
Induction of Apoptosis in Human Pancreatic Cancer Stem Cells by the Endoplasmic Reticulum-Targeted Alkylphospholipid Analog Edelfosine and Potentiation by Autophagy Inhibition

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Pancreatic Cancer Spheroid Formation
2.4. Sorting of Cancer Stem Cells by Means of Flow Cytometry
2.5. Soft Agar Colony Assay
2.6. Xenograft Mouse Models
2.7. Apoptosis Analysis by Means of Flow Cytometry
2.8. Confocal Microscopy of Pancreatic Cancer Spheroids
2.9. Analysis of Subcellular Edelfosine Localization by Confocal Microscopy
2.10. Western Blotting
2.11. Primary Cultures from Pancreatic Cancer Patients
2.12. Chemograms
2.13. Statistical Analysis
3. Results
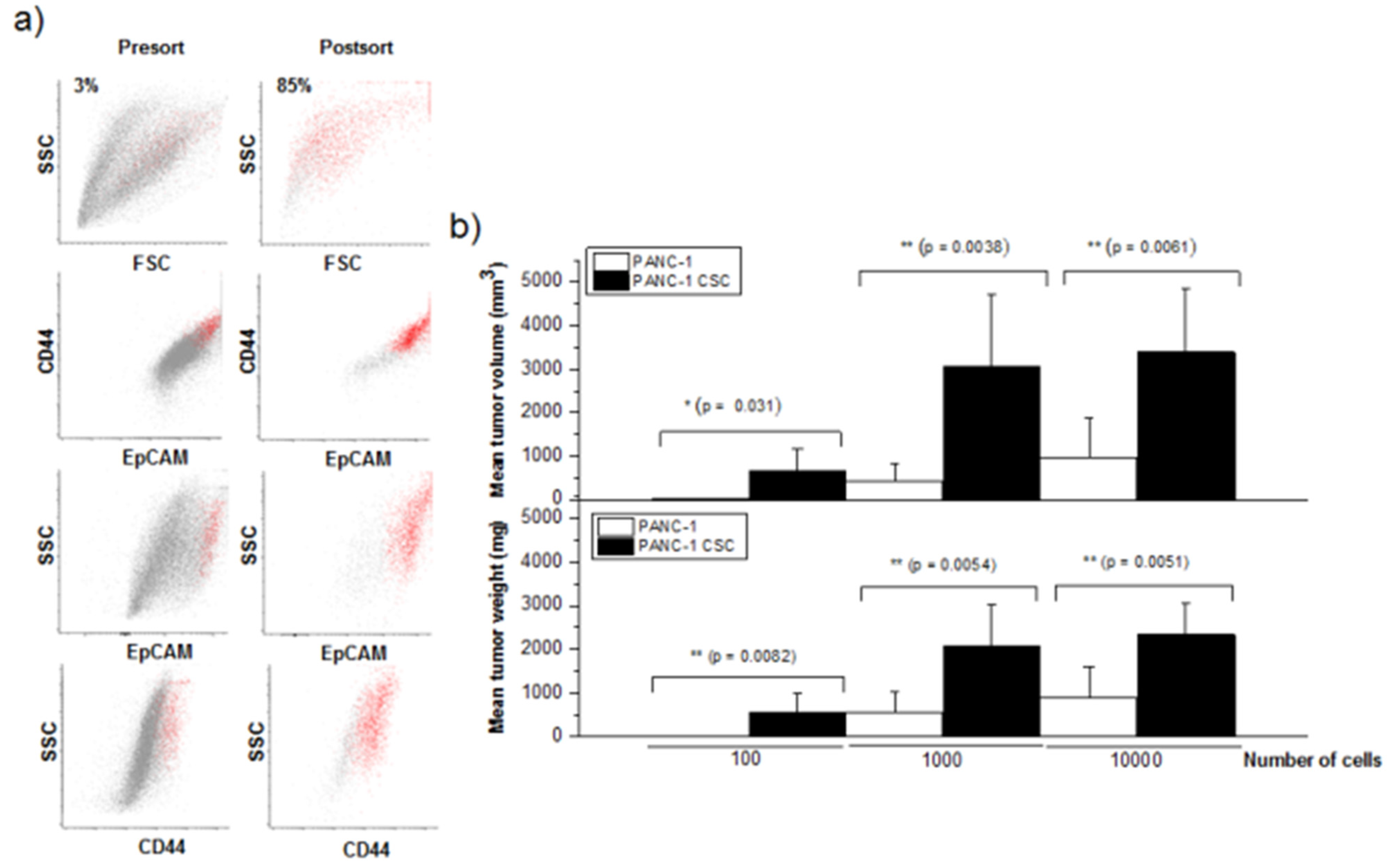
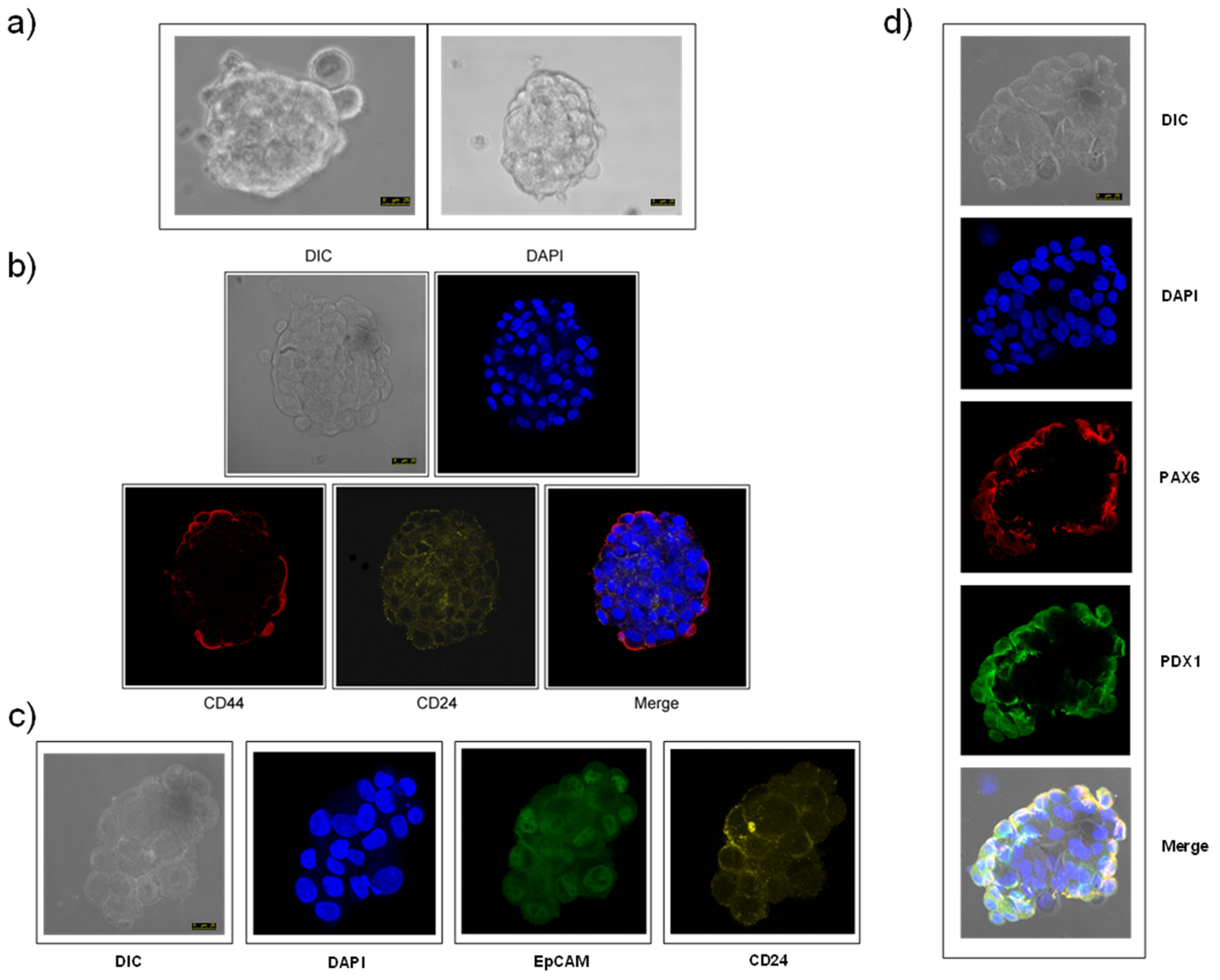
3.1. Isolation and Characterization of Human Pancreatic CSCs from the PANC-1 Cell Line
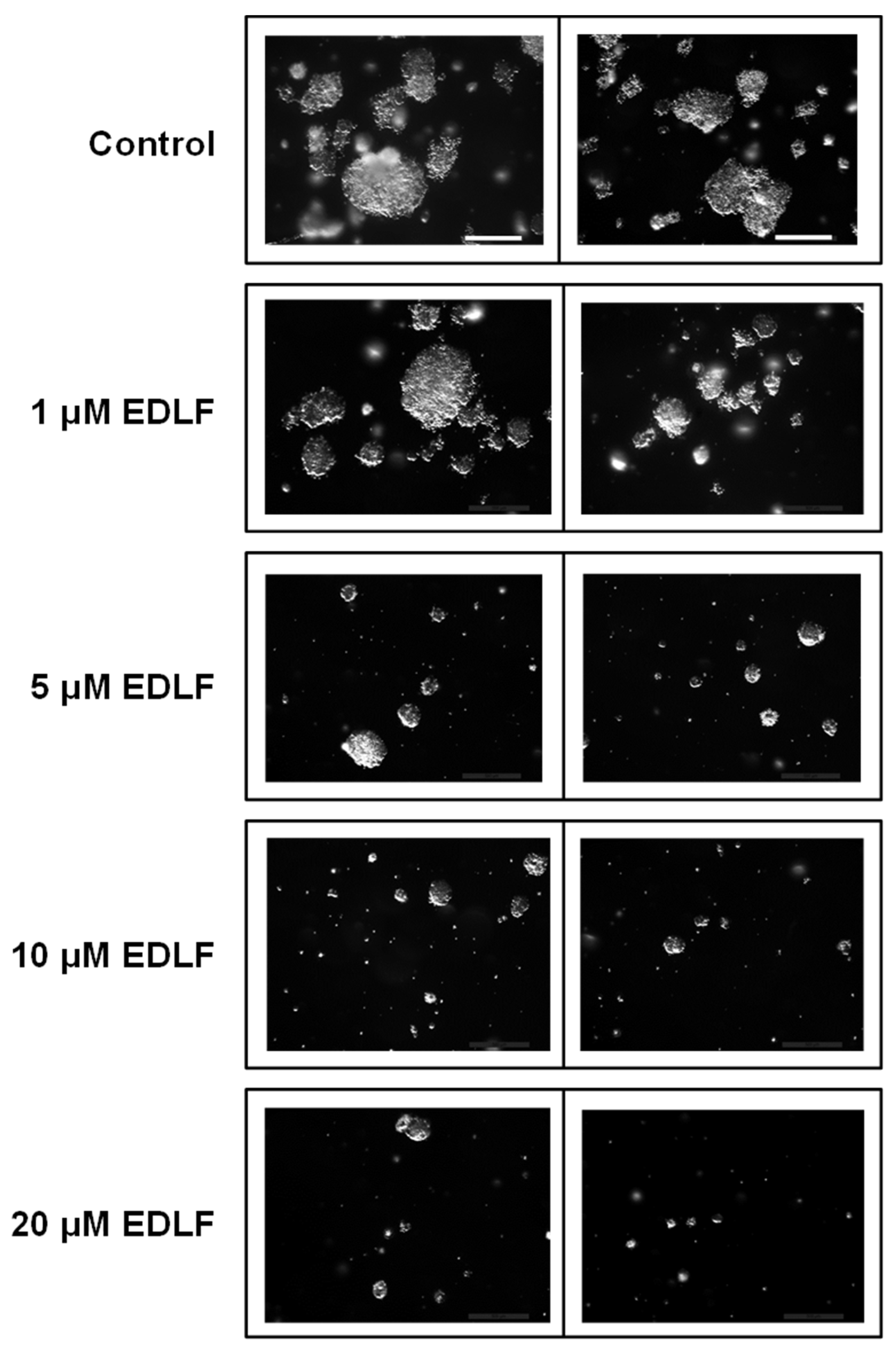
3.2. Edelfosine Inhibits Formation of CSC Pancreatic Cancer Spheroids
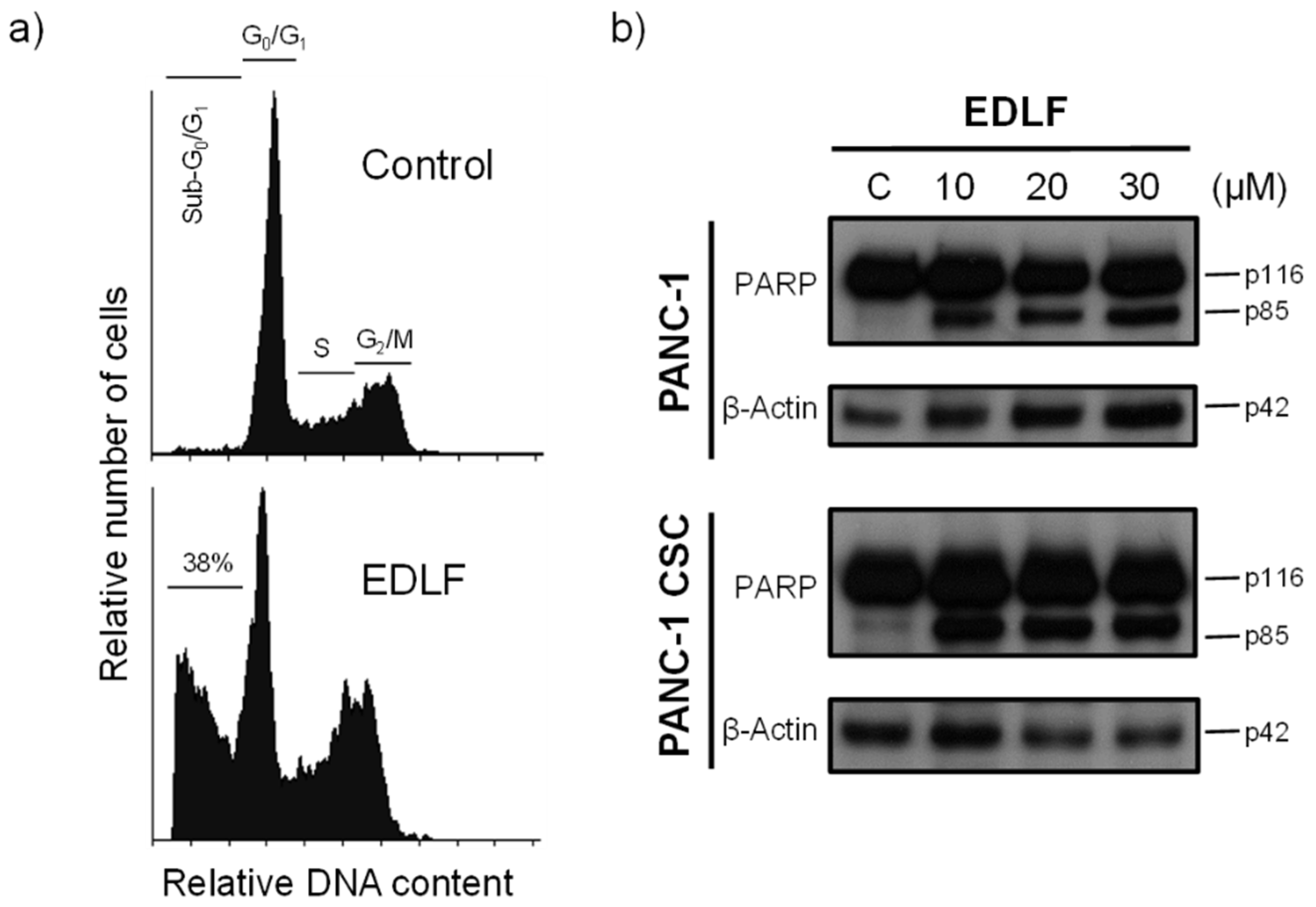
3.3. Edelfosine Induces Apoptosis in PANC-1 CSCs
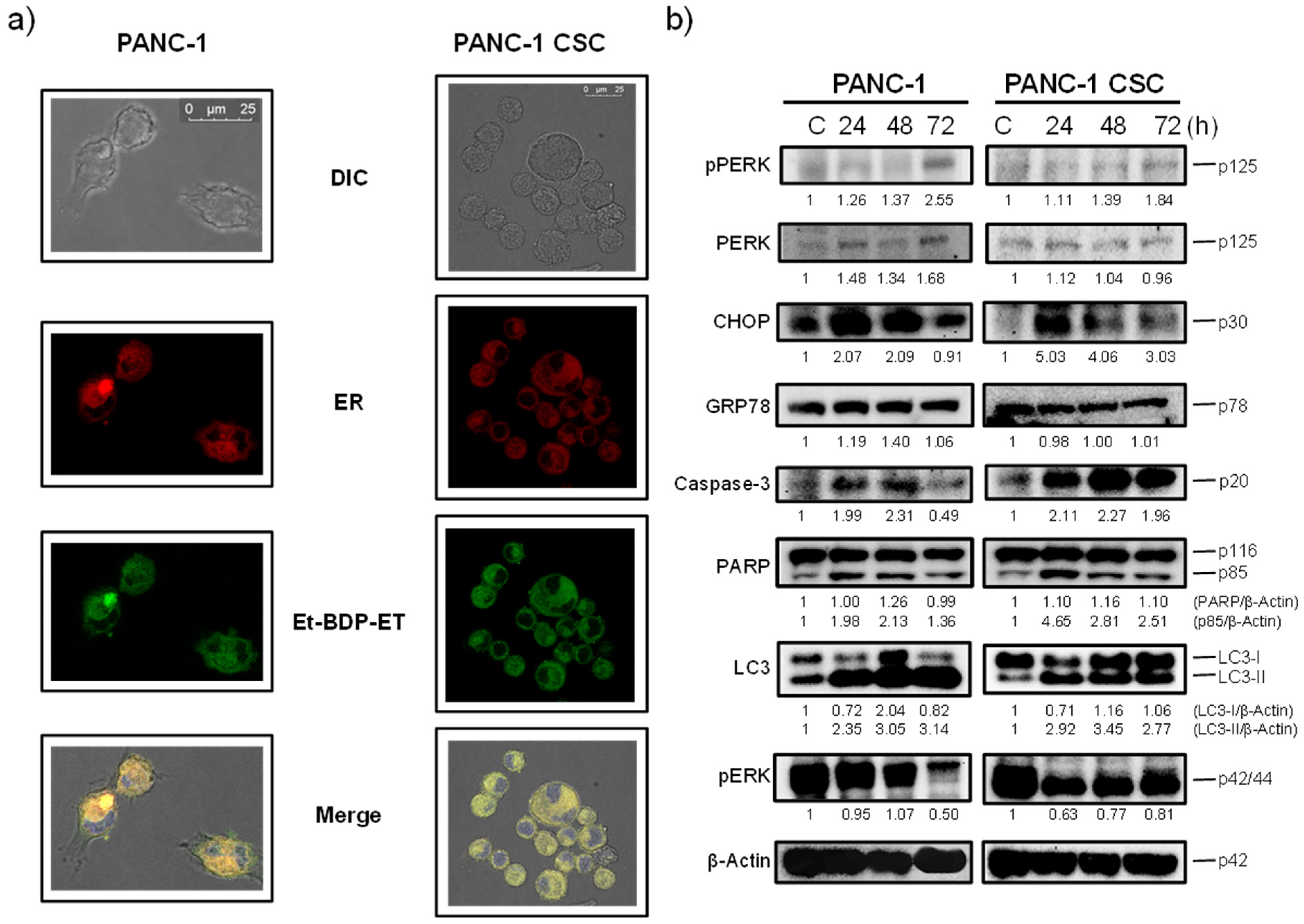
3.4. Edelfosine Accumulates in the ER of Parental PANC-1 and PANC-1 CSCs and Induces an ER Stress Response
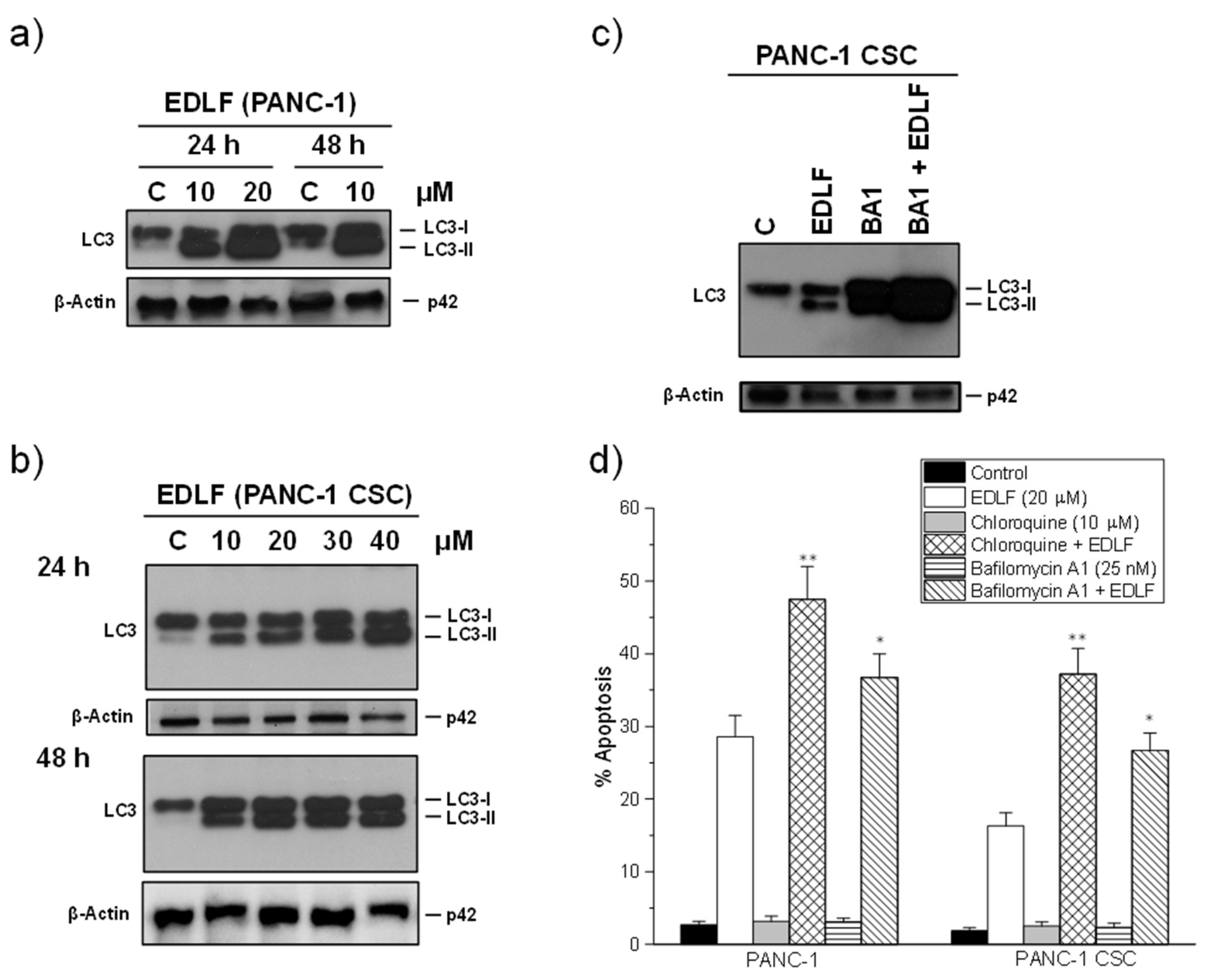
3.5. Autophagy Inhibitors Potentiate Edelfosine-Induced Apoptosis in PANC-1 and PANC-1 CSCs
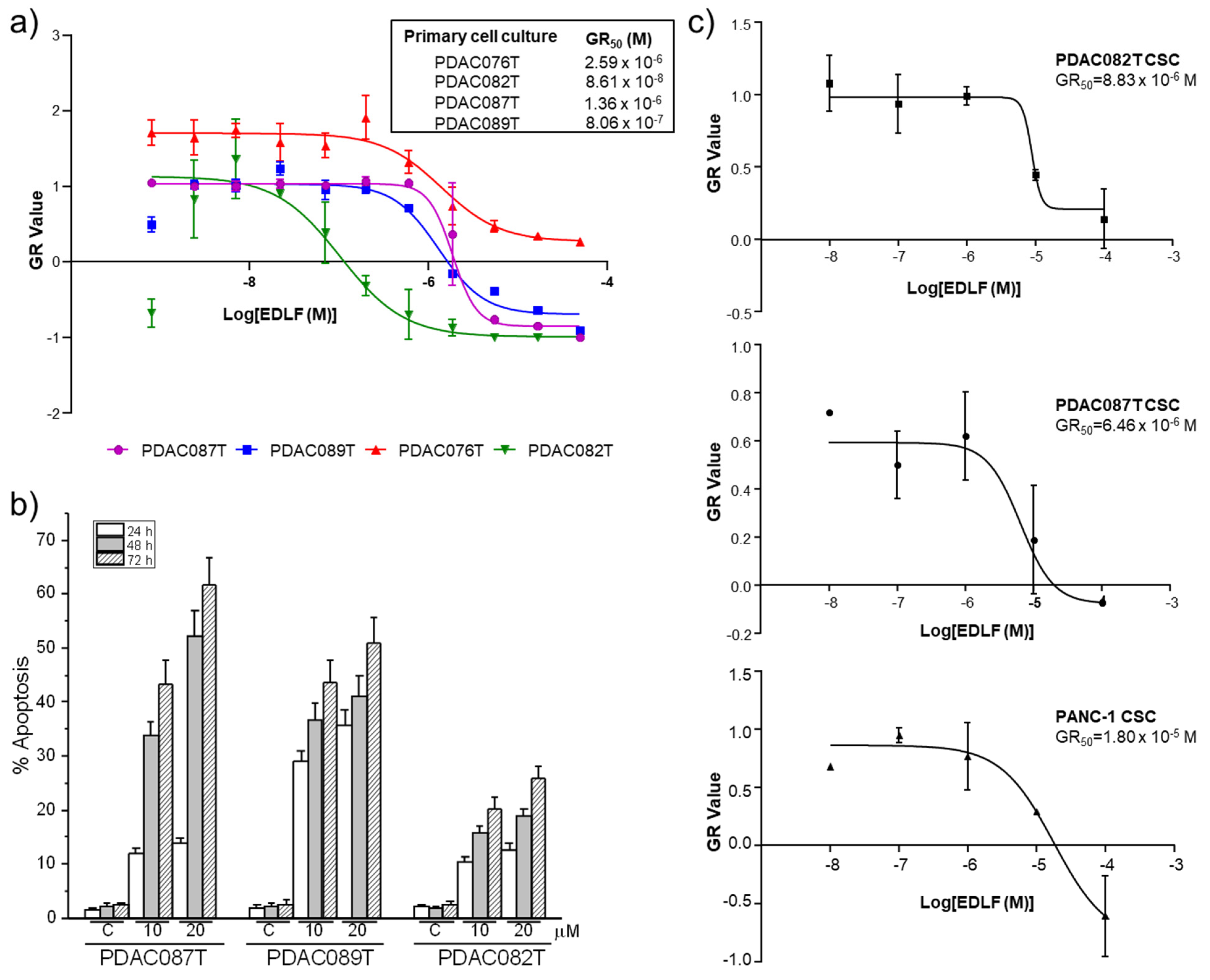
3.6. Edelfosine Inhibits Cell Proliferation and Induces Cell Death in Patient-Derived Primary Pancreatic Cancer Cell Cultures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mollinedo, F.; Gajate, C. Direct Endoplasmic Reticulum Targeting by the Selective Alkylphospholipid Analog and Antitumor Ether Lipid Edelfosine as a Therapeutic Approach in Pancreatic Cancer. Cancers 2021, 13, 4173. [Google Scholar] [CrossRef]
- WHO International Agency for Research on Cancer. The Global Cancer Observatory. Globocan 2020. 2021. Available online: https://gco.iarc.fr/today/data/factsheets/populations/900-world-fact-sheets.pdf (accessed on 20 August 2021).
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Li, D.; Xie, K.; Wolff, R.; Abbruzzese, J.L. Pancreatic cancer. Lancet 2004, 363, 1049–1057. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G. Cancer Stem Cells: Current Status and Evolving Complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2019, 17, 204–232. [Google Scholar] [CrossRef] [PubMed]
- Makena, M.R.; Ranjan, A.; Thirumala, V.; Reddy, A.P. Cancer stem cells: Road to therapeutic resistance and strategies to overcome resistance. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165339. [Google Scholar] [CrossRef]
- Zhou, H.-M.; Zhang, J.-G.; Zhang, X.; Li, Q. Targeting cancer stem cells for reversing therapy resistance: Mechanism, signaling, and prospective agents. Signal Transduct. Target. Ther. 2021, 6, 62. [Google Scholar] [CrossRef]
- Patil, K.; Khan, F.B.; Akhtar, S.; Ahmad, A.; Uddin, S. The plasticity of pancreatic cancer stem cells: Implications in therapeutic resistance. Cancer Metastasis Rev. 2021, 40, 691–720. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Crews, L.; Holm, F.; Jamieson, Q.J.L.A.C.F.H.C.H.M. RNA editing-dependent epitranscriptome diversity in cancer stem cells. Nat. Rev. Cancer 2017, 17, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Vila, M.; Takahashi, R.-U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [Green Version]
- Dallas, N.A.; Xia, L.; Fan, F.; Gray, M.J.; Gaur, P.; van Buren, G.; Samuel, S.; Kim, M.P.; Lim, S.J.; Ellis, L.M. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009, 69, 1951–1957. [Google Scholar] [CrossRef] [Green Version]
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.D.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.-Y.; et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajate, C.; Mollinedo, F. Biological Activities, Mechanisms of Action and Biomedical Prospect of the Antitumor Ether Phospholipid ET-18-OCH3 (Edelfosine), A Proapoptotic Agent in Tumor Cells. Curr. Drug Metab. 2002, 3, 491–525. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F.; Gajate, C.; Martin-Santamaria, S.; Gago, F. ET-18-OCH3 (Edelfosine): A Selective Antitumour Lipid Targeting Apoptosis Through Intracellular Activation of Fas/CD95 Death Receptor. Curr. Med. Chem. 2004, 11, 3163–3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollinedo, F. Antitumor ether lipids: Proapoptotic agents with multiple therapeutic indications. Expert Opin. Ther. Pat. 2007, 17, 385–405. [Google Scholar] [CrossRef]
- Mollinedo, F. Editorial (Thematic Issue: Antitumor Alkylphospholipid Analogs: A Promising and Growing Family of Synthetic Cell Membrane-Targeting Molecules for Cancer Treatment). Anti-Cancer Agents Med. Chem. 2014, 14, 495–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajate, C.; Mollinedo, F. The antitumor ether lipid ET-18-OCH3 induces apoptosis through translocation and capping of Fas/CD95 into membrane rafts in human leukemic cells. Blood 2001, 98, 3860–3863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajate, C.; Del Canto-Jañez, E.; Acuña, A.U.; Amat-Guerri, F.; Geijo-Barrientos, E.; Santos-Beneit, A.M.; Veldman, R.J.; Mollinedo, F. Intracellular Triggering of Fas Aggregation and Recruitment of Apoptotic Molecules into Fas-enriched Rafts in Selective Tumor Cell Apoptosis. J. Exp. Med. 2004, 200, 353–365. [Google Scholar] [CrossRef] [Green Version]
- Gajate, C.; Mollinedo, F. Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts. Blood 2007, 109, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto-Miguel, T.; Fonteriz, R.; Vay, L.; Gajate, C.; López-Hernández, S.; Mollinedo, F. Endoplasmic Reticulum Stress in the Proapoptotic Action of Edelfosine in Solid Tumor Cells. Cancer Res. 2007, 67, 10368–10378. [Google Scholar] [CrossRef] [Green Version]
- Gajate, C.; Matos-Da-Silva, M.; Dakir, E.H.; Fonteriz, R.; Alvarez, J.; Mollinedo, F. Antitumor alkyl-lysophospholipid analog edelfosine induces apoptosis in pancreatic cancer by targeting endoplasmic reticulum. Oncogene 2012, 31, 2627–2639. [Google Scholar] [CrossRef] [Green Version]
- Gajate, C.; Mollinedo, F. Lipid Rafts, Endoplasmic Reticulum and Mitochondria in the Antitumor Action of the Alkylphospholipid Analog Edelfosine. Anti-Cancer Agents Med. Chem. 2014, 14, 509–527. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, X.; Dakir, E.H.; Mollinedo, F.; Gajate, C. Endoplasmic reticulum targeting in Ewing’s sarcoma by the alkylphospholipid analog edelfosine. Oncotarget 2015, 6, 14596–14613. [Google Scholar] [CrossRef] [Green Version]
- Gajate, C.; Mollinedo, F. Lipid rafts and raft-mediated supramolecular entities in the regulation of CD95 death receptor apoptotic signaling. Apoptosis 2015, 20, 584–606. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid rafts as signaling hubs in cancer cell survival/death and invasion: Implications in tumor progression and therapy. J. Lipid Res. 2020, 61, 611–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollinedo, F.; de la Iglesia-Vicente, J.; Gajate, C.; Estella-Hermoso de Mendoza, A.; Villa-Pulgarin, J.; Campanero, M.; Blanco-Prieto, M.J. Lipid raft-targeted therapy in multiple myeloma. Oncogene 2010, 29, 3748–3757. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F.; de la Iglesia-Vicente, J.; Gajate, C.; Estella-Hermoso de Mendoza, A.; Villa-Pulgarin, J.A.; de Frias, M.; Roué, G.; Gil, J.; Colomer, D.; Campanero, M.A.; et al. In vitro and In vivo Selective Antitumor Activity of Edelfosine against Mantle Cell Lymphoma and Chronic Lymphocytic Leukemia Involving Lipid Rafts. Clin. Cancer Res. 2010, 16, 2046–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollinedo, F.; Fernández-Luna, J.L.; Gajate, C.; Martín-Martín, B.; Benito, A.; Martínez-Dalmau, R.; Modolell, M. Selective induction of apoptosis in cancer cells by the ether lipid ET-18-OCH3 (Edelfosine): Molecular structure requirements, cellular uptake, and protection by Bcl-2 and Bcl-X(L). Cancer Res. 1997, 57, 1320–1328. [Google Scholar]
- Gajate, C.; Fonteriz, R.; Cabaner, C.; Alvarez-Noves, G.; Alvarez-Rodriguez, Y.; Modolell, M.; Mollinedo, F. Intracellular triggering of Fas, independently of FasL, as a new mechanism of antitumor ether lipid-induced apoptosis. Int. J. Cancer 2000, 85, 674–682. [Google Scholar] [CrossRef]
- Oyadomari, S.; Araki, E.; Mori, M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis 2002, 7, 335–345. [Google Scholar] [CrossRef]
- Hu, Y.; Fu, L. Targeting cancer stem cells: A new therapy to cure cancer patients. Am. J. Cancer Res. 2012, 2, 340–356. [Google Scholar]
- Chen, L.-S.; Wang, A.-X.; Dong, B.; Pu, K.-F.; Yuan, L.-H.; Zhu, Y.-M. A new prospect in cancer therapy: Targeting cancer stem cells to eradicate cancer. Chin. J. Cancer 2012, 31, 564–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Nguyen, C.; Ulrich, A.; Pour, P.; Ouellette, M. Immortalization with telomerase of the Nestin-positive cells of the human pancreas. Biochem. Biophys. Res. Commun. 2003, 301, 1038–1044. [Google Scholar] [CrossRef]
- Gajate, C.; Barasoain, I.; Andreu, J.M.; Mollinedo, F. Induction of apoptosis in leukemic cells by the reversible microtubule-disrupting agent 2-methoxy-5-(2′,3′,4′-trimethoxyphenyl)-2,4,6-cycloheptatrien-1-one: Protection by Bcl-2 and Bcl-X(L) and cell cycle arrest. Cancer Res. 2000, 60, 2651–2659. [Google Scholar] [PubMed]
- Gajate, C.; Santos-Beneit, A.M.; Macho, A.; Lazaro, M.D.C.; de Rojas, A.H.; Modolell, M.; Mollinedo, F. Involvement of mitochondria and caspase-3 in ET-18-OCH3-induced apoptosis of human leukemic cells. Int. J. Cancer 2000, 86, 208–218. [Google Scholar] [CrossRef]
- Gajate, C.; Mollinedo, F. Lipid Raft Isolation by Sucrose Gradient Centrifugation and Visualization of Raft-Located Proteins by Fluorescence Microscopy: The Use of Combined Techniques to Assess Fas/CD95 Location in Rafts During Apoptosis Triggering. Methods Mol. Biol. 2021, 2187, 147–186. [Google Scholar] [CrossRef] [PubMed]
- Martin-Martin, B.; Nabokina, S.M.; Blasi, J.; Lazo, P.A.; Mollinedo, F. Involvement of SNAP-23 and syntaxin 6 in human neutrophil exocytosis. Blood 2000, 96, 2574–2583. [Google Scholar] [CrossRef]
- Mollinedo, F.; Fernández, M.; Hornillos, V.; Delgado, J.; Amat-Guerri, F.; Acuña, A.U.; Nieto-Miguel, T.; Villa-Pulgarín, J.; González-García, C.; Ceña, V.; et al. Involvement of lipid rafts in the localization and dysfunction effect of the antitumor ether phospholipid edelfosine in mitochondria. Cell Death Dis. 2011, 2, e158. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- García-Navas, R.; Gajate, C.; Mollinedo, F. Neutrophils drive endoplasmic reticulum stress-mediated apoptosis in cancer cells through arginase-1 release. Sci. Rep. 2021, 11, 12574. [Google Scholar] [CrossRef] [PubMed]
- Gayet, O.; Loncle, C.; Duconseil, P.; Gilabert, M.; Lopez, M.B.; Moutardier, V.; Turrini, O.; Calvo, E.; Ewald, J.; Giovannini, M.; et al. A subgroup of pancreatic adenocarcinoma is sensitive to the 5-aza-dC DNA methyltransferase inhibitor. Oncotarget 2011, 6, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, F.S.; Deramaudt, T.B.; Brunner, T.B.; Boretti, M.I.; Gooch, K.J.; Stoffers, D.A.; Bernhard, E.J.; Rustgi, A.K. Successful growth and characterization of mouse pancreatic ductal cells: Functional properties of the Ki-RAS(G12V) oncogene. Gastroenterology 2004, 127, 250–260. [Google Scholar] [CrossRef]
- David-Cordonnier, M.-H.; Gajate, C.; Olmea, O.; Laine, W.; de la Iglesia-Vicente, J.; Perez, C.; Cuevas, C.; Otero, G.; Manzanares, I.; Bailly, C.; et al. DNA and Non-DNA Targets in the Mechanism of Action of the Antitumor Drug Trabectedin. Chem. Biol. 2005, 12, 1201–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner, M.; Niepel, M.; Chung, M.; Sorger, P.K. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods 2016, 13, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, E.A.; Galarza, S.; Gencoglu, M.F.; Cornelison, R.C.; Munson, J.M.; Peyton, S.R. Applicability of drug response metrics for cancer studies using biomaterials. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180226. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Lee, C.J.; Simeone, D.M. Identification of Human Pancreatic Cancer Stem Cells. Methods Mol. Biol. 2009, 568, 161–173. [Google Scholar] [CrossRef]
- Simeone, D.M. Pancreatic Cancer Stem Cells: Implications for the Treatment of Pancreatic Cancer: Figure 1. Clin. Cancer Res. 2008, 14, 5646–5648. [Google Scholar] [CrossRef] [Green Version]
- Mascarenhas, J.B.; Young, K.P.; Littlejohn, E.; Yoo, B.K.; Salgia, R.; Lang, D. PAX6 Is Expressed in Pancreatic Cancer and Actively Participates in Cancer Progression through Activation of the MET Tyrosine Kinase Receptor Gene. J. Biol. Chem. 2009, 284, 27524–27532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, D.; Mascarenhas, J.B.; Powell, S.K.; Halegoua, J.; Nelson, M.; Ruggeri, B.A. PAX6 is expressed in pancreatic adenocarcinoma and is downregulated during induction of terminal differentiation. Mol. Carcinog. 2007, 47, 148–156. [Google Scholar] [CrossRef]
- Ooki, A.; Dinalankara, W.; Marchionni, L.; Tsay, J.-C.; Goparaju, C.; Maleki, Z.; Rom, W.; Pass, H.; Hoque, M.O. Epigenetically regulated PAX6 drives cancer cells toward a stem-like state via GLI-SOX2 signaling axis in lung adenocarcinoma. Oncogene 2018, 37, 5967–5981. [Google Scholar] [CrossRef]
- Ashizawa, S.; Brunicardi, F.C.; Wang, X.-P. PDX-1 and the Pancreas. Pancreas 2004, 28, 109–120. [Google Scholar] [CrossRef]
- Yu, J.; Liu, S.-H.; Sanchez, R.; Nemunaitis, J.; Rozengurt, E.; Brunicardi, F.C. PDX1 associated therapy in translational medicine. Ann. Transl. Med. 2016, 4, 214. [Google Scholar] [CrossRef] [Green Version]
- Estella-Hermoso de Mendoza, A.; Campanero, M.A.; De La Iglesia-Vicente, J.; Gajate, C.; Mollinedo, F.; Blanco-Prieto, M.J. Antitumor Alkyl Ether Lipid Edelfosine: Tissue Distribution and Pharmacokinetic Behavior in Healthy and Tumor-Bearing Immunosuppressed Mice. Clin. Cancer Res. 2009, 15, 858–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuesta-Marbán, A.; Botet, J.; Czyz, O.; Cacharro, L.M.; Gajate, C.; Hornillos, V.; Delgado, J.; Zhang, H.; Amat-Guerri, F.; Acuña, A.U.; et al. Drug Uptake, Lipid Rafts, and Vesicle Trafficking Modulate Resistance to an Anticancer Lysophosphatidylcholine Analogue in Yeast. J. Biol. Chem. 2013, 288, 8405–8418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2003, 11, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Mao, C.; Lee, B.; Lee, A.S. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol. Cell. Biol. 2006, 26, 5688–5697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-L.; Chen, J.; Wang, Y.-P.; Zheng, H. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy 2011, 7, 1035–1044. [Google Scholar] [CrossRef] [Green Version]
- Yonekawa, T.; Thorburn, A. Autophagy and cell death. Essays Biochem. 2013, 55, 105–117. [Google Scholar]
- Wu, J.; Kong, F.; Pan, Q.; Du, Y.; Ye, J.; Zheng, F.; Li, H.; Zhou, J. Autophagy protects against cholesterol-induced apoptosis in pancreatic beta-cells. Biochem. Biophys. Res. Commun. 2017, 482, 678–685. [Google Scholar] [CrossRef]
- García-Navas, R.; Munder, M.; Mollinedo, F. Depletion ofL-arginine induces autophagy as a cytoprotective response to endoplasmic reticulum stress in human T lymphocytes. Autophagy 2012, 8, 1557–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 2015, 11, 1437–1438. [Google Scholar] [CrossRef] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Melo-Lima, S.; Lopes, M.C.; Mollinedo, F. Necroptosis is associated with low procaspase-8 and active RIPK1 and -3 in human glioma cells. Oncoscience 2014, 1, 649–664. [Google Scholar] [CrossRef] [PubMed]
- Melo-Lima, S.; Gajate, C.; Mollinedo, F. Triggers and signaling cross-talk controlling cell death commitment. Cell Cycle 2015, 14, 465–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo-Lima, S.; Lopes, M.C.; Mollinedo, F. ERK1/2 acts as a switch between necrotic and apoptotic cell death in ether phospholipid edelfosine-treated glioblastoma cells. Pharmacol. Res. 2015, 95, 2–11. [Google Scholar] [CrossRef]
- Trédan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug Resistance and the Solid Tumor Microenvironment. JNCI J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wang, Z.; Ajani, J.A.; Song, S. Drug resistance and Cancer stem cells. Cell Commun. Signal. 2021, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef]
- Bello, C.; Bai, J.; Zambron, B.K.; Elías-Rodríguez, P.; Gajate, C.; Robina, I.; Caffa, I.; Cea, M.; Montecucco, F.; Nencioni, A.; et al. Induction of cell killing and autophagy by amphiphilic pyrrolidine derivatives on human pancreatic cancer cells. Eur. J. Med. Chem. 2018, 150, 457–478. [Google Scholar] [CrossRef]
- Marín-Ramos, N.I.; Balabasquer, M.; Ortega-Nogales, F.J.; Torrecillas, I.R.; Gil-Ordóñez, A.; Ramiro, B.M.; Garrido, A.R.; Cushman, I.; Romero, A.; Medrano, F.J.; et al. A Potent Isoprenylcysteine Carboxylmethyltransferase (ICMT) Inhibitor Improves Survival in Ras-Driven Acute Myeloid Leukemia. J. Med. Chem. 2019, 62, 6035–6046. [Google Scholar] [CrossRef]
- Villa-Pulgarín, J.A.; Gajate, C.; Botet, J.; Jiménez, A.; Justies, N.; Varela-M, R.E.; Cuesta-Marbán, A.; Müller, I.; Modolell, M.; Revuelta, J.L.; et al. Mitochondria and lipid raft-located FOF1-ATP synthase as major therapeutic targets in the antileishmanial and anticancer activities of ether lipid edelfosine. PLOS Negl. Trop. Dis. 2017, 11, e0005805. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Gajate, C. Mitochondrial Targeting Involving Cholesterol-Rich Lipid Rafts in the Mechanism of Action of the Antitumor Ether Lipid and Alkylphospholipid Analog Edelfosine. Pharmaceutics 2021, 13, 763. [Google Scholar] [CrossRef]
- Czyz, O.; Bitew, T.; Cuesta-Marbán, A.; McMaster, C.R.; Mollinedo, F.; Zaremberg, V. Alteration of Plasma Membrane Organization by an Anticancer Lysophosphatidylcholine Analogue Induces Intracellular Acidification and Internalization of Plasma Membrane Transporters in Yeast. J. Biol. Chem. 2013, 288, 8419–8432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Novel therapeutic approaches for pancreatic cancer by combined targeting of RAF→MEK→ERK signaling and autophagy survival response. Ann. Transl. Med. 2019, 7, S153. [Google Scholar] [CrossRef]
- Mollinedo, F. Neutrophil Degranulation, Plasticity, and Cancer Metastasis. Trends Immunol. 2019, 40, 228–242. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C.; Morales, A.I.; del Canto-Jañez, E.; Justies, N.; Collía, F.; Rivas, J.V.; Modolell, M.; Iglesias, A. Novel Anti-Inflammatory Action of Edelfosine Lacking Toxicity with Protective Effect in Experimental Colitis. J. Pharmacol. Exp. Ther. 2009, 329, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Dong, Q.; Li, J.; Zhang, K.; Qin, J.; Zhao, J.; Sun, Q.; Wang, Z.; Wartmann, T.; Jauch, K.W.; et al. Targeting cancer stem cells and their niche: Perspectives for future therapeutic targets and strategies. Semin. Cancer Biol. 2018, 53, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tao, H.; Wang, H.; Yang, Y.; Yang, R.; Dai, X.; Ding, X.; Wu, H.; Chen, S.; Sun, T. Doxycycline Inhibits Cancer Stem Cell-Like Properties via PAR1/FAK/PI3K/AKT Pathway in Pancreatic Cancer. Front. Oncol. 2021, 10, 619317. [Google Scholar] [CrossRef]
- Wang, H.; Ning, Z.; Li, Y.; Zhu, X.; Meng, Z. Bufalin suppresses cancer stem-like cells in gemcitabine-resistant pancreatic cancer cells via Hedgehog signaling. Mol. Med. Rep. 2016, 14, 1907–1914. [Google Scholar] [CrossRef]
- Shankar, S.; Nall, D.; Tang, S.-N.; Meeker, D.; Passarini, J.; Sharma, J.; Srivastava, R.K. Resveratrol Inhibits Pancreatic Cancer Stem Cell Characteristics in Human and KrasG12D Transgenic Mice by Inhibiting Pluripotency Maintaining Factors and Epithelial-Mesenchymal Transition. PLoS ONE 2011, 6, e16530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, X.; Wu, W.; Fan, C.; Xu, C.; Liao, J.; Rich, L.J.; Huang, R.-Y.; Repasky, E.A.; Wang, X.; Li, F. An ABCG2 non-substrate anticancer agent FL118 targets drug-resistant cancer stem-like cells and overcomes treatment resistance of human pancreatic cancer. J. Exp. Clin. Cancer Res. 2018, 37, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker | Number of Cells | ||
|---|---|---|---|
| 102 | 103 | 104 | |
| Unsorted PANC-1 | 0/6 (0%) | 2/6 (33.3%) | 4/6 (66.6%) |
| CD44+CD24+EpCAM+ PANC-1 CSCs | 6/10 (60%) | 10/10 (100%) | 10/10 (100%) |
| Treatment | Perimeter (AU) | p | Spheroid Colony Numbers | p |
|---|---|---|---|---|
| Control | 5.46 ± 2.58 | 145 ± 18 | ||
| 1 μM EDLF | 4.01 ± 2.20 | 0.25 | 127 ± 6 | 0.1013 |
| 5 μM EDLF | 1.95 ± 0.73 | 0.0005 | 70 ± 12 | 0.0004 |
| 10 μM EDLF | 1.46 ± 0.70 | 0.0001 | 46 ± 13 | 0.0001 |
| 20 μM EDLF | 1.44 ± 0.60 | 0.0002 | 32 ± 7 | <0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gajate, C.; Gayet, O.; Fraunhoffer, N.A.; Iovanna, J.; Dusetti, N.; Mollinedo, F. Induction of Apoptosis in Human Pancreatic Cancer Stem Cells by the Endoplasmic Reticulum-Targeted Alkylphospholipid Analog Edelfosine and Potentiation by Autophagy Inhibition. Cancers 2021, 13, 6124. https://doi.org/10.3390/cancers13236124
Gajate C, Gayet O, Fraunhoffer NA, Iovanna J, Dusetti N, Mollinedo F. Induction of Apoptosis in Human Pancreatic Cancer Stem Cells by the Endoplasmic Reticulum-Targeted Alkylphospholipid Analog Edelfosine and Potentiation by Autophagy Inhibition. Cancers. 2021; 13(23):6124. https://doi.org/10.3390/cancers13236124
Chicago/Turabian StyleGajate, Consuelo, Odile Gayet, Nicolas A. Fraunhoffer, Juan Iovanna, Nelson Dusetti, and Faustino Mollinedo. 2021. "Induction of Apoptosis in Human Pancreatic Cancer Stem Cells by the Endoplasmic Reticulum-Targeted Alkylphospholipid Analog Edelfosine and Potentiation by Autophagy Inhibition" Cancers 13, no. 23: 6124. https://doi.org/10.3390/cancers13236124
APA StyleGajate, C., Gayet, O., Fraunhoffer, N. A., Iovanna, J., Dusetti, N., & Mollinedo, F. (2021). Induction of Apoptosis in Human Pancreatic Cancer Stem Cells by the Endoplasmic Reticulum-Targeted Alkylphospholipid Analog Edelfosine and Potentiation by Autophagy Inhibition. Cancers, 13(23), 6124. https://doi.org/10.3390/cancers13236124