]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mier, J.W. The tumor microenvironment in renal cell cancer. Curr. Opin. Oncol. 2019, 31, 194–199. [Google Scholar] [CrossRef]
- Komohara, Y.; Hasita, H.; Ohnishi, K.; Fujiwara, Y.; Suzu, S.; Eto, M.; Takeya, M. Macrophage infiltration and its prognostic relevance in clear cell renal cell carcinoma. Cancer Sci. 2011, 102, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Rosser, E.C.; Mauri, C. Regulatory B cells: Origin, phenotype, and function. Immunity 2015, 42, 607–612. [Google Scholar] [CrossRef] [Green Version]
- Sarvaria, A.; Madrigal, J.A.; Saudemont, A. B cell regulation in cancer and anti-tumor immunity. Cell Mol. Immunol. 2017, 14, 662–674. [Google Scholar] [CrossRef] [Green Version]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.-M.; Hsiao, L.-P.; Lacroix, L.; Bougoüin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef]
- Finkin, S.; Yuan, D.; Stein, I.; Taniguchi, K.; Weber, A.; Unger, K.; Browning, J.L.; Goossens, N.; Nakagawa, S.; Gunasekaran, G.; et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat. Immunol. 2015, 16, 1235–1244. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Bui, J.D.; Schreiber, R.D. Cancer immunosurveillance, immunoediting and inflammation: Independent or interdependent processes? Curr. Opin. Immunol. 2007, 19, 203–208. [Google Scholar] [CrossRef]
- Stubbs, M.; McSheehy, P.M.; Griffiths, J.R.; Bashford, C.L. Causes and consequences of tumour acidity and implications for treatment. Mol. Med. Today 2000, 6, 15–19. [Google Scholar] [CrossRef]
- Sormendi, S.; Wielockx, B. Hypoxia pathway proteins as central mediators of metabolism in the tumor cells and their microenvironment. Front Immunol. 2018, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.A.; Kerbel, R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef]
- Garcia-Lora, A.; Algarra, I.; Garrido, F. MHC class I antigens, immune surveillance, and tumor immune escape. J. Cell Physiol. 2003, 195, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Tatli Dogan, H.; Kiran, M.; Bilgin, B.; Kiliçarslan, A.; Sendur, M.A.N.; Yalçin, B.; Ardiçoglu, A.; Atmaca, A.F.; Gumuskaya, B. Prognostic significance of the programmed death ligand 1 expression in clear cell renal cell carcinoma and correlation with the tumor microenvironment and hypoxia-inducible factor expression. Diagn. Pathol. 2018, 13, 60. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, Z.; Xu, X.; Yu, Z.; Mi, J. The influence of microenvironment on tumor immunotherapy. FEBS J. 2019, 286, 4160–4175. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Moreno-Altamirano, M.M.B.; Prado-Garcia, H.; Sánchez-García, F.J. Lactate contribution to the tumor microenvironment: Mechanisms, effects on immune cells and therapeutic relevance. Front Immunol. 2016, 7, 52. [Google Scholar] [CrossRef] [Green Version]
- Pan, D.; Kobayashi, A.; Jiang, P.; Ferrari de Andrade, L.; Tay, R.E.; Luoma, A.M.; Tsoucas, D.; Qiu, X.; Lim, K.; Rao, P.; et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 2018, 359, 770–775. [Google Scholar] [CrossRef] [Green Version]
- Varela, I.; Tarpey, P.; Raine, K.; Huang, D.; Ong, C.K.; Stephens, P.; Davies, H.; Jones, D.; Lin, M.-L.; Teague, J.; et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011, 469, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoni, M.; Massari, F.; Amantini, C.; Nabissi, M.; Maines, F.; Burattini, L.; Berardi, R.; Santoni, G.; Montironi, R.; Tortora, G.; et al. Emerging role of tumor-associated macrophages as therapeutic targets in patients with metastatic renal cell carcinoma. Cancer Immunol. Immunother. 2013, 62, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Li, W.; Martini, D.J.; Norton, C.; Bossé, D.; Wankowicz, S.M.; Cullen, D.; et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, D.A.; Ishii, Y.; Walsh, A.M.; Van Allen, E.M.; Wu, C.J.; Shukla, S.A.; Choueiri, T.K. Clinical validation of PBRM1 alterations as a marker of immune checkpoint inhibitor response in renal cell carcinoma. JAMA Oncol. 2019, 5, 1631–1633. [Google Scholar] [CrossRef] [PubMed]
- Aili, A.; Jie, W.; Lixiang, X.; Junjie, W. Mutational Analysis of PBRM1 and Significance of PBRM1 Mutation in Anti-PD-1 Immunotherapy of Clear Cell Renal Cell Carcinoma. Front Oncol. 2021, 11, 712765. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 1, 97–111. [Google Scholar] [CrossRef] [Green Version]
- Acharya, N.; Sabatos-Peyton, C.; Carrizosa Anderson, A. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 2020, 8, e000911. [Google Scholar] [CrossRef]
- Granier, C.; Dariane, C.; Combe, P.; Verkarre, V.; Urien, S.; Badoual, C.; Roussel, H.; Mandavit, M.; Ravel, P.; Sibony, M.; et al. Tim-3 expression on tumor-infiltrating PD-1+CD8+ T cells correlates with poor clinical outcome in renal cell carcinoma. Cancer Res. 2017, 77, 1075–1082. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, M.V.; Drake, C.G. LAG-3 in Cancer Immunotherapy. Curr. Top. Microbiol. Immunol. 2011, 344, 269–278. [Google Scholar]
- Sittig, S.P.; Kollgaard, T.; Gronbaek, K.; Idorn, M.; Hennenlotter, J.; Stenzl, A.; Gouttefangeas, C.; Thor Straten, P. Clonal expansion of renal cell carcinoma-infiltrating T lymphocytes. Oncoimmunology 2013, 2, e26014. [Google Scholar] [CrossRef] [Green Version]
- Miao, W.; Qi, D.; Jiangtao, J.; Yuhan, W.; Yuting, L.; Qin, L. LAG3 and its emerging role in cancer immunotherapy. Clin. Transl. Med. 2021, 3, e365. [Google Scholar]
- Long, L.; Xue, Z.; Fuchun, C.; Qi, P.; Phiphatwatchara, P.; Yuyang, Z.; Honglei, C. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marhelava, K.; Pilch, Z.; Bajor, M.; Graczyk-Jarzynka, A.; Zagozdzon, R. Targeting negative and positive immune checkpoints with monoclonal antibodies in therapy of cancer. Cancers 2019, 11, 1756. [Google Scholar] [CrossRef] [Green Version]
- Solomon, B.L.; Garrido-Laguna, I. Tigit: A novel immunotherapy target moving from bench to bedside. Cancer Immunol. Immunother. 2018, 67, 1659–1667. [Google Scholar] [CrossRef]
- Brenner, W.; Farber, G.; Herget, T.; Lehr, H.A.; Hengstler, J.G.; Thuroff, J.W. Loss of tumor suppressor protein PTEN during renal carcinogenesis. Int. J. Cancer 2002, 99, 53–57. [Google Scholar] [CrossRef]
- Makhov, P.B.; Golovine, K.; Kutikov, A.; Teper, E.; Canter, D.J.; Simhan, J.; Uzzo, R.G.; Kolenko, V.M. Modulation of Akt/mTOR Signaling Overcomes Sunitinib Resistance in Renal and Prostate Cancer Cells. Mol. Cancer Ther. 2012, 11, 1510–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, A.M.; Feldman, D.R.; Voss, M.H.; Ginsberg, M.S.; Baum, M.S.; Brocks, D.R.; Fischer, P.M.; Trinos, M.J.; Patil, S.; Motzer, R.J. Phase 1 trial of everolimus plus sunitinib in patients with metastatic renal cell carcinoma. Cancer 2012, 118, 1868–1876. [Google Scholar] [CrossRef] [Green Version]
- Allen, E.; Walters, I.B.; Hanahan, D. Brivanib, a dual FGF/VEGF inhibitor, is active both first and second line against mouse pancreatic neuroendocrine tumors developing adaptive/evasive resistance to VEGF inhibition. Clin. Cancer Res. 2011, 17, 5299–5310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, H.; Brave, M.; Beaver, J.A.; Cheng, J.; Tang, S.; Zahalka, E.; Palmby, T.R.; Venugopal, R.; Song, P.; Liu, Q.; et al. U.S. Food and Drug Administration Approval: Cabozantinib for the Treatment of Advanced Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 330–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powles, T.; Albiges, L.; Bex, A.; Grünwald, V.; Porta, C.; Procopio, G.; Schmidinger, M.; Suárez, C.; De Velasco, G.; On behalf of the Esmo Guidelines Committee. Esmo clinical practice guideline update on the use of immunotherapy in early stage and advanced renal cell carcinoma. Ann. Oncol Off. J Eur. Soc. Med. Oncol. 2021, 32, 1511–1519. [Google Scholar] [CrossRef]
- Ciamporcero, E.; Miles, K.M.; Adelaiye, R.; Ramakrishnan, S.; Shen, L.; Ku, S.; Pizzimenti, S.; Sennino, B.; Barrera, G.; Pili, R. Combination strategy targeting VEGF and HGF/c-met in human renal cell carcinoma models. Mol. Cancer Ther. 2015, 14, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.K.; Tangen, C.; Thompson, I.M., Jr.; Balzer-Haas, N.; George, D.J.; Heng, D.; Shuch, B.; Stein, M.; Tretiakova, M.; Humphrey, P.; et al. A comparison of sunitinib with cabozantinib, crizotinib, and savolitinib for treatment of advanced papillary renal cell carcinoma: A randomised, open-label, phase 2 trial. Lancet 2021, 397, 695–703. [Google Scholar] [CrossRef]
- Atkins, M.B.; Gravis, G.; Drosik, K.; Demkow, T.; Tomczak, P.; Wong, S.S.; Michaelson, M.D.; Choueiri, T.K.; Wu, B.; Navale, L.; et al. Trebananib (AMG 386) in Combination with Sunitinib in Patients with Metastatic Renal Cell Cancer: An Open-Label, Multicenter, Phase II Study. J. Clin. Oncol. 2015, 33, 3431–3438. [Google Scholar] [CrossRef]
- Huang, H.; Lai, J.Y.; Do, J.; Liu, D.; Li, L.; Del Rosario, J.; Doppalapudi, V.R.; Pirie-Shepherd, S.; Levin, N.; Bradshaw, C.; et al. Specifically targeting angiopoietin-2 inhibits angiogenesis, Tie2-expressing monocyte infiltration, and tumor growth. Clin. Cancer Res. 2011, 17, 1001–1011. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Zhang, Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Genet. Dev. 2004, 14, 155–164. [Google Scholar] [CrossRef]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Morschhauser, F.; Tilly, H.; Chaidos, A.; McKay, P.; Phillips, T.; Assouline, S.; Batlevi, C.L.; Campbell, P.; Ribrag, V.; Damaj, G.L.; et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: An open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020, 21, 1433–1442. [Google Scholar] [CrossRef]
- Hoy, S.M. Tazemetostat: First Approval. Drugs 2020, 80, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Huang, L.; Shen, R.; Bernard-Cacciarella, M.; Zhou, P.; Hu, C.; Di Benedetto, M.; Janin, A.; Bousquet, G.; Li, H.; et al. Drug resistance-related sunitinib sequestration in autophagolysosomes of endothelial cells. Int. J. Oncol. 2020, 56, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Wiedmer, T.; Blank, A.; Pantasis, S.; Normand, L.; Bill, R.; Krebs, P.; Tschan, M.P.; Miranoni, I.; Perren, A. Autophagy inhibition improves sunitinib efficacy in pancreatic neuroendocrine tumors via a lysosome-dependent mechanism. Mol. Cancer Ther. 2017, 16, 2502–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, M.; Pàez-Ribes, M.; Cortez, E.; Casanovas, O.; Pietras, K. Use of a Mouse Model of Pancreatic Neuroendocrine Tumors to Find Pericyte Biomarkers of Resistance to Anti-angiogenic Therapy. Horm. Metab. Res. 2011, 43, 884–889. [Google Scholar] [CrossRef] [PubMed]
- Mita, A.C.; Takimoto, C.H.; Mita, M.; Tolcher, A.; Sankhala, K.; Sarantopoulos, J.; Valdivieso, M.; Wood, L.; Rasmussen, E.; Sun, Y.N.; et al. Phase 1 study of AMG 386, a selective angiopoietin 1/2−neutralizing peptibody, in combination with chemotherapy in adults with advanced solid tumors. Clin. Cancer Res. 2010, 16, 3044–3056. [Google Scholar] [CrossRef] [Green Version]
- Kreamer, K.M. Immune Checkpoint Blockade: A New Paradigm in Treating Advanced Cancer. J. Adv. Pract. Oncol. 2014, 5, 418–431. [Google Scholar]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Frontera, O.A.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a promising target for cancer immunotherapy. Oncol. Targets Ther. 2018, 11, 7005–7009. [Google Scholar] [CrossRef] [Green Version]
- Zelba, H.; Bedke, J.; Hennenlotter, J.; Mostböck, S.; Zettl, M.; Zichner, T.; Chandran, A.; Stenzl, A.; Rammensee, H.; Gouttefangeas, C. PD-1 and LAG-3 Dominate Checkpoint Receptor-Mediated T-cell Inhibition in Renal Cell Carcinoma. Cancer. Immunol. Res. 2019, 7, 1891–1899. [Google Scholar] [CrossRef]
- Brignone, C.; Escudier, B.; Grygar, C.; Marcu, M.; Triebel, F. A phase I pharmacokinetic and biological correlative study of IMP321, a novel MHC class II agonist, in patients with advanced renal cell carcinoma. Clin. Cancer Res. 2009, 15, 6225–6231. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.A.; Bakouny, Z.; Hirsch, L.; Flippot, R.; Van Allen, E.M.; Wu, C.J.; Choueiri, T.K. Beyond conventional immune-checkpoint inhibition—Novel immunotherapies for renal cell carcinoma. Nat. Rev. Clin. Oncol. 2021, 18, 199–214. [Google Scholar] [CrossRef]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat plus pembrolizumab in patients with advanced solid tumors: Phase I results from a multicenter, open-label phase I/II trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, 3223–3230. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poeck, H.; Besch, R.; Maihoefer, C.; Renn, M.; Tormo, D.; Morskaya, S.S.; Kirschnek, S.; Gaffal, E.; Landsberg, J.; Hellmuth, J.; et al. 5′-Triphosphate-siRNA: Turning gene silencing and Rig-I activation against melanoma. Nat. Med. 2008, 14, 1256–1263. [Google Scholar] [CrossRef]
- Sullivan, R.J. Back to the future: Rethinking and retooling il2 in the immune checkpoint inhibitor era. Cancer Discov. 2019, 9, 694–695. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Khong, H.; Fa’ak, F.; Bentebibel, S.E.; Janssen, L.; Chesson, B.C.; Creasy, C.A.; Forget, M.A.; Kahn, L.; Pazdrak, B.; et al. bempegaldesleukin selectively depletes intratumoral tregs and potentiates t cell-mediated cancer therapy. Nat. Commun. 2020, 11, 661. [Google Scholar] [CrossRef]
- Vano, Y.A.; Rioux-Leclercq, N.; Dalban, C.; Sautes-Fridman, C.; Bougoüin, A.; Chaput, N.; Chouaib, S.; Beuselinck, B.; Chevreau, C.; Gross-Goupil, M.; et al. NIVOREN GETUG-AFU 26 translational study: Association of PD-1, AXL, and PBRM-1 with outcomes in patients (pts) with metastatic clear cell renal cell carcinoma (mccRCC) treated with nivolumab (N). J. Clin. Oncol. 2020, 38, 618. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Blass, E.; Ott, P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef]
- Graham, J.; Dudani, S.; Heng, D. Prognostication in kidney cancer: Recent advances and future directions. J. Clin. Oncol. 2018, 36, 3567–3573. [Google Scholar] [CrossRef]
- Rodriguez-Vida, A.; Strijbos, M.; Hutson, T. Predictive and prognostic biomarkers of targeted agents and modern immunotherapy in renal cell carcinoma. ESMO Open 2016, 1, e00001. [Google Scholar] [CrossRef] [Green Version]
- Templeton, A.J.; Knox, J.J.; Lin, X.; Simantov, R.; Xie, W.; Lawrence, N.; Broom, R.; Fay, A.P.; Rini, B.; Donskov, F.; et al. Change in neutrophil-to-lymphocyte ratio in response to targeted therapy for metastatic renal cell carcinoma as a prognosticator and biomarker of efficacy. Eur. Urol. 2016, 70, 358–364. [Google Scholar] [CrossRef]
- Viers, B.R.; Houston Thompson, R.; Boorjian, S.A.; Boorjian, S.A.; Lohse, C.M.; Leibovich, B.C.; Tollefson, M.K. Preoperative neutrophil-lymphocyte ratio predicts death among patients with localized clear cell renal carcinoma undergoing nephrectomy. Urol. Oncol. 2014, 32, 1277–1284. [Google Scholar] [CrossRef]
- Beuselinck, B.; Job, S.; Becht, E.; Karadimou, A.; Verkarre, V.; Couchy, G.; Giraldo, N.; Rioux-Leclercq, N.; Molinié, V.; Sibony, M.; et al. Molecular subtypes of clear cell renal cell carcinoma are associated with sunitinib response in the metastatic setting. Clin. Cancer Res. 2015, 21, 1329–1339. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Zhou, C. The past, present and future of immunotherapy against tumor. Transl. Lung Cancer Res. 2015, 4, 253–264. [Google Scholar]
- Rini, B.I.; Huseni, M.; Atkins, M.; McDermott, D.; Powles, T.; Escudier, B.; Banchereau, R.; Liu, L.-F.; Leng, N.; Fan, J.; et al. Molecular correlates differentiate response to atezolizumab (atezo) + bevacizumab (bev) vs sunitinib (sun): Results from a phase iii study (immotion151) in untreated metastatic renal cell carcinoma (mrcc). Ann. Oncol. 2018, 29, viii724–viii725. [Google Scholar] [CrossRef]
- Rini, B. Vascular endothelial growth factor-targeted therapy in renal cell carcinoma: Current status and future directions. Clin. Cancer Res. 2007, 13, 1098–1106. [Google Scholar] [CrossRef]
- Jubb, M.; Pham, T.; Hanby, A.; Frantz, G.; Peale, F.; Wu, T.; Koeppen, H.; Hillan, K. Expression of vascular endothelial growth factor, hypoxia inducible factor 1alpha, and carbonic anhydrase IX in human tumours. J. Clin. Pathol. 2004, 57, 504–512. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ballesteros, P.Á.; Chamorro, J.; Román-Gil, M.S.; Pozas, J.; Gómez Dos Santos, V.; Granados, Á.R.; Grande, E.; Alonso-Gordoa, T.; Molina-Cerrillo, J. Molecular Mechanisms of Resistance to Immunotherapy and Antiangiogenic Treatments in Clear Cell Renal Cell Carcinoma. Cancers 2021, 13, 5981. https://doi.org/10.3390/cancers13235981
Ballesteros PÁ, Chamorro J, Román-Gil MS, Pozas J, Gómez Dos Santos V, Granados ÁR, Grande E, Alonso-Gordoa T, Molina-Cerrillo J. Molecular Mechanisms of Resistance to Immunotherapy and Antiangiogenic Treatments in Clear Cell Renal Cell Carcinoma. Cancers. 2021; 13(23):5981. https://doi.org/10.3390/cancers13235981
Chicago/Turabian StyleBallesteros, Pablo Álvarez, Jesús Chamorro, María San Román-Gil, Javier Pozas, Victoria Gómez Dos Santos, Álvaro Ruiz Granados, Enrique Grande, Teresa Alonso-Gordoa, and Javier Molina-Cerrillo. 2021. "Molecular Mechanisms of Resistance to Immunotherapy and Antiangiogenic Treatments in Clear Cell Renal Cell Carcinoma" Cancers 13, no. 23: 5981. https://doi.org/10.3390/cancers13235981
APA StyleBallesteros, P. Á., Chamorro, J., Román-Gil, M. S., Pozas, J., Gómez Dos Santos, V., Granados, Á. R., Grande, E., Alonso-Gordoa, T., & Molina-Cerrillo, J. (2021). Molecular Mechanisms of Resistance to Immunotherapy and Antiangiogenic Treatments in Clear Cell Renal Cell Carcinoma. Cancers, 13(23), 5981. https://doi.org/10.3390/cancers13235981