
Molecular Alterations Associated with Acquired Drug Resistance during Combined Treatment with Encorafenib and Binimetinib in Melanoma Cell Lines




Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell lines and Culture Reagents
2.2. Establishment of Resistant Cell Lines
2.3. Cell Proliferation Assay
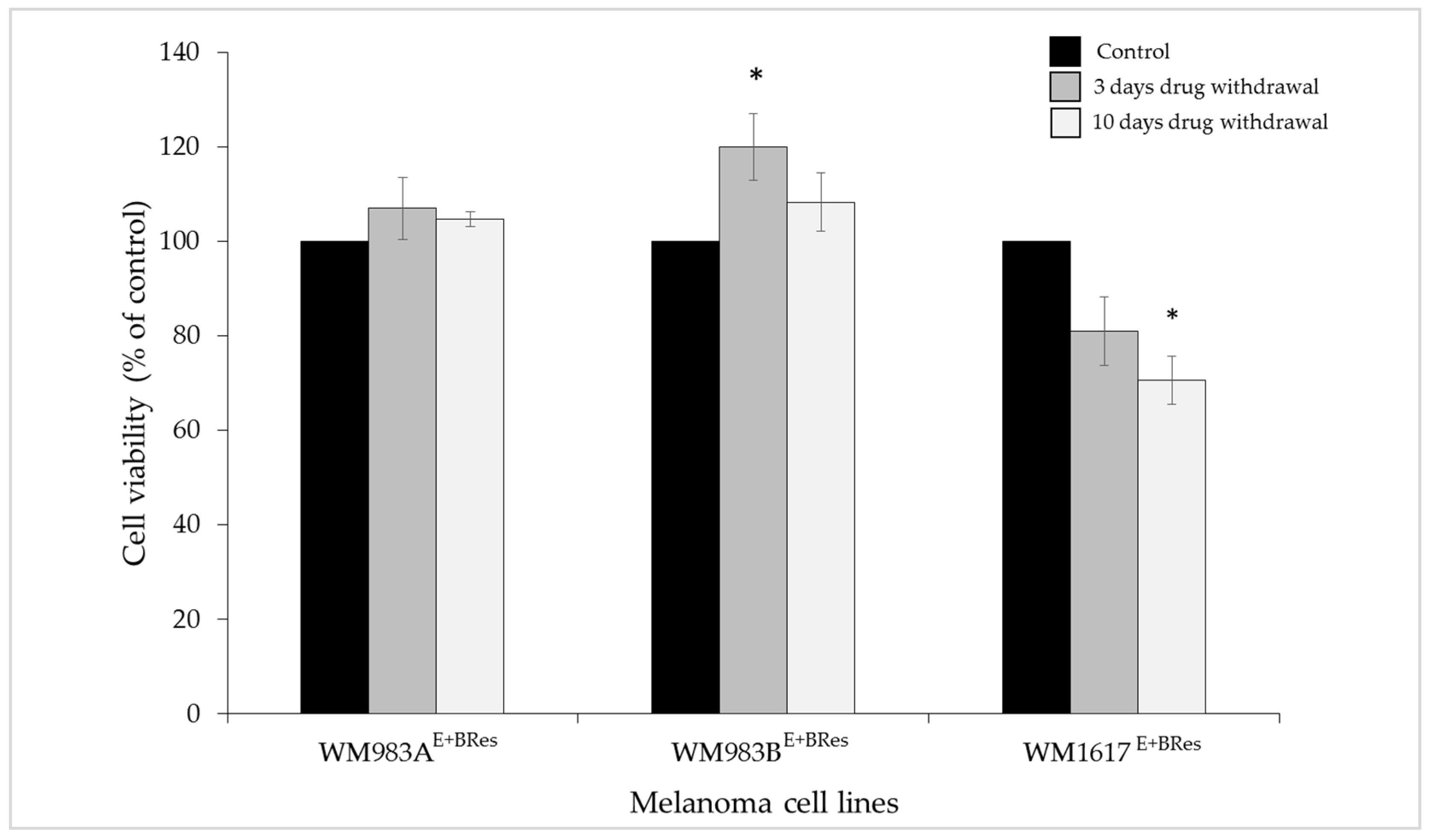
2.4. Drug Holiday Experiment
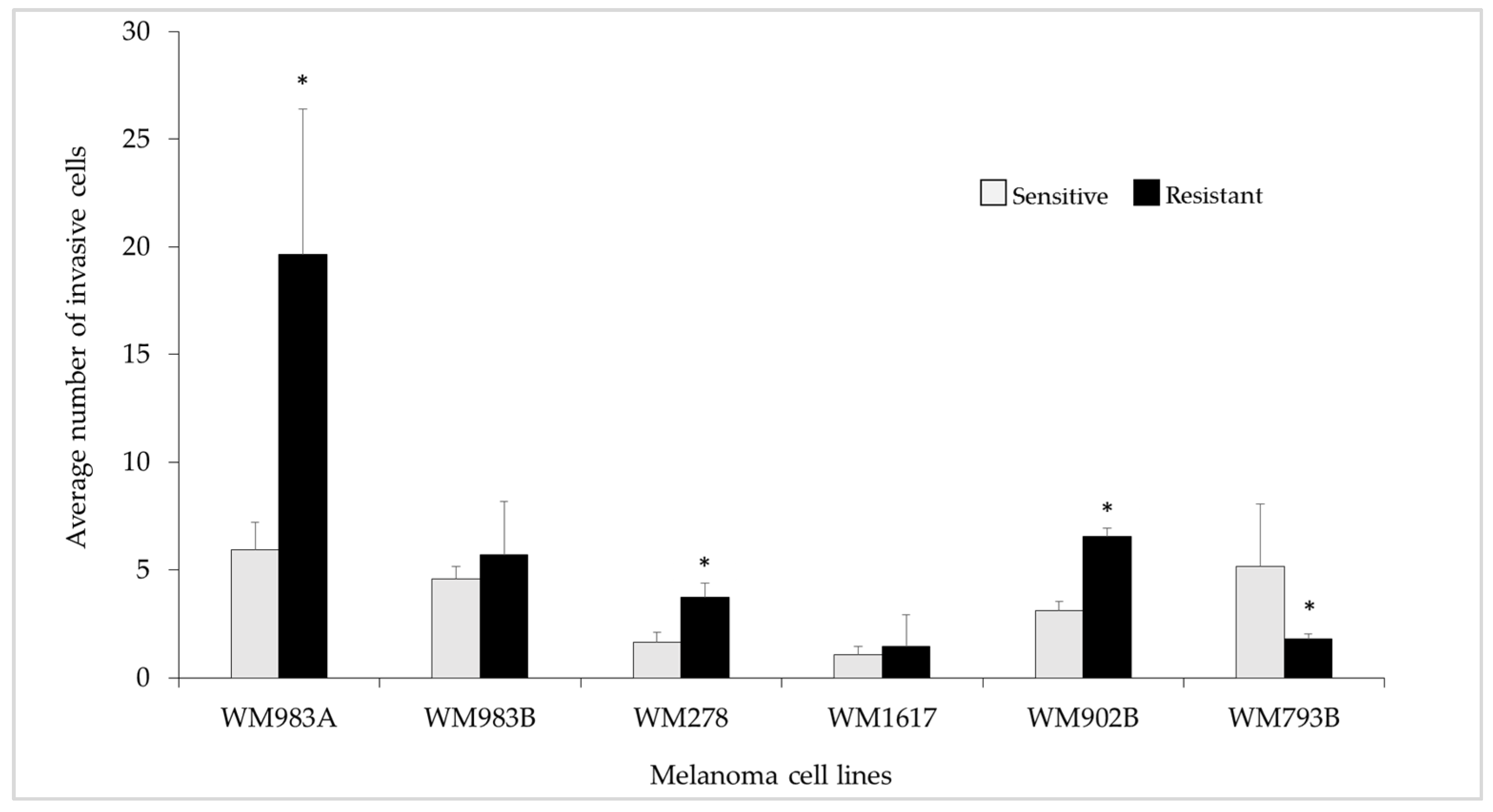
2.5. In Vitro Invasion Assay
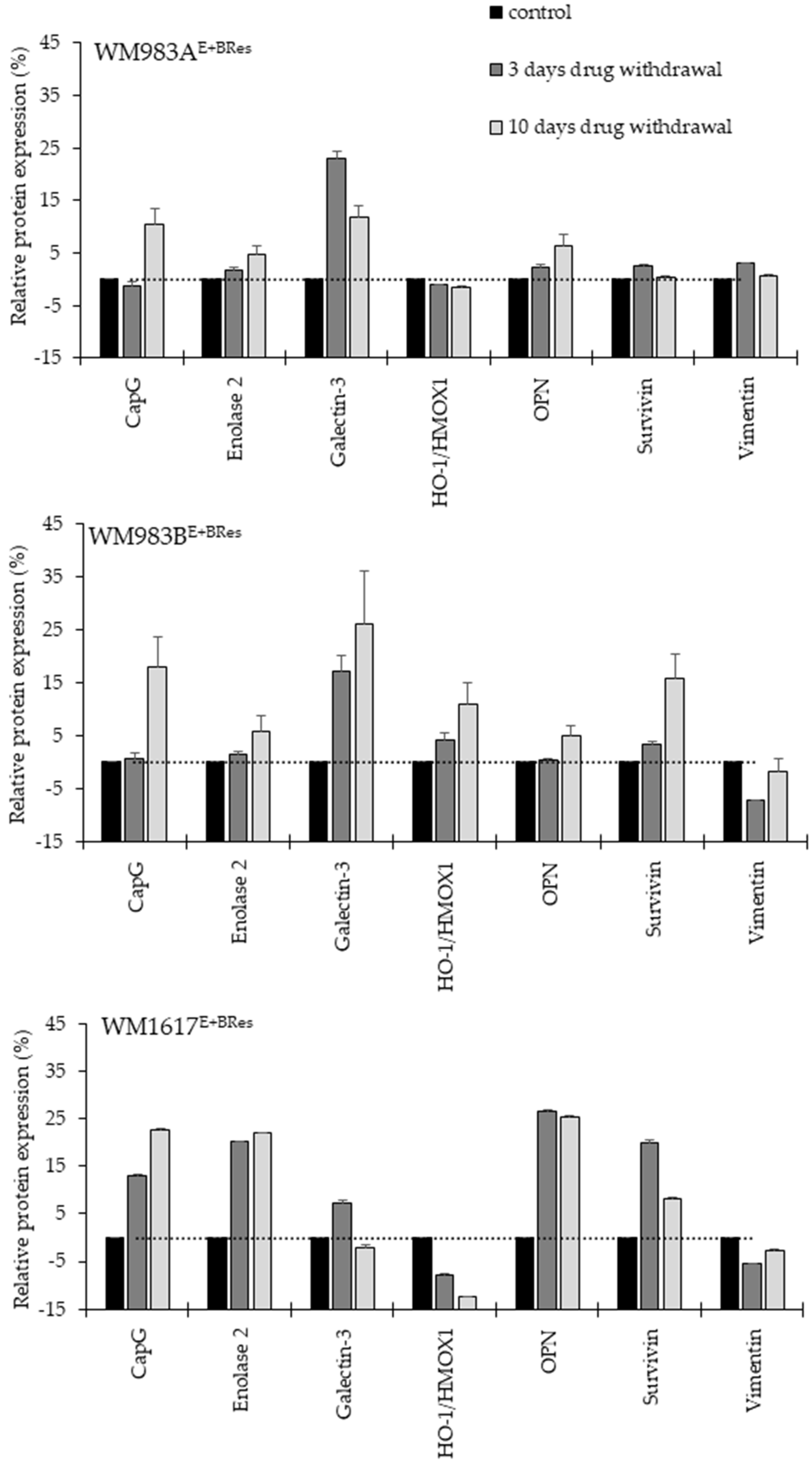
2.6. Protein Expression Analysis
2.7. RNA Isolation
2.8. RNA Sequencing and Data Analyses
2.9. Gene Expression Analysis
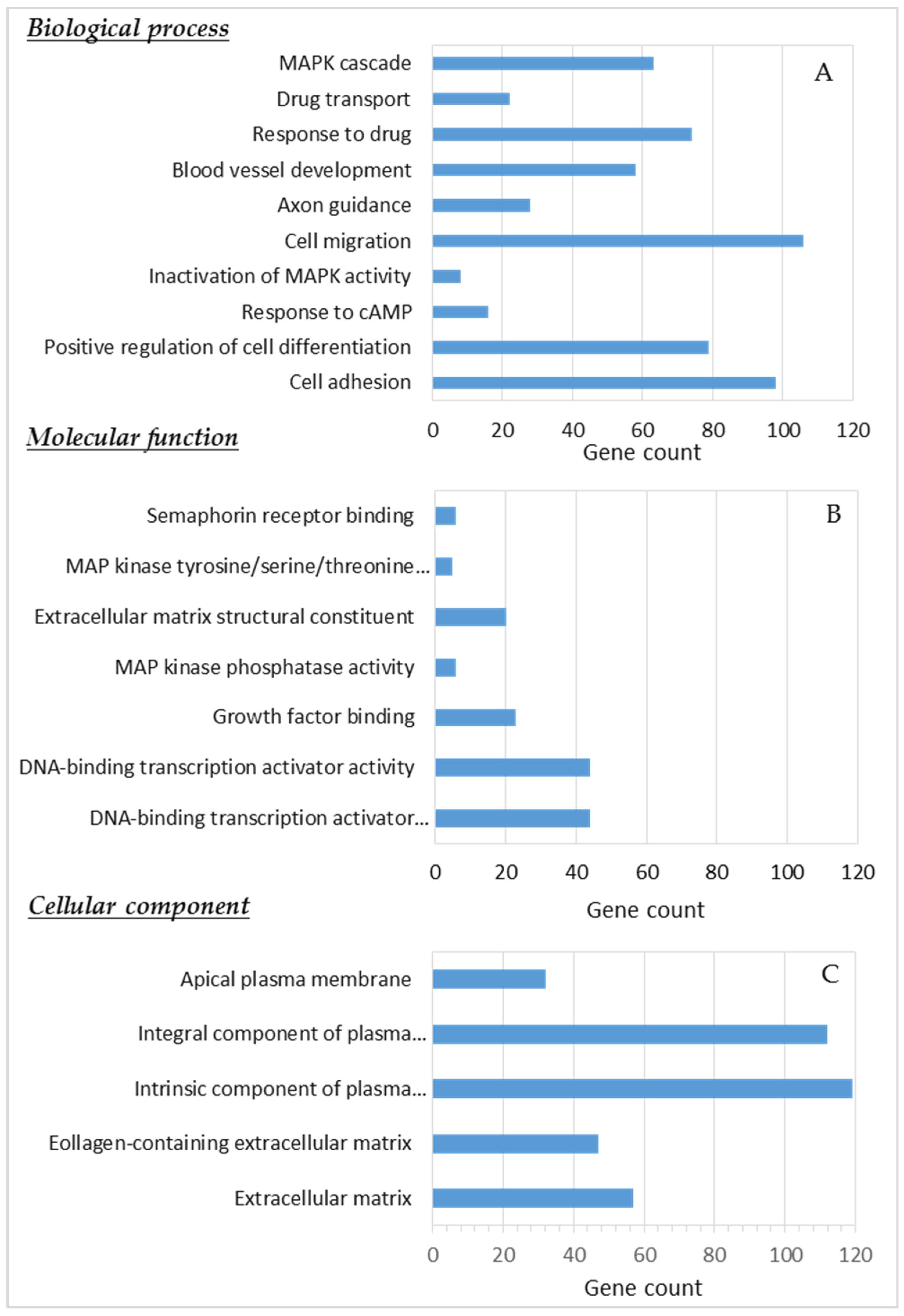
2.10. Gene Ontology Functional Analysis and Gene Set Enrichment Analysis (GSEA)
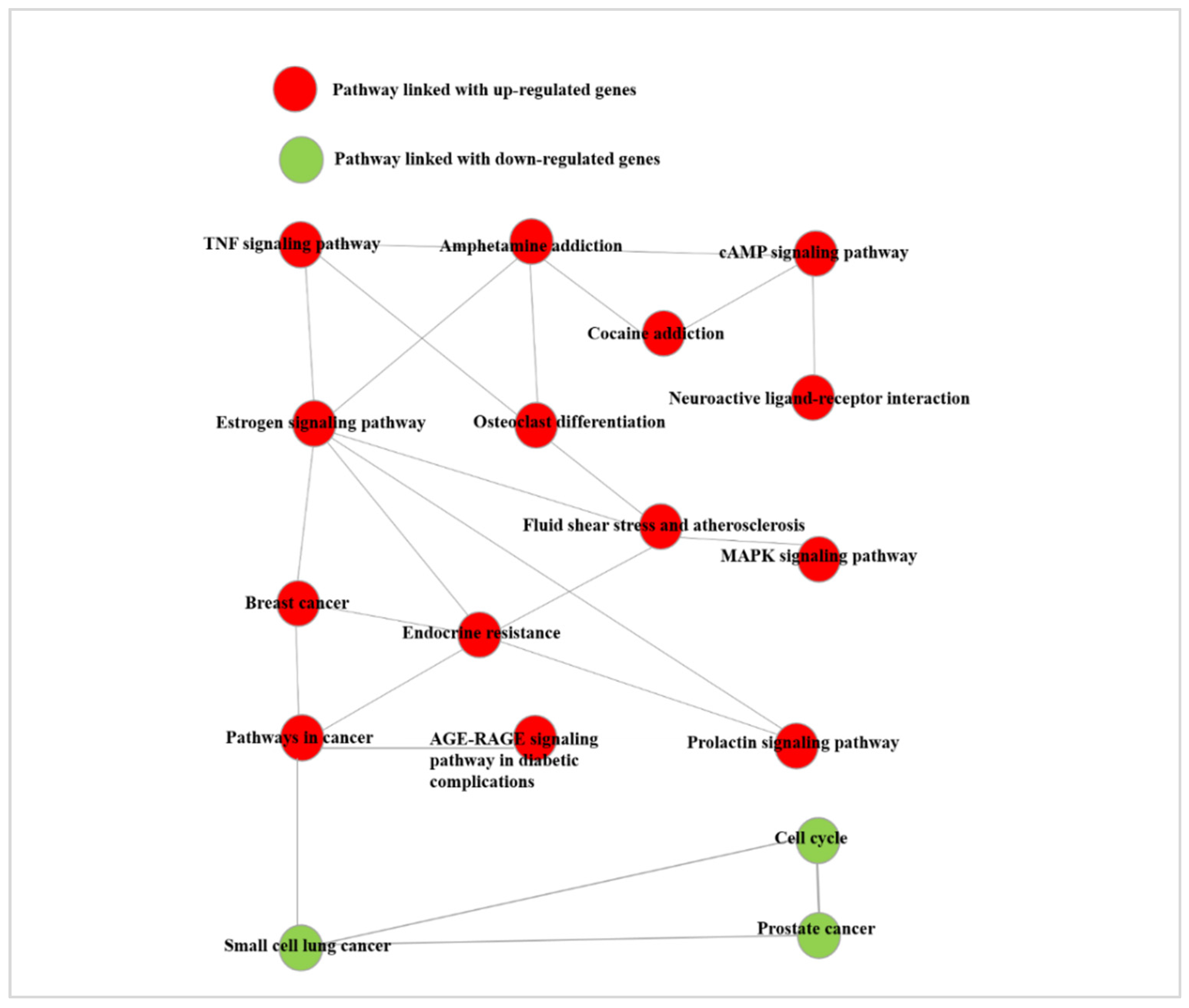
2.11. Pathway Analysis
2.12. Quantitative Real-Time PCR
2.13. Statistical Analysis
3. Results
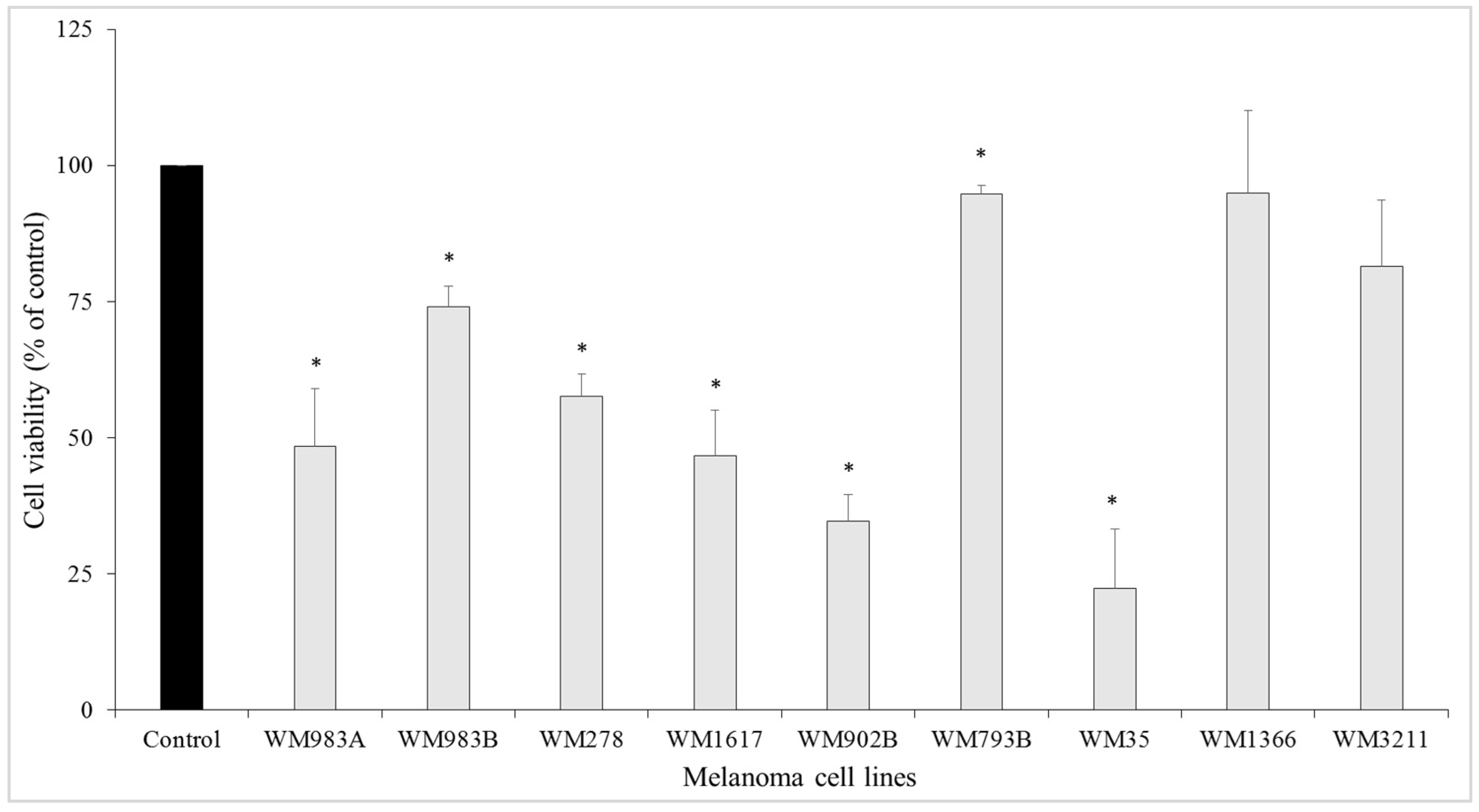
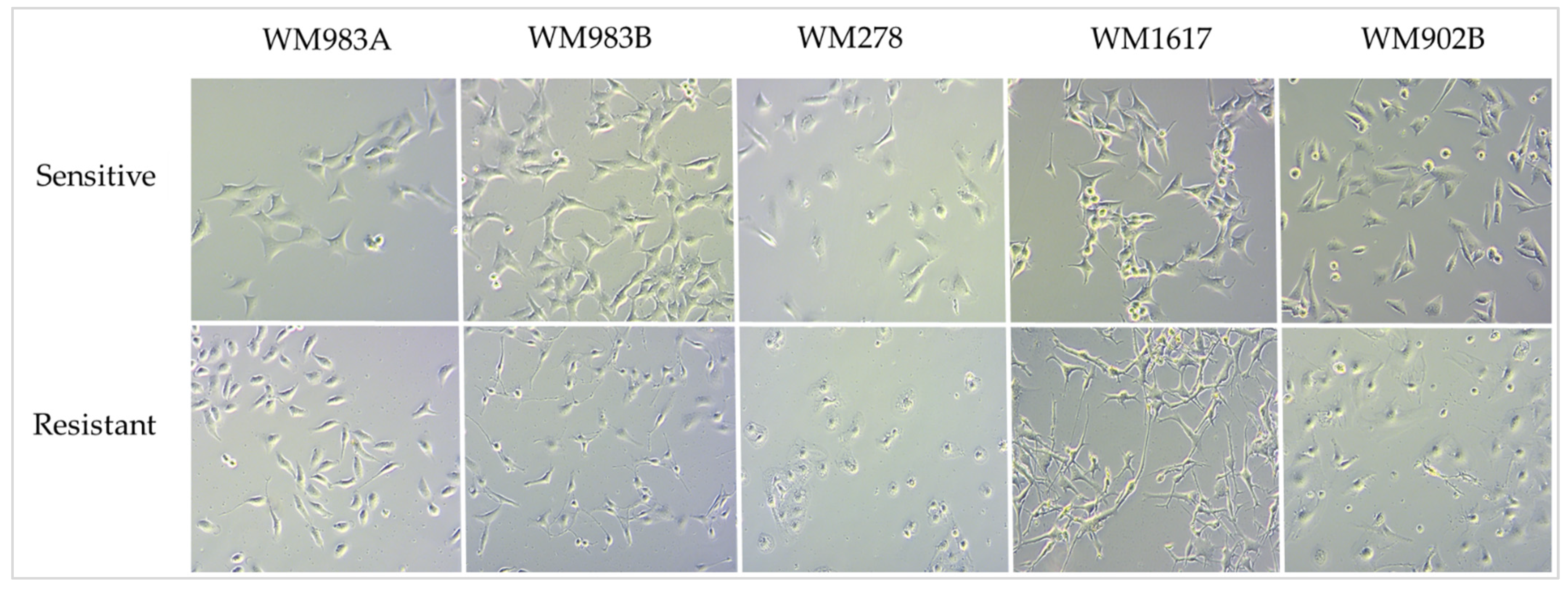
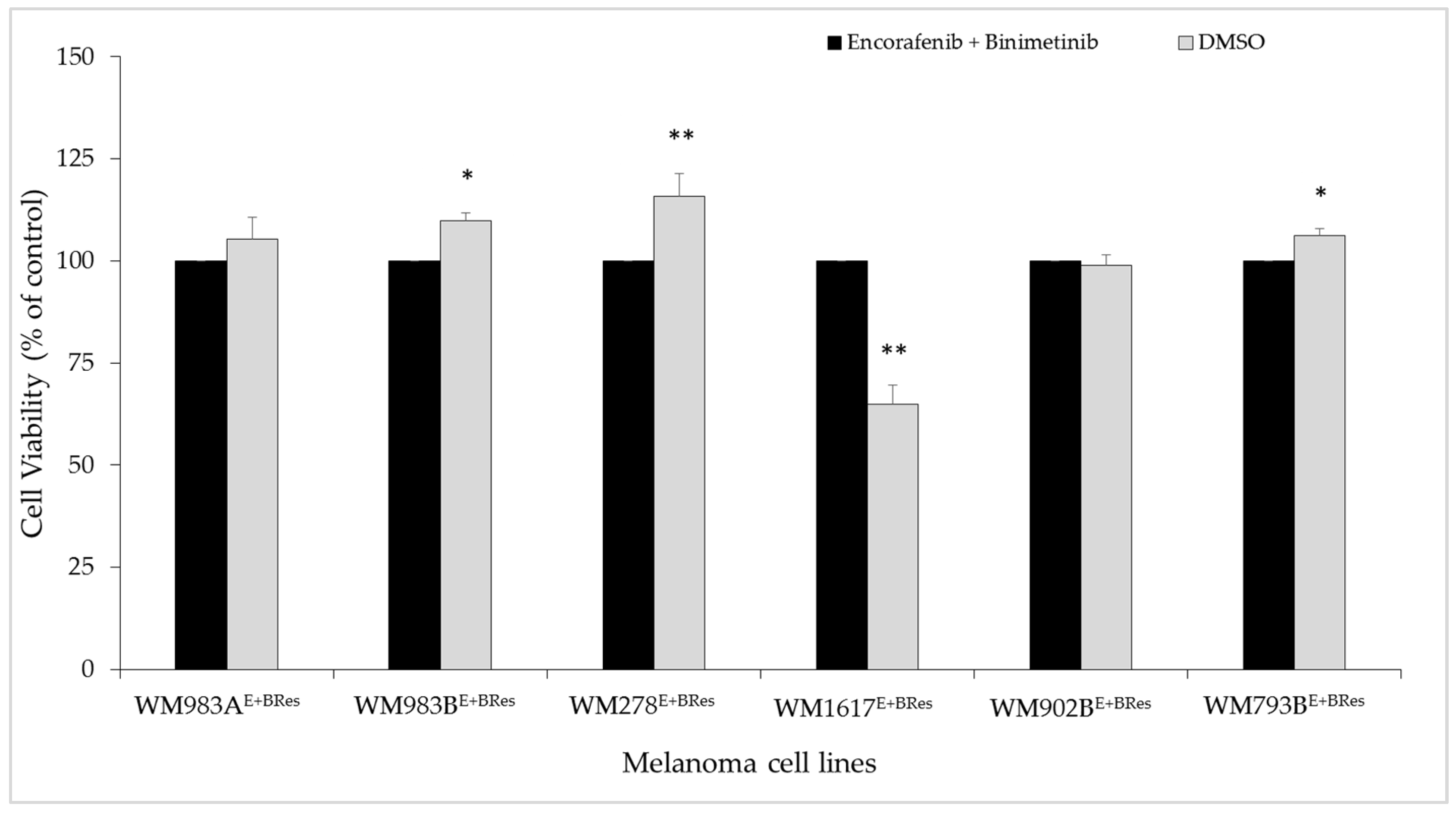
3.1. Growth-Inhibitory Effect of the ENCO+BINI Combination on Melanoma Cell Lines and the Development of Resistant Cell Lines
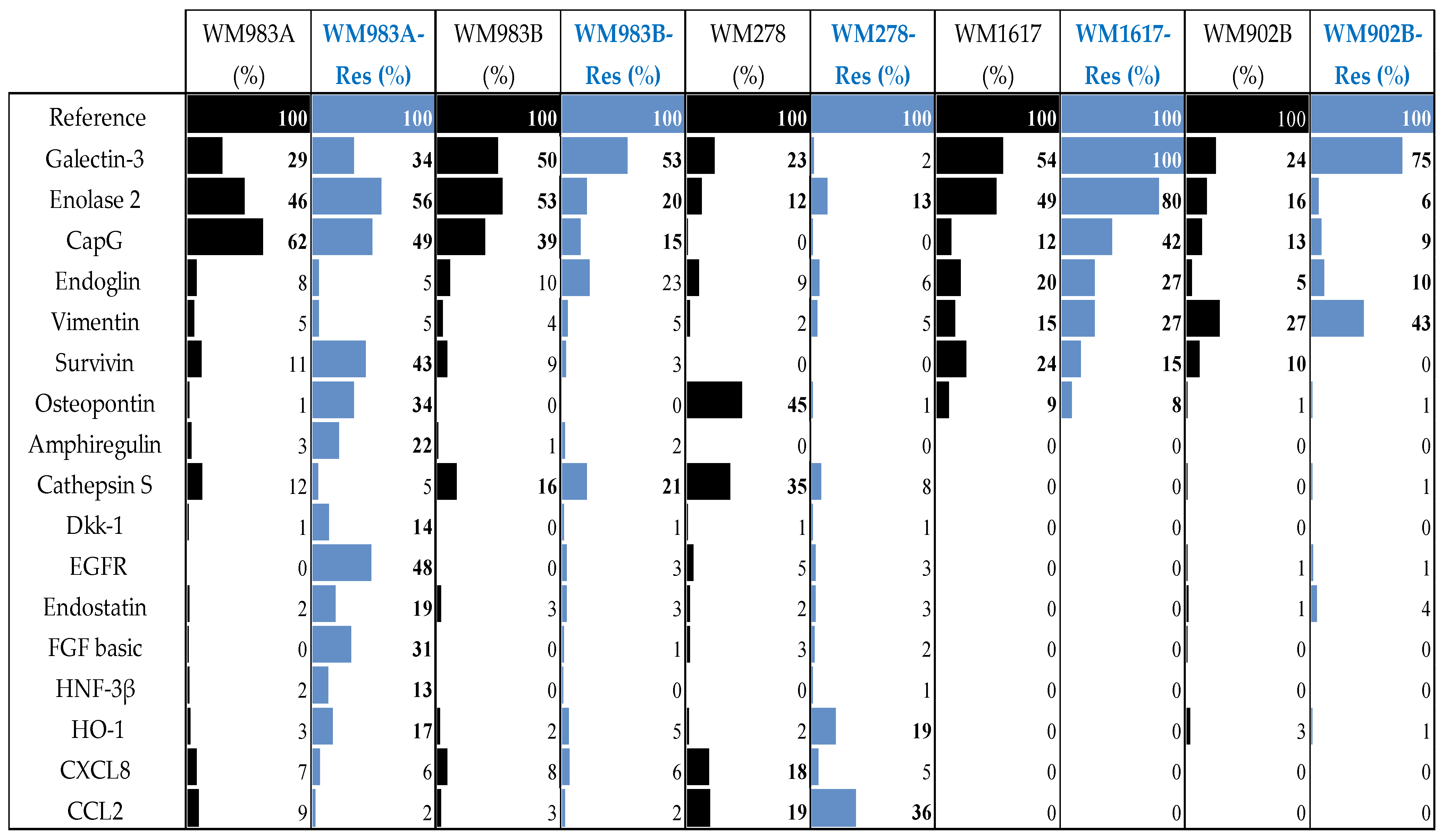
3.2. Protein Array Analysis of the Parental and Resistant Cell Lines
3.3. Effect of Drug Withdrawal on the Viability and Protein Expression of the Brafi/Meki Resistant Melanoma Cell Lines
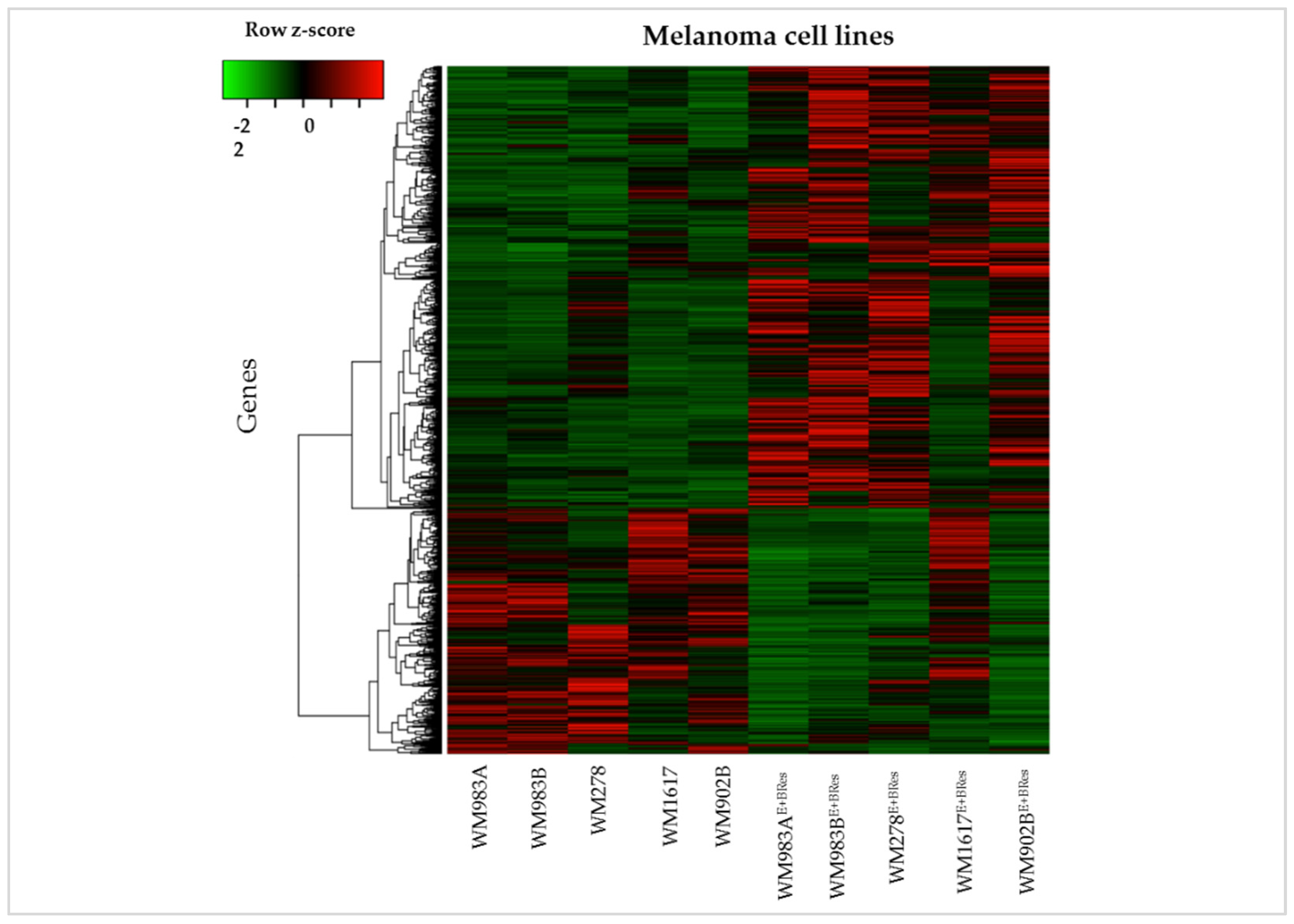
3.4. Identification of Differentially Expressed Genes in Resistant Melanoma Cell Lines Using RNA-Seq Analysis
3.5. GSEA of Differentially Expressed Genes
3.6. Pathway Analysis of Differentially Expressed Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Helgadottir, H.; Rocha Trocoli Drakensjo, I.; Girnita, A. Personalized Medicine in Malignant Melanoma: Towards Patient Tailored Treatment. Front. Oncol. 2018, 8, 202. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef] [Green Version]
- Garber, K. Melanoma Drug Vindicates Targeted Approach. Science 2009, 326, 1619. [Google Scholar] [CrossRef]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Tanda, E.T.; Vanni, I.; Boutros, A.; Andreotti, V.; Bruno, W.; Ghiorzo, P.; Spagnolo, F. Current State of Target Treatment in BRAF Mutated Melanoma. Front. Mol. Biosci. 2020, 7, 154. [Google Scholar] [CrossRef]
- Trojaniello, C.; Festino, L.; Vanella, V.; Ascierto, P.A. Encorafenib in combination with binimetinib for unresectable or metastatic melanoma with BRAF mutations. Expert Rev. Clin. Pharmacol. 2019, 12, 259–266. [Google Scholar] [CrossRef]
- Delord, J.P.; Robert, C.; Nyakas, M.; McArthur, G.A.; Kudchakar, R.; Mahipal, A.; Yamada, Y.; Sullivan, R.; Arance, A.; Kefford, R.F.; et al. Phase I Dose-Escalation and -Expansion Study of the BRAF Inhibitor Encorafenib (LGX818) in Metastatic BRAF-Mutant Melanoma. Clin. Cancer Res. 2017, 23, 5339–5348. [Google Scholar] [CrossRef] [Green Version]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.; Yacoub, N.; Mishra, R.; White, A.; Long, Y.; Alanazi, S.; Garrett, J.T. Current Advances in the Treatment of BRAF-Mutant Melanoma. Cancers 2020, 12, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Eroglu, Z.; Infante, J.; Patel, S.; Daud, A.; Johnson, D.B.; Gonzalez, R.; Kefford, R.; Hamid, O.; Schuchter, L.; et al. Long-Term Outcomes in Patients with BRAF V600-Mutant Metastatic Melanoma Who Received Dabrafenib Combined With Trametinib. J. Clin. Oncol. 2018, 36, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Proietti, I.; Skroza, N.; Bernardini, N.; Tolino, E.; Balduzzi, V.; Marchesiello, A.; Michelini, S.; Volpe, S.; Mambrin, A.; Mangino, G.; et al. Mechanisms of Acquired BRAF Inhibitor Resistance in Melanoma: A Systematic Review. Cancers 2020, 12, 2801. [Google Scholar] [CrossRef] [PubMed]
- Szasz, I.; Koroknai, V.; Kiss, T.; Vizkeleti, L.; Adany, R.; Balazs, M. Molecular alterations associated with acquired resistance to BRAFV600E targeted therapy in melanoma cells. Melanoma Res. 2019, 29, 390–400. [Google Scholar] [CrossRef]
- Koroknai, V.; Ecsedi, S.; Vízkeleti, L.; Kiss, T.; Szász, I.; Lukács, A.; Papp, O.; Ádány, R.; Balázs, M. Genomic profiling of invasive melanoma cell lines by array comparative genomic hybridization. Melanoma Res. 2016, 26, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth, B.B.; Arianti, R.; Shaw, A.; Vamos, A.; Vereb, Z.; Poliska, S.; Gyory, F.; Bacso, Z.; Fesus, L.; Kristof, E. FTO Intronic SNP Strongly Influences Human Neck Adipocyte Browning Determined by Tissue and PPARgamma Specific Regulation: A Transcriptome Analysis. Cells 2020, 9, 987. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.H.; Hwang, S.H.; Cho, H.S.; Lee, M. Differential Gene Expression Common to Acquired and Intrinsic Resistance to BRAF Inhibitor Revealed by RNA-Seq Analysis. Biomol. Ther. 2019, 27, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Czarnecka, A.M.; Bartnik, E.; Fiedorowicz, M.; Rutkowski, P. Targeted Therapy in Melanoma and Mechanisms of Resistance. Int. J. Mol. Sci. 2020, 21, 4576. [Google Scholar] [CrossRef]
- Moreira, A.; Heinzerling, L.; Bhardwaj, N.; Friedlander, P. Current Melanoma Treatments: Where Do We Stand? Cancers 2021, 13, 221. [Google Scholar] [CrossRef]
- Ziogas, D.C.; Konstantinou, F.; Bouros, S.; Theochari, M.; Gogas, H. Combining BRAF/MEK Inhibitors with Immunotherapy in the Treatment of Metastatic Melanoma. Am. J. Clin. Dermatol. 2021, 22, 301–314. [Google Scholar] [CrossRef]
- McClure, E.; Patel, A.; Carr, M.J.; Sun, J.; Zager, J.S. The combination of encorafenib and binimetinib for the treatment of patients with BRAF-mutated advanced, unresectable, or metastatic melanoma: An update. Expert Rev. Precis. Med. Drug Dev. 2021, 6, 19–29. [Google Scholar] [CrossRef]
- Haugh, A.M.; Salama, A.K.S.; Johnson, D.B. Advanced Melanoma: Resistance Mechanisms to Current Therapies. Hematol. Oncol. Clin. N. Am. 2021, 35, 111–128. [Google Scholar] [CrossRef]
- Dratkiewicz, E.; Simiczyjew, A.; Pietraszek-Gremplewicz, K.; Mazurkiewicz, J.; Nowak, D. Characterization of Melanoma Cell Lines Resistant to Vemurafenib and Evaluation of Their Responsiveness to EGFR- and MET-Inhibitor Treatment. Int. J. Mol. Sci. 2019, 21, 113. [Google Scholar] [CrossRef] [Green Version]
- Pouliquen, D.L.; Boissard, A.; Coqueret, O.; Guette, C. Biomarkers of tumor invasiveness in proteomics (Review). Int. J. Oncol. 2020, 57, 409–432. [Google Scholar] [CrossRef]
- Yu, C.F.; Chen, F.H.; Lu, M.H.; Hong, J.H.; Chiang, C.S. Dual roles of tumour cells-derived matrix metalloproteinase 2 on brain tumour growth and invasion. Br. J. Cancer 2017, 117, 1828–1836. [Google Scholar] [CrossRef] [Green Version]
- Das Thakur, M.; Salangsang, F.; Landman, A.S.; Sellers, W.R.; Pryer, N.K.; Levesque, M.P.; Dummer, R.; McMahon, M.; Stuart, D.D. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013, 494, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Tetu, P.; Vercellino, L.; Reger de Moura, C.; Baroudjian, B.; Dumaz, N.; Mourah, S.; Lebbe, C. Mitogen-activated protein kinase blockade in melanoma: Intermittent versus continuous therapy, from preclinical to clinical data. Curr. Opin. Oncol. 2021, 33, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Algazi, A.P.; Othus, M.; Daud, A.I.; Lo, R.S.; Mehnert, J.M.; Truong, T.-G.; Conry, R.; Kendra, K.; Doolittle, G.C.; Clark, J.I.; et al. Continuous versus intermittent BRAF and MEK inhibition in patients with BRAF-mutated melanoma: A randomized phase 2 trial. Nat. Med. 2020, 26, 1564–1568. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Kuilman, T.; Shahrabi, A.; Boshuizen, J.; Kemper, K.; Song, J.Y.; Niessen, H.W.M.; Rozeman, E.A.; Geukes Foppen, M.H.; Blank, C.U.; et al. Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature 2017, 550, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Mourad-Zeidan, A.A.; Melnikova, V.O.; Wang, H.; Raz, A.; Bar-Eli, M. Expression profiling of Galectin-3-depleted melanoma cells reveals its major role in melanoma cell plasticity and vasculogenic mimicry. Am. J. Pathol. 2008, 173, 1839–1852. [Google Scholar] [CrossRef] [Green Version]
- Braeuer, R.R.; Shoshan, E.; Kamiya, T.; Bar-Eli, M. The sweet and bitter sides of galectins in melanoma progression. Pigment Cell Melanoma Res. 2012, 25, 592–601. [Google Scholar] [CrossRef]
- Glinsky, V.V.; Kiriakova, G.; Glinskii, O.V.; Mossine, V.V.; Mawhinney, T.P.; Turk, J.R.; Glinskii, A.B.; Huxley, V.H.; Price, J.E.; Glinsky, G.V. Synthetic galectin-3 inhibitor increases metastatic cancer cell sensitivity to taxol-induced apoptosis in vitro and in vivo. Neoplasia 2009, 11, 901–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, S.; Kato, J.; Sawada, M.; Horimoto, K.; Okura, M.; Hida, T.; Uhara, H. Usefulness of neuron-specific enolase as a serum marker of metastatic melanoma. J. Dermatol. 2020, 47, 1141–1148. [Google Scholar] [CrossRef]
- Berthier-Vergnes, O.; El Kharbili, M.; de la Fouchardière, A.; Pointecouteau, T.; Verrando, P.; Wierinckx, A.; Lachuer, J.; Le Naour, F.; Lamartine, J. Gene expression profiles of human melanoma cells with different invasive potential reveal TSPAN8 as a novel mediator of invasion. Br. J. Cancer 2011, 104, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Molnar, E.; Garay, T.; Donia, M.; Baranyi, M.; Rittler, D.; Berger, W.; Timar, J.; Grusch, M.; Hegedus, B. Long-Term Vemurafenib Exposure Induced Alterations of Cell Phenotypes in Melanoma: Increased Cell Migration and Its Association with EGFR Expression. Int. J. Mol. Sci. 2019, 20, 4484. [Google Scholar] [CrossRef] [Green Version]
- Jain, T.; Bryce, A. Intermittent BRAF Inhibition Can Achieve Prolonged Disease Control in BRAF Mutant Melanoma. Cureus 2015, 7, e410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dooley, A.J.; Gupta, A.; Middleton, M.R. Ongoing Response in BRAF V600E-Mutant Melanoma after Cessation of Intermittent Vemurafenib Therapy: A Case Report. Target. Oncol. 2016, 11, 557–563. [Google Scholar] [CrossRef] [Green Version]
- Bradley, C.A. Understanding tumour drug addiction. Nat. Rev. Cancer 2017, 17, 634–635. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.; Moriceau, G.; Sun, L.; Lomeli, S.; Piva, M.; Damoiseaux, R.; Holmen, S.L.; Sharpless, N.E.; Hugo, W.; Lo, R.S. Exploiting Drug Addiction Mechanisms to Select against MAPKi-Resistant Melanoma. Cancer Discov. 2018, 8, 74–93. [Google Scholar] [CrossRef] [Green Version]
- Chi, Y.; Xue, J.; Huang, S.; Xiu, B.; Su, Y.; Wang, W.; Guo, R.; Wang, L.; Li, L.; Shao, Z.; et al. CapG promotes resistance to paclitaxel in breast cancer through transactivation of PIK3R1/P50. Theranostics 2019, 9, 6840–6855. [Google Scholar] [CrossRef]
- Fukumori, T.; Kanayama, H.-O.; Raz, A. The role of galectin-3 in cancer drug resistance. Drug Resist. Updates 2007, 10, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhe, N.; Wang, J.; Chen, S.; Lin, X.; Chai, Q.; Zhang, Y.; Zhao, J.; Fang, Q. Heme oxygenase-1 plays a crucial role in chemoresistance in acute myeloid leukemia. Hematology 2015, 20, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Krishnakumar, S.; Kanwar, R.K.; Cheung, C.H.A.; Kanwar, J.R. Clinical aspects for survivin: A crucial molecule for targeting drug-resistant cancers. Drug Discov. Today 2015, 20, 578–587. [Google Scholar] [CrossRef]
- Zhao, K.; Lu, Y.; Chen, Y.; Cheng, J.; Zhang, W. RNA sequencing data of Vemurafenib-resistant melanoma cells and parental cells. Data Brief. 2020, 30, 105610. [Google Scholar] [CrossRef]
- Anastas, J.N.; Kulikauskas, R.M.; Tamir, T.; Rizos, H.; Long, G.V.; von Euw, E.M.; Yang, P.T.; Chen, H.W.; Haydu, L.; Toroni, R.A.; et al. WNT5A enhances resistance of melanoma cells to targeted BRAF inhibitors. J. Clin. Investig. 2014, 124, 2877–2890. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Yan, S.; Xiao, S.; Xue, M. Knockdown of ALPK2 inhibits the development and progression of Ovarian Cancer. Cancer Cell Int. 2020, 20, 267. [Google Scholar] [CrossRef]
- Junnila, S.; Kokkola, A.; Karjalainen-Lindsberg, M.L.; Puolakkainen, P.; Monni, O. Genome-wide gene copy number and expression analysis of primary gastric tumors and gastric cancer cell lines. BMC Cancer 2010, 10, 73. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Lin, F.; Deng, X.; Shen, L.; Fang, Y.; Fei, Z.; Zhao, L.; Zhang, X.; Pan, H.; Xie, D.; et al. Profiling and bioinformatics analyses reveal differential circular RNA expression in radioresistant esophageal cancer cells. J. Transl. Med. 2016, 14, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Tun, H.W.; Marlow, L.A.; von Roemeling, C.A.; Cooper, S.J.; Kreinest, P.; Wu, K.; Luxon, B.A.; Sinha, M.; Anastasiadis, P.Z.; Copland, J.A. Pathway signature and cellular differentiation in clear cell renal cell carcinoma. PLoS ONE 2010, 5, e10696. [Google Scholar] [CrossRef]
- Sinnberg, T.; Levesque, M.P.; Krochmann, J.; Cheng, P.F.; Ikenberg, K.; Meraz-Torres, F.; Niessner, H.; Garbe, C.; Busch, C. Wnt-signaling enhances neural crest migration of melanoma cells and induces an invasive phenotype. Mol. Cancer 2018, 17, 59. [Google Scholar] [CrossRef] [Green Version]
- Shathasivam, P.; Kollara, A.; Spybey, T.; Park, S.; Clarke, B.; Ringuette, M.J.; Brown, T.J. VEPH1 expression decreases vascularisation in ovarian cancer xenografts and inhibits VEGFA and IL8 expression through inhibition of AKT activation. Br. J. Cancer 2017, 116, 1065–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Jia, X.M.; Gao, N.N.; Tang, H.; Huang, W.; Ning, N. Overexpressed VEPH1 inhibits epithelial-mesenchymal transition, invasion, and migration of human cutaneous melanoma cells through inactivating the TGF-β signaling pathway. Cell Cycle 2019, 18, 2860–2875. [Google Scholar] [CrossRef]
- Chen, D.; Lu, T.; Tan, J.; Li, H.; Wang, Q.; Wei, L. Long Non-coding RNAs as Communicators and Mediators Between the Tumor Microenvironment and Cancer Cells. Front. Oncol. 2019, 9, 739. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Xing, L.; Zhang, X.; Zhang, X. A Four-Pseudogene Classifier Identified by Machine Learning Serves as a Novel Prognostic Marker for Survival of Osteosarcoma. Genes 2019, 10, 414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, Z.; Xu, J.; Li, J.; Li, S.; Zhang, M.; Yang, D. Systematic identification of non-coding pharmacogenomic landscape in cancer. Nat. Commun. 2018, 9, 3192. [Google Scholar] [CrossRef]
- Dolcino, M.; Pelosi, A.; Fiore, P.F.; Patuzzo, G.; Tinazzi, E.; Lunardi, C.; Puccetti, A. Long Non-Coding RNAs Play a Role in the Pathogenesis of Psoriatic Arthritis by Regulating MicroRNAs and Genes Involved in Inflammation and Metabolic Syndrome. Front. Immunol. 2018, 9, 1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Forst, C.V.; Gordon, A.; Gussin, G.; Geber, A.B.; Fernandez, P.J.; Ding, T.; Lashua, L.; Wang, M.; Balmaseda, A.; et al. Characterization of antibiotic resistance and host-microbiome interactions in the human upper respiratory tract during influenza infection. Microbiome 2020, 8, 39. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line. | Sex/Age (Years) | Origin a | Growth Phase b | BRAF | NRAS | |
|---|---|---|---|---|---|---|
| Type c | Mutation | Mutation | ||||
| WM983A p1 | M/54 | Primary | VGP | NM | V600E | wt |
| WM983B p2 | - | Metastasis | - | - | V600E | wt |
| WM278 m1 | F/62 | Primary | VGP | NM | V600E | wt |
| WM1617 m2 | F/62 | Metastasis | - | - | V600E | wt |
| WM902B | F | Primary | VGP | SSM | V600E | wt |
| WM793B | M/37 | Primary | RGP/VGP | SSM | V600E | wt |
| WM35 | F/24 | Primary | RGP/VGP | SSM | V600E | wt |
| WM1366 | M/79 | Primary | VGP | - | wt | Q61L |
| WM3211 | M/74 | Primary | RGP | SSM | wt | wt |
| Upregulated Genes | Downregulated Genes | ||||
|---|---|---|---|---|---|
| Gene Symbol | Fold Change | p-Value | Gene Symbol | Fold Change | p-Value |
| CXCL12 | 73.055 | 0.030 | DMRT2 | −36.360 | 0.004 |
| COL5A1 | 45.342 | 0.009 | MRGPRX4 | −26.686 | 0.002 |
| ALPK2 | 37.803 | 0.005 | TRIM51 | −23.160 | 0.003 |
| ABCC3 | 22.176 | 0.005 | CTD-2207A17.1 | −23.002 | 0.049 |
| CHST15 | 21.430 | 0.021 | RP4-718J7.4 | −21.898 | 0.018 |
| RP11-326A19.5 | 21.394 | 0.033 | VEPH1 | −20.866 | 0.000 |
| LAMA5 | 21.264 | 0.010 | RP11-459E5.1 | −20.773 | 0.011 |
| SAMD11 | 20.976 | 0.003 | GJB1 | −20.765 | 0.028 |
| RP11-54O7.3 | 20.856 | 0.004 | ART3 | −20.169 | 0.015 |
| HHIPL2 | 20.509 | 0.004 | FABP7 | −19.772 | 0.022 |
| Name | ES | p-Value |
|---|---|---|
| REGULATION OF CELL POPULATION PROLIFERATION a | 0.219 | 0.013 |
| BIOLOGICAL ADHESION a | 0.190 | 0.027 |
| APOPTOTIC PROCESS a | 0.205 | 0.027 |
| REGULATION OF CELL DEATH a | 0.204 | 0.030 |
| RESPONSE TO DRUG a | 0.408 | 0.002 |
| RESPONSE TO OXYGEN LEVELS a | 0.367 | 0.008 |
| VASCULATURE DEVELOPMENT a | 0.251 | 0.010 |
| PROTEIN KINASE ACTIVITY a | 0.350 | 0.005 |
| REGULATION OF CELL DEVELOPMENT b | −0.202 | 0.016 |
| REGULATION OF CHROMOSOME ORGANIZATION b | −0.343 | 0.035 |
| REGENERATION b | −0.365 | 0.039 |
| GLIAL CELL DIFFERENTIATION b | −0.304 | 0.049 |
| Molecular Pathway. | p-Value | Genes Included |
|---|---|---|
| ATF-2 transcription factor network a | 1.71E-06 | JDP2, MMP2, FOS, ATF3, SOCS3, JUN, JUNB, JUND, DUSP1, DUSP8 |
| Ensemble of genes encoding the extracellular matrix and extracellular matrix-associated proteins a | 3.14E-06 | A2M, FSTL3, ADAM8, CCL26, SVEP1, FBLN1, SPON2, AGT, MMP2, MMP11, VEGFD, LTBP4, CXCL12, AMBP, NID2, SEMA4B, SEMA3C, SFRP4, SEMA4G, POSTN, MUC1, BDNF, C1QL1, MMP24, GPC1, NTF4, ARTN, MMP25, COL18A1, OVGP1, PCSK6, ANXA8L1, WNT4, NTN5, COL1A1, COL5A1, WNT6, TSKU, CSF1, IGFBP5, IGFBP6, SRGN, EGLN3, PLXDC1, LAMA5, HTRA3, EDIL3, LGALS9, MMRN2 |
| AP-1 transcription factor network a | 8.49E-06 | AGT, FOS, FOSB, ATF3, HLA-A, JUN, JUNB, JUND, BCL2L11, DUSP1 |
| LPA receptor-mediated events a | 2.01E-04 | MAPT, MMP2, FOS, SRC, LPAR2, SLC9A3R2, JUN, LPAR1 |
| Epithelial mesenchymal transition a | 6.13E-04 | IL32, OXTR, JUN, TAGLN, POSTN, NNMT, MAGEE1, GADD45B, BDNF, MMP2, TPM1, FBLN1, NID2, MYLK, FSTL3, COL1A1, SFRP4, CXCL12, COL5A1, BASP1, GPC1, EDIL3 |
| Reactive oxygen species pathway a | 1.18E-02 | PDLIM1, G6PD, ABCC1, GPX3, TXNRD1, JUNB, FTL |
| TNF-alpha signalling via NF-kB a | 1.46E-02 | JUN, SMAD3, GADD45B, CSF1, CEBPD, DUSP1, TNFRSF9, FOS, SOCS3, ZFP36, NFIL3, NINJ1, BCL3, FOSB, MAP3K8, JUNB, ATF3 |
| KRAS signalling up b | 6.06E-04 | ST6GAL1, MAP3K1, CCL20, IL10RA, RGS16, LIF, EMP1, PLAT, ETV1, ETV4, DUSP6, ETV5, PCSK1N, TSPAN13, C3AR1 |
| IL-2/STAT5 signalling b | 1.68E-03 | CD83, S100A1, IL10RA, RGS16, LIF, EMP1, ETV4, TIAM1, MAFF, BCL2, CTLA4, ITGA6, ICOS, SMPDL3A |
| TNF-alpha signalling via NF-kB b | 1.20E-02 | DUSP4, NR4A1, DUSP2, CD83, NR4A3, BCL2A1, CCL20, SPHK1, MAFF, LIF, TNC, PHLDA2 |
| Coagulation b | 1.69E-02 | ACOX2, S100A1, MAFF, S100A13, PLAT, CTSE, DUSP6, MBL2, CPN1 |
| Early oestrogen response b | 2.78E-02 | TIAM1, MREG, INPP5F, ELOVL2, MYB, TFF3, BCL2, HR, RAB17, NBL1, SLC19A2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, V.; Szász, I.; Koroknai, V.; Kiss, T.; Balázs, M. Molecular Alterations Associated with Acquired Drug Resistance during Combined Treatment with Encorafenib and Binimetinib in Melanoma Cell Lines. Cancers 2021, 13, 6058. https://doi.org/10.3390/cancers13236058
Patel V, Szász I, Koroknai V, Kiss T, Balázs M. Molecular Alterations Associated with Acquired Drug Resistance during Combined Treatment with Encorafenib and Binimetinib in Melanoma Cell Lines. Cancers. 2021; 13(23):6058. https://doi.org/10.3390/cancers13236058
Chicago/Turabian StylePatel, Vikas, István Szász, Viktória Koroknai, Tímea Kiss, and Margit Balázs. 2021. "Molecular Alterations Associated with Acquired Drug Resistance during Combined Treatment with Encorafenib and Binimetinib in Melanoma Cell Lines" Cancers 13, no. 23: 6058. https://doi.org/10.3390/cancers13236058
APA StylePatel, V., Szász, I., Koroknai, V., Kiss, T., & Balázs, M. (2021). Molecular Alterations Associated with Acquired Drug Resistance during Combined Treatment with Encorafenib and Binimetinib in Melanoma Cell Lines. Cancers, 13(23), 6058. https://doi.org/10.3390/cancers13236058