
Challenges for the Development of Extracellular Vesicle-Based Nucleic Acid Medicines

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Therapeutic Ability of Oligonucleotides Encapsulated EVs
2.1. siRNAs
2.2. Antisense Oligonucleotides
2.3. miRNAs
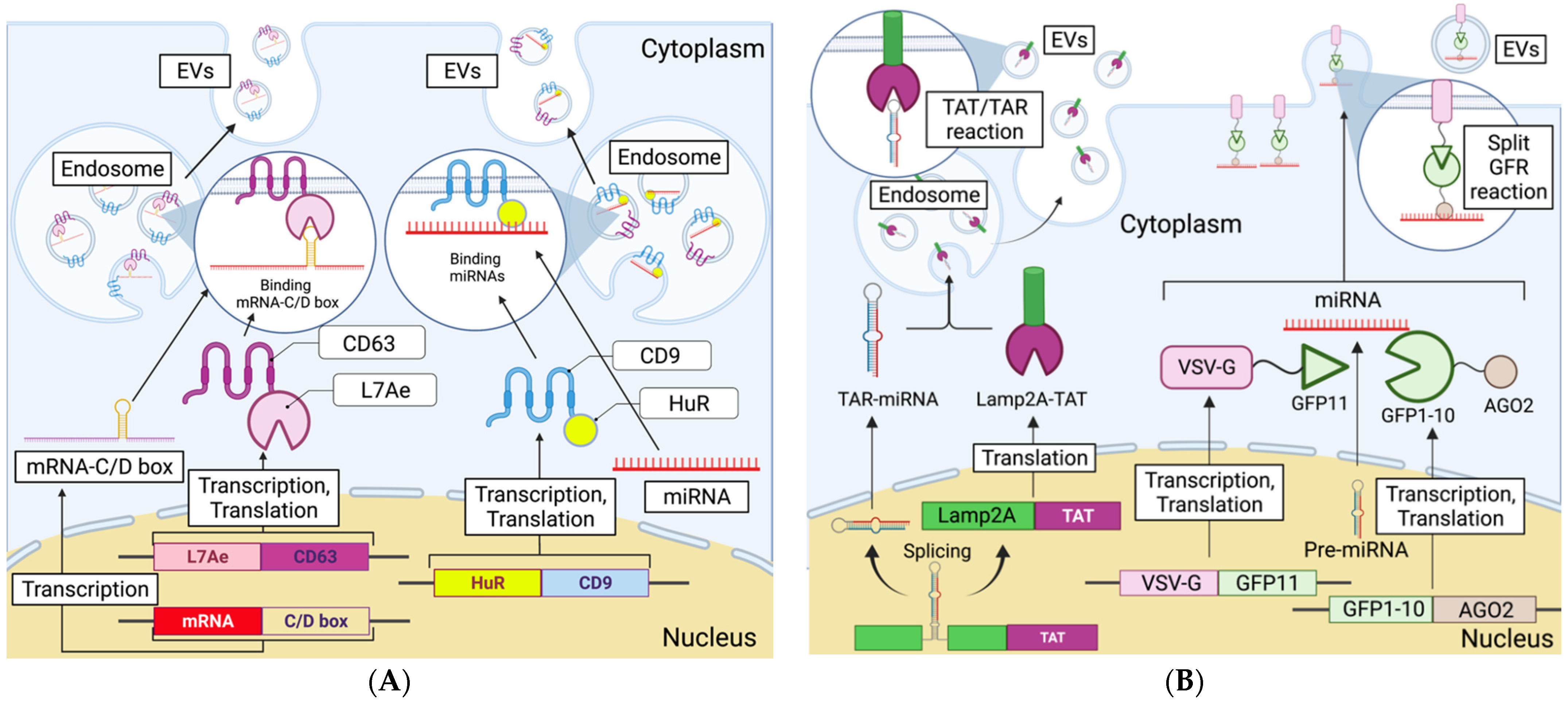
3. Loading of Nucleic Acid Medicine into EVs
3.1. Pre-Secretion Loading
3.2. Post-Secretion Loading
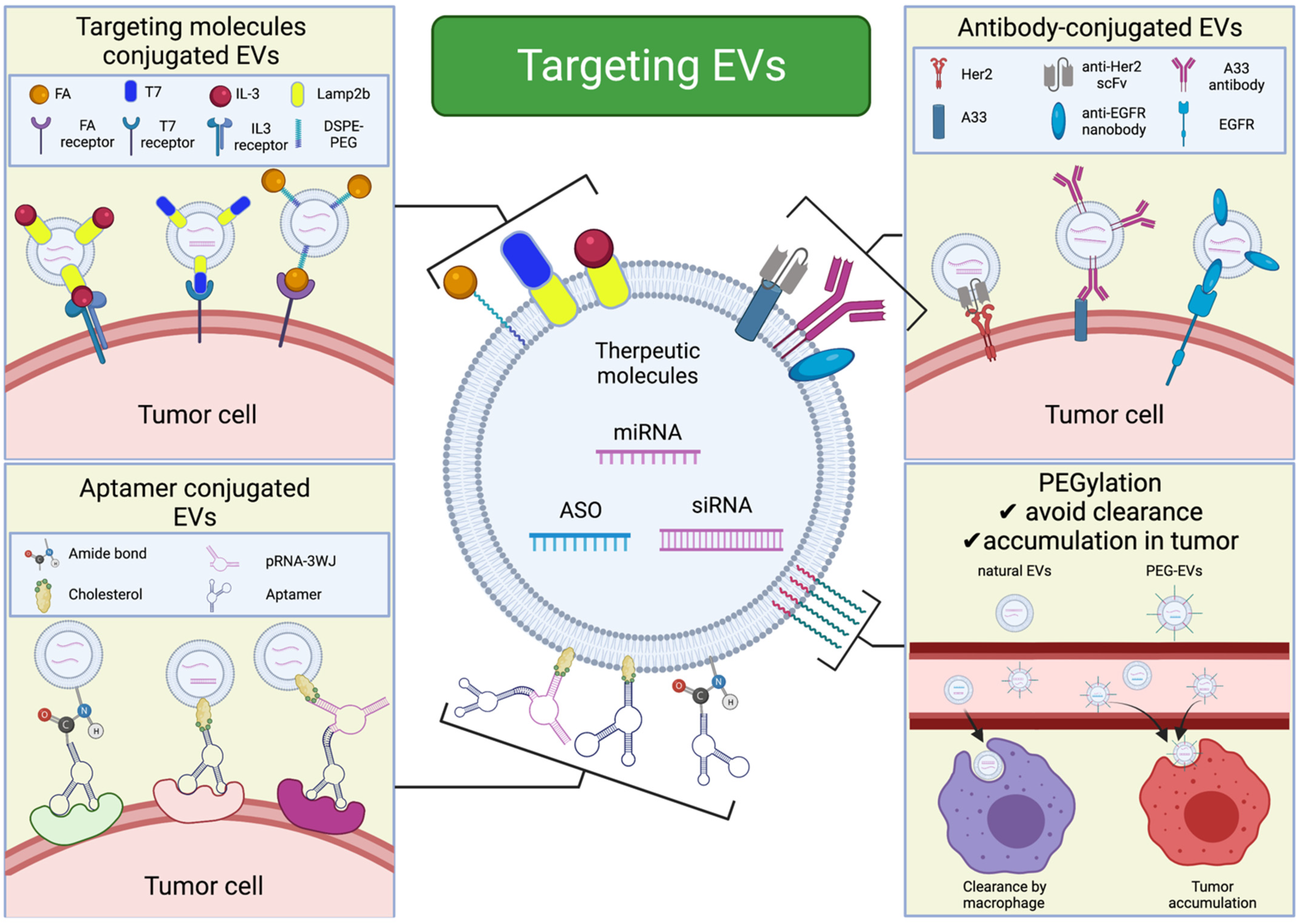
4. Tumor-Targeting EVs
4.1. EVs Biodistribution
4.2. Passive Targeting
4.3. Active Targeting
4.4. Active Targeting Using Aptamers
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navajas, R.; Corrales, F.J.; Paradela, A. Serum Exosome Isolation by Size-Exclusion Chromatography for the Discovery and Validation of Preeclampsia-Associated Biomarkers. Methods Mol. Biol. 2019, 1959, 39–50. [Google Scholar] [PubMed]
- Chen, C.K.; Liao, J.; Li, M.S.; Khoo, B.L. Urine biopsy technologies: Cancer and beyond. Theranostics 2020, 10, 7872–7888. [Google Scholar] [CrossRef] [PubMed]
- Kaczor-Urbanowicz, K.E.; Wei, F.; Rao, S.L.; Kim, J.; Shin, H.; Cheng, J.; Tu, M.; Wong, D.T.W.; Kim, Y. Clinical validity of saliva and novel technology for cancer detection. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Schartz, N.E.; Movassagh, M.; Flament, C.; Pautier, P.; Morice, P.; Pomel, C.; Lhomme, C.; Escudier, B.; Le Chevalier, T.; et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet 2002, 360, 295–305. [Google Scholar] [CrossRef]
- Song, Z.; Cai, Z.; Yan, J.; Shao, Y.W.; Zhang, Y. Liquid biopsies using pleural effusion-derived exosomal DNA in advanced lung adenocarcinoma. Transl. Lung Cancer Res. 2019, 8, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Qiu, Q.; Zhang, H.; Chu, L.; Du, Y.; Zhang, J.; Zhou, C.; Liang, F.; Shi, S.; Wang, S.; et al. Concordance between the assessment of Aβ42, T-tau, and P-T181-tau in peripheral blood neuronal-derived exosomes and cerebrospinal fluid. Alzheimers Dement. 2019, 15, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.; Rai, A.J. Isolation and Characterization of Amniotic Fluid-Derived Extracellular Vesicles for Biomarker Discovery. Methods Mol. Biol. 2019, 1885, 287–294. [Google Scholar] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bi, J.; Huang, J.; Tang, Y.; Du, S.; Li, P. Exosome: A Review of Its Classification, Isolation Techniques, Storage, Diagnostic and Targeted Therapy Applications. Int. J. Nanomed. 2020, 15, 6917–6934. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, E.M.; Schultz, S.W.; Schink, K.O.; Pedersen, N.M.; Nähse, V.; Carlson, A.; Brech, A.; Stenmark, H.; Raiborg, C. Concerted ESCRT and clathrin recruitment waves define the timing and morphology of intraluminal vesicle formation. Nat. Commun. 2018, 9, 2932. [Google Scholar] [CrossRef] [Green Version]
- Teng, F.; Fussenegger, M. Shedding Light on Extracellular Vesicle Biogenesis and Bioengineering. Adv. Sci. 2021, 8, 2003505. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42. [Google Scholar] [CrossRef]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Perrotta, I.; Aquila, S. Exosomes in human atherosclerosis: An ultrastructural analysis study. Ultrastruct. Pathol. 2016, 40, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, C.M.; Loyer, X.; Rautou, P.E.; Amabile, N. Extracellular vesicles in coronary artery disease. Nat. Rev. Cardiol. 2017, 14, 259–272. [Google Scholar] [CrossRef]
- Xiao, Y.; Zheng, L.; Zou, X.; Wang, J.; Zhong, J.; Zhong, T. Extracellular vesicles in type 2 diabetes mellitus: Key roles in pathogenesis, complications, and therapy. J. Extracell. Vesicles 2019, 8, 1625677. [Google Scholar] [CrossRef] [Green Version]
- Hill, A.F. Extracellular Vesicles and Neurodegenerative Diseases. J. Neurosci. 2019, 39, 9269–9273. [Google Scholar] [CrossRef]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef]
- Tutrone, R.; Donovan, M.J.; Torkler, P.; Tadigotla, V.; McLain, T.; Noerholm, M.; Skog, J.; McKiernan, J. Clinical utility of the exosome based ExoDx Prostate(IntelliScore) EPI test in men presenting for initial Biopsy with a PSA 2–10 ng/mL. Prostate Cancer Prostatic Dis. 2020, 23, 607–614. [Google Scholar] [CrossRef]
- Hoshino, A.; Kim, H.S.; Bojmar, L.; Gyan, K.E.; Cioffi, M.; Hernandez, J.; Zambirinis, C.P.; Rodrigues, G.; Molina, H.; Heissel, S.; et al. Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers. Cell 2020, 182, 1044–1061. [Google Scholar] [CrossRef]
- Li, K.; Chen, Y.; Li, A.; Tan, C.; Liu, X. Exosomes play roles in sequential processes of tumor metastasis. Int. J. Cancer 2019, 144, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Wahlgren, J.; Karlson, T.D.L.; Brisslert, M.; Vaziri Sani, F.; Telemo, E.; Sunnerhagen, P.; Valadi, H. Plasma exosomes can deliver exogenous short interfering RNA to monocytes and lymphocytes. Nucleic Acids Res. 2012, 40, e130. [Google Scholar] [CrossRef] [Green Version]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-Targeted Therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Trivedi, P.; Jain, N.K. Advances in siRNA delivery in cancer therapy. Artif. Cells Nanomed. Biotechnol. 2018, 46, 274–283. [Google Scholar] [CrossRef]
- Behlke, M.A. Chemical modification of siRNAs for in vivo use. Oligonucleotides 2008, 18, 305–319. [Google Scholar] [CrossRef] [Green Version]
- Turturici, G.; Tinnirello, R.; Sconzo, G.; Geraci, F. Extracellular membrane vesicles as a mechanism of cell-to-cell communication: Advantages and disadvantages. Am. J. Physiol. Cell Physiol. 2014, 306, C621–C633. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, S.; Danielson, S.; Clements, V.; Edwards, N.; Ostrand-Rosenberg, S.; Fenselau, C. Surface Glycoproteins of Exosomes Shed by Myeloid-Derived Suppressor Cells Contribute to Function. J. Proteome Res. 2017, 16, 238–246. [Google Scholar] [CrossRef]
- Haraszti, R.A.; Didiot, M.-C.; Sapp, E.; Leszyk, J.; Shaffer, S.A.; Rockwell, H.E.; Gao, F.; Narain, N.R.; DiFiglia, M.; Kiebish, M.A.; et al. High-resolution proteomic and lipidomic analysis of exosomes and microvesicles from different cell sources. J. Extracell. Vesicles 2016, 5, 32570. [Google Scholar] [CrossRef] [PubMed]
- El Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J.A. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- He, C.; Zheng, S.; Luo, Y.; Wang, B. Exosome Theranostics: Biology and Translational Medicine. Theranostics 2018, 8, 237–255. [Google Scholar] [CrossRef]
- Rana, S.; Yue, S.; Stadel, D.; Zöller, M. Toward tailored exosomes: The exosomal tetraspanin web contributes to target cell selection. Int. J. Biochem. Cell Biol. 2012, 44, 1574–1584. [Google Scholar] [CrossRef] [PubMed]
- Hannon, G.J. RNA interference. Nature 2002, 418, 244–251. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Aqil, F.; Munagala, R.; Jeyabalan, J.; Agrawal, A.K.; Kyakulaga, A.H.; Wilcher, S.A.; Gupta, R.C. Milk exosomes—Natural nanoparticles for siRNA delivery. Cancer Lett. 2019, 449, 186–195. [Google Scholar] [CrossRef]
- Zhao, L.; Gu, C.; Gan, Y.; Shao, L.; Chen, H.; Zhu, H. Exosome-mediated siRNA delivery to suppress postoperative breast cancer metastasis. J. Control. Release 2020, 318, 1–15. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Huang, B.; Zhang, K.; Liu, M.; Xu, T. Long non-coding RNA in cervical cancer: From biology to therapeutic opportunity. Biomed. Pharmacother. 2020, 127, 110209. [Google Scholar] [CrossRef]
- Yang, J.; Luo, S.; Zhang, J.; Yu, T.; Fu, Z.; Zheng, Y.; Xu, X.; Liu, C.; Fan, M.; Zhang, Z. Exosome-mediated delivery of antisense oligonucleotides targeting α-synuclein ameliorates the pathology in a mouse model of Parkinson’s disease. Neurobiol. Dis. 2021, 148, 105218. [Google Scholar] [CrossRef]
- Xu, H.; Liao, C.; Liang, S.; Ye, B.C. A Novel Peptide-Equipped Exosomes Platform for Delivery of Antisense Oligonucleotides. ACS Appl. Mater. Interfaces 2021, 13, 10760–10767. [Google Scholar] [CrossRef] [PubMed]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarshad, A.A.; Juan, A.H.; Muler, A.I.C.; Anastasakis, D.G.; Wang, X.; Genzor, P.; Feng, X.; Tsai, P.F.; Sun, H.W.; Haase, A.D.; et al. Argonaute-miRNA Complexes Silence Target mRNAs in the Nucleus of Mammalian Stem Cells. Mol. Cell 2018, 71, 1040–1050.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Shi, K.; Yang, S.; Liu, J.; Zhou, Q.; Wang, G.; Song, J.; Li, Z.; Zhang, Z.; Yuan, W. Effect of exosomal miRNA on cancer biology and clinical applications. Mol. Cancer 2018, 17, 147. [Google Scholar] [CrossRef]
- Gilligan, K.E.; Dwyer, R.M. Engineering Exosomes for Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boca, S.; Gulei, D.; Zimta, A.A.; Onaciu, A.; Magdo, L.; Tigu, A.B.; Ionescu, C.; Irimie, A.; Buiga, R.; Berindan-Neagoe, I. Nanoscale delivery systems for microRNAs in cancer therapy. Cell. Mol. Life Sci. 2020, 77, 1059–1086. [Google Scholar] [CrossRef] [PubMed]
- Kogure, T.; Lin, W.L.; Yan, I.K.; Braconi, C.; Patel, T. Intercellular nanovesicle-mediated microRNA transfer: A mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology 2011, 54, 1237–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Wang, Q.; Zhang, X.; Yan, N.; Li, X. Exosomal miR-126 blocks the development of non-small cell lung cancer through the inhibition of ITGA6. Cancer Cell Int. 2020, 20, 574. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Tan, X.; Liu, P.; Yang, Y.; Huang, Y.; Liu, X.; Meng, X.; Yu, B.; Wu, M.; Jin, H. ITGA6 and RPSA synergistically promote pancreatic cancer invasion and metastasis via PI3K and MAPK signaling pathways. Exp. Cell Res. 2019, 379, 30–47. [Google Scholar] [CrossRef]
- Akhtar, S.; Benter, I.F. Nonviral delivery of synthetic siRNAs in vivo. J. Clin. Investig. 2007, 117, 3623–3632. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, H.; Ning, T.; Liu, D.; Deng, T.; Liu, R.; Bai, M.; Zhu, K.; Li, J.; Fan, Q.; et al. Exosome-Delivered c-Met siRNA Could Reverse Chemoresistance to Cisplatin in Gastric Cancer. Int. J. Nanomed. 2020, 15, 2323–2335. [Google Scholar] [CrossRef] [Green Version]
- Bellavia, D.; Raimondo, S.; Calabrese, G.; Forte, S.; Cristaldi, M.; Patinella, A.; Memeo, L.; Manno, M.; Raccosta, S.; Diana, P.; et al. Interleukin 3-receptor targeted exosomes inhibit in vitro and in vivo Chronic Myelogenous Leukemia cell growth. Theranostics 2017, 7, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Pi, F.; Binzel, D.W.; Lee, T.J.; Li, Z.; Sun, M.; Rychahou, P.; Li, H.; Haque, F.; Wang, S.; Croce, C.M.; et al. Nanoparticle orientation to control RNA loading and ligand display on extracellular vesicles for cancer regression. Nat. Nanotechnol. 2018, 13, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Kim, M.; Lee, Y.; Byun, J.W.; Hwang, D.W.; Lee, M. Systemic delivery of microRNA-21 antisense oligonucleotides to the brain using T7-peptide decorated exosomes. J. Control. Release 2020, 317, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Sawada, K.; Miyamoto, M.; Shimizu, A.; Yamamoto, M.; Kinose, Y.; Nakamura, K.; Kawano, M.; Kodama, M.; Hashimoto, K.; et al. Exploring the potential of engineered exosomes as delivery systems for tumor-suppressor microRNA replacement therapy in ovarian cancer. Biochem. Biophys. Res. Commun. 2020, 527, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Monfared, H.; Jahangard, Y.; Nikkhah, M.; Mirnajafi-Zadeh, J.; Mowla, S.J. Potential Therapeutic Effects of Exosomes Packed With a miR-21-Sponge Construct in a Rat Model of Glioblastoma. Front. Oncol. 2019, 9, 782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhang, X.; Du, L.; Wang, Y.; Liu, X.; Tian, H.; Wang, L.; Li, P.; Zhao, Y.; Duan, W.; et al. Exosome-transmitted miR-128-3p increase chemosensitivity of oxaliplatin-resistant colorectal cancer. Mol. Cancer 2019, 18, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Li, L.; Piontek, K.; Sakaguchi, M.; Selaru, F.M. Exosome miR-335 as a novel therapeutic strategy in hepatocellular carcinoma. Hepatology 2018, 67, 940–954. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.P.; Khan, S.; Gilligan, K.E.; Zafar, H.; Lalor, P.; Glynn, C.; O’Flatharta, C.; Ingoldsby, H.; Dockery, P.; De Bhulbh, A.; et al. Employing mesenchymal stem cells to support tumor-targeted delivery of extracellular vesicle (EV)-encapsulated microRNA-379. Oncogene 2018, 37, 2137–2149. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Kan, S.; Zhu, Y.; Feng, S.; Feng, W.; Gao, S. Engineered exosome-mediated delivery of functionally active miR-26a and its enhanced suppression effect in HepG2 cells. Int. J. Nanomed. 2018, 13, 585–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, F.M.; Hossain, A.; Gumin, J.; Momin, E.N.; Shimizu, Y.; Ledbetter, D.; Shahar, T.; Yamashita, S.; Parker Kerrigan, B.; Fueyo, J.; et al. Mesenchymal stem cells as natural biofactories for exosomes carrying miR-124a in the treatment of gliomas. Neuro Oncol. 2018, 20, 380–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, R.; Lee, S.; Lee, J.; Kim, M.; Kim, W.J.; Lee, H.W.; Lee, M.Y.; Kim, J.; Chang, W. Exosomes derived from microRNA-584 transfected mesenchymal stem cells: Novel alternative therapeutic vehicles for cancer therapy. BMB Rep. 2018, 51, 406–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, G.; Song, X.; Yang, F.; Wu, S.; Wang, J.; Chen, Z.; Liu, Y. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Q.; Ramakrishnaiah, V.; Henry, S.; Fouraschen, S.; de Ruiter, P.E.; Kwekkeboom, J.; Tilanus, H.W.; Janssen, H.L.; van der Laan, L.J. Hepatic cell-to-cell transmission of small silencing RNA can extend the therapeutic reach of RNA interference (RNAi). Gut 2012, 61, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Kojima, R.; Bojar, D.; Rizzi, G.; Hamri, G.C.-E.; El-Baba, M.D.; Saxena, P.; Ausländer, S.; Tan, K.R.; Fussenegger, M. Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nat. Commun. 2018, 9, 1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhou, X.; Wei, M.; Gao, X.; Zhao, L.; Shi, R.; Sun, W.; Duan, Y.; Yang, G.; Yuan, L. In Vitro and in Vivo RNA Inhibition by CD9-HuR Functionalized Exosomes Encapsulated with miRNA or CRISPR/dCas9. Nano Lett. 2019, 19, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Sutaria, D.S.; Jiang, J.; Elgamal, O.A.; Pomeroy, S.M.; Badawi, M.; Zhu, X.; Pavlovicz, R.; Azevedo-Pouly, A.C.P.; Chalmers, J.; Li, C.; et al. Low active loading of cargo into engineered extracellular vesicles results in inefficient miRNA mimic delivery. J. Extracell. Vesicles 2017, 6, 1333882. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, Q.; Zi, Z.; Liu, Z.; Wan, C.; Crisman, L.; Shen, J.; Liu, X. Programmable Extracellular Vesicles for Macromolecule Delivery and Genome Modifications. Dev. Cell. 2020, 55, 784–801.e789. [Google Scholar] [CrossRef] [PubMed]
- Cabantous, S.; Terwilliger, T.C.; Waldo, G.S. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 2005, 23, 102–107. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, A.J.; Mäger, I.; de Jong, O.G.; Varela, M.A.; Schiffelers, R.M.; El Andaloussi, S.; Wood, M.J.A.; Vader, P. Functional Delivery of Lipid-Conjugated siRNA by Extracellular Vesicles. Mol. Ther. 2017, 25, 1580–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didiot, M.C.; Hall, L.M.; Coles, A.H.; Haraszti, R.A.; Godinho, B.M.; Chase, K.; Sapp, E.; Ly, S.; Alterman, J.F.; Hassler, M.R.; et al. Exosome-mediated Delivery of Hydrophobically Modified siRNA for Huntingtin mRNA Silencing. Mol. Ther. 2016, 24, 1836–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haraszti, R.A.; Miller, R.; Didiot, M.C.; Biscans, A.; Alterman, J.F.; Hassler, M.R.; Roux, L.; Echeverria, D.; Sapp, E.; DiFiglia, M.; et al. Optimized Cholesterol-siRNA Chemistry Improves Productive Loading onto Extracellular Vesicles. Mol. Ther. 2018, 26, 1973–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shandilya, S.; Rani, P.; Onteru, S.K.; Singh, D. Natural ligand-receptor mediated loading of siRNA in milk derived exosomes. J. Biotechnol. 2020, 318, 1–9. [Google Scholar] [CrossRef]
- Lamichhane, T.N.; Jeyaram, A.; Patel, D.B.; Parajuli, B.; Livingston, N.K.; Arumugasaamy, N.; Schardt, J.S.; Jay, S.M. Oncogene Knockdown via Active Loading of Small RNAs into Extracellular Vesicles by Sonication. Cell. Mol. Bioeng. 2016, 9, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Sidu, R.K.; Zou, H.; Alam, M.K.; Yang, M.; Lee, Y. Inhibition of Glioma Cells’ Proliferation by Doxorubicin-Loaded Exosomes via Microfluidics. Int. J. Nanomed. 2020, 15, 8331–8343. [Google Scholar] [CrossRef] [PubMed]
- Kooijmans, S.A.A.; Stremersch, S.; Braeckmans, K.; de Smedt, S.C.; Hendrix, A.; Wood, M.J.A.; Schiffelers, R.M.; Raemdonck, K.; Vader, P. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Control. Release 2013, 172, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Ran, N.; Dong, X.; Zuo, B.; Yang, R.; Zhou, Q.; Moulton, H.M.; Seow, Y.; Yin, H. Anchor peptide captures, targets, and loads exosomes of diverse origins for diagnostics and therapy. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Zhang, D.; Zhu, Z.; Dela Cruz, C.S.; Jin, Y. Epithelial cell-derived microvesicles activate macrophages and promote inflammation via microvesicle-containing microRNAs. Sci. Rep. 2016, 6, 35250. [Google Scholar] [CrossRef] [Green Version]
- de Abreu, R.C.; Ramos, C.V.; Becher, C.; Lino, M.; Jesus, C.; da Costa Martins, P.A.; Martins, P.A.T.; Moreno, M.J.; Fernandes, H.; Ferreira, L. Exogenous loading of miRNAs into small extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12111. [Google Scholar] [CrossRef] [PubMed]
- Wiklander, O.P.B.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lázaro-Ibáñez, E.; Faruqu, F.N.; Saleh, A.F.; Silva, A.M.; Tzu-Wen Wang, J.; Rak, J.; Al-Jamal, K.T.; Dekker, N. Selection of Fluorescent, Bioluminescent, and Radioactive Tracers to Accurately Reflect Extracellular Vesicle Biodistribution in vivo. ACS Nano 2021, 15, 3212–3227. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Barenholz, Y. Doxil®--the first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, M.J.; Soon-Shiong, P.; Desai, N. Protein nanoparticles as drug carriers in clinical medicine. Adv. Drug Deliv. Rev. 2008, 60, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Zhao, L.; Lu, F.; Gao, X.; Dong, Y.; Zhao, Y.; Wei, M.; Yang, G.; Xing, C.; Liu, L. Mononuclear phagocyte system blockade improves therapeutic exosome delivery to the myocardium. Theranostics 2020, 10, 218–230. [Google Scholar] [CrossRef]
- Arvizo, R.R.; Miranda, O.R.; Moyano, D.F.; Walden, C.A.; Giri, K.; Bhattacharya, R.; Robertson, J.D.; Rotello, V.M.; Reid, J.M.; Mukherjee, P. Modulating Pharmacokinetics, Tumor Uptake and Biodistribution by Engineered Nanoparticles. PLoS ONE 2011, 6, e24374. [Google Scholar] [CrossRef] [Green Version]
- Midekessa, G.; Godakumara, K.; Ord, J.; Viil, J.; Lättekivi, F.; Dissanayake, K.; Kopanchuk, S.; Rinken, A.; Andronowska, A.; Bhattacharjee, S.; et al. Zeta Potential of Extracellular Vesicles: Toward Understanding the Attributes that Determine Colloidal Stability. ACS Omega 2020, 5, 16701–16710. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.L.; Wang, L.; Chow, E.K.; Lim, C.T.; Goh, B.C. Exosomes in Cancer Nanomedicine and Immunotherapy: Prospects and Challenges. Trends Biotechnol. 2017, 35, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Harris, C.L.; Court, J.; Mason, M.D.; Morgan, B.P. Antigen-presenting cell exosomes are protected from complement-mediated lysis by expression of CD55 and CD59. Eur. J. Immunol. 2003, 33, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Nishikawa, M.; Shinotsuka, H.; Matsui, Y.; Ohara, S.; Imai, T.; Takakura, Y. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. J. Biotechnol. 2013, 165, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Chonn, A.; Semple, S.C.; Cullis, P.R. Association of blood proteins with large unilamellar liposomes in vivo. Relation to circulation lifetimes. J. Biol. Chem. 1992, 267, 18759–18765. [Google Scholar] [CrossRef]
- Shi, S.; Li, T.; Wen, X.; Wu, S.Y.; Xiong, C.; Zhao, J.; Lincha, V.R.; Chow, D.S.; Liu, Y.; Sood, A.K.; et al. Copper-64 Labeled PEGylated Exosomes for In Vivo Positron Emission Tomography and Enhanced Tumor Retention. Bioconjugate Chem. 2019, 30, 2675–2683. [Google Scholar] [CrossRef]
- Hoang Thi, T.T.; Pilkington, E.H.; Nguyen, D.H.; Lee, J.S.; Park, K.D.; Truong, N.P. The Importance of Poly(ethylene glycol) Alternatives for Overcoming PEG Immunogenicity in Drug Delivery and Bioconjugation. Polymers 2020, 12, 298. [Google Scholar] [CrossRef] [Green Version]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Dai, W.; He, B.; Zhang, H.; Wang, X.; Wang, Y.; Zhang, Q. Current Multistage Drug Delivery Systems Based on the Tumor Microenvironment. Theranostics 2017, 7, 538–558. [Google Scholar] [CrossRef]
- Nie, W.; Wu, G.; Zhang, J.; Huang, L.L.; Ding, J.; Jiang, A.; Zhang, Y.; Liu, Y.; Li, J.; Pu, K.; et al. Responsive Exosome Nano-bioconjugates for Synergistic Cancer Therapy. Angew. Chem. 2020, 59, 2018–2022. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Hu, S.; Huang, K.; Su, T.; Li, Z.; Vandergriff, A.; Cores, J.; Dinh, P.U.; Allen, T.; Shen, D.; et al. Tumor cell-derived exosomes home to their cells of origin and can be used as Trojan horses to deliver cancer drugs. Theranostics 2020, 10, 3474–3487. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Ríos, A.J.; Molina-Crespo, Á.; Bouzo, B.L.; López-López, R.; Moreno-Bueno, G.; de la Fuente, M. Exosome-mimetic nanoplatforms for targeted cancer drug delivery. J. Nanobiotechnol. 2019, 17, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Gai, C.; Li, Z.; Ding, D.; Zheng, J.; Zhang, W.; Lv, S.; Li, W. Targeted exosome-encapsulated erastin induced ferroptosis in triple negative breast cancer cells. Cancer Sci. 2019, 110, 3173–3182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longatti, A.; Schindler, C.; Collinson, A.; Jenkinson, L.; Matthews, C.; Fitzpatrick, L.; Blundy, M.; Minter, R.; Vaughan, T.; Shaw, M.; et al. High affinity single-chain variable fragments are specific and versatile targeting motifs for extracellular vesicles. Nanoscale 2018, 10, 14230–14244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaj, L.; Lessard, R.; Dai, L.; Cho, Y.J.; Pomeroy, S.L.; Breakefield, X.O.; Skog, J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011, 2, 180. [Google Scholar] [CrossRef] [PubMed]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol. Sin. 2017, 38, 754–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Gao, Y.; Gong, C.; Wang, Z.; Xia, Q.; Gu, F.; Hu, C.; Zhang, L.; Guo, H.; Gao, S. A33 antibody-functionalized exosomes for targeted delivery of doxorubicin against colorectal cancer. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 1973–1985. [Google Scholar] [CrossRef]
- Si, Y.; Kim, S.; Zhang, E.; Tang, Y.; Jaskula-Sztul, R.; Markert, J.M.; Chen, H.; Zhou, L.; Liu, X.M. Targeted Exosomes for Drug Delivery: Biomanufacturing, Surface Tagging, and Validation. Biotechnol. J. 2020, 15, e1900163. [Google Scholar] [CrossRef]
- Pham, T.C.; Jayasinghe, M.K.; Pham, T.T.; Yang, Y.; Wei, L.; Usman, W.M.; Chen, H.; Pirisinu, M.; Gong, J.; Kim, S.; et al. Covalent conjugation of extracellular vesicles with peptides and nanobodies for targeted therapeutic delivery. J. Extracell. Vesicles 2021, 10, e12057. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Ling, X.; Yang, Y.; Zhang, J.; Li, Q.; Niu, X.; Hu, G.; Chen, B.; Li, H.; Wang, Y.; et al. Embryonic Stem Cells-Derived Exosomes Endowed with Targeting Properties as Chemotherapeutics Delivery Vehicles for Glioblastoma Therapy. Adv. Sci. 2019, 6, 1801899. [Google Scholar] [CrossRef]
- Jia, G.; Han, Y.; An, Y.; Ding, Y.; He, C.; Wang, X.; Tang, Q. NRP-1 targeted and cargo-loaded exosomes facilitate simultaneous imaging and therapy of glioma in vitro and in vivo. Biomaterials 2018, 178, 302–316. [Google Scholar] [CrossRef]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Wang, L.; Zhu, C.; Zheng, Q.; Wang, G.; Tong, J.; Fang, Y.; Xia, Y.; Cheng, G.; He, X.; et al. Aptamer-Conjugated Extracellular Nanovesicles for Targeted Drug Delivery. Cancer Res. 2018, 78, 798–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagheri, E.; Abnous, K.; Farzad, S.A.; Taghdisi, S.M.; Ramezani, M.; Alibolandi, M. Targeted doxorubicin-loaded mesenchymal stem cells-derived exosomes as a versatile platform for fighting against colorectal cancer. Life Sci. 2020, 261, 118369. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Shi, M.; Liu, X.; Jin, C.; Xing, X.; Qiu, L.; Tan, W. Aptamer-Functionalized Exosomes: Elucidating the Cellular Uptake Mechanism and the Potential for Cancer-Targeted Chemotherapy. Anal. Chem. 2019, 91, 2425–2430. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Gragoudas, E.S.; Adamis, A.P.; Cunningham, E.T., Jr.; Feinsod, M.; Guyer, D.R. Pegaptanib for neovascular age-related macular degeneration. N. Engl. J. Med. 2004, 351, 2805–2816. [Google Scholar] [CrossRef] [Green Version]
- Romano, S.; Fonseca, N.; Simões, S.; Gonçalves, J.; Moreira, J.N. Nucleolin-based targeting strategies for cancer therapy: From targeted drug delivery to cytotoxic ligands. Drug Discov. Today 2019, 24, 1985–2001. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, S.; Vaught, J.D.; Bock, C.; Gold, L.; Katilius, E.; Keeney, T.R.; Kim, N.; Saccomano, N.A.; Wilcox, S.K.; Zichi, D.; et al. From SOMAmer-Based Biomarker Discovery to Diagnostic and Clinical Applications: A SOMAmer-Based, Streamlined Multiplex Proteomic Assay. PLoS ONE 2011, 6, e26332. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Types of Oligonucleotide | Cargo Mediators | Cancer Type | Parent Cell | Loading Method | Function | References |
|---|---|---|---|---|---|---|
| siRNA | siS100A4 | breast cancer | breast cancer cell | Coincubation and extrusion | Involved in various pathways | [40] |
| si-c-Met | gastric cancer | HEK293T cell | Parental cells transfection using Lipofectamine | Reverse chemoresistance to Cisplatin | [53] | |
| siKRASG12S | lung cancer | Milk | Electroporation and Exo-fect | Antiproliferative effect via silencing KRASG12S | [39] | |
| BCR-ABL siRNA | Chronic myeloid leukemia | HEK293T cell | Parental cells transfection using Lipofectamine | Chronic myeloid leukemia cell growth in vitro and in vivo | [54] | |
| siSurvivin | Prostate cancer | HEK293T cell | Binding of Cholesterol to the arrowhead of pRNA-3WJ fused with siSurvivin | Inhibition cell apoptosis | [55] | |
| ASO | G3139 (BCL-2 ASO) | Hepatocellular carcinoma | HepG2 cells | Cholesterol-conjugated ASO was loaded onto the EVs | Downregulation of anti-apoptotic Bcl-2 | [43] |
| Antisense miRNA oligonucleotide against miR-21 | Glioblastoma | 293T cells | Electroporation | Reduction of tumor size via upregulation of PDCD4 and PTEN | [56] | |
| miRNA | miR-126 | non-small cell lung carcinoma | patient serum | Exo-fect | Inhibiting tumor proliferation and migration via downregulation of ITGA6 | [50] |
| miR-199 | Ovarian cancer | Omental fibroblast derived from ovarian cancer patients | Electroporation | Inhibition of cell proliferation and invasion via suppression of c-Met | [57] | |
| miR-21-sponge | Glioblastoma | HEK293T cells | Parental cells transfection using Lipofectamine | Declining cell proliferation and elevation in apoptotic rates via upregulation of PDCD4 and RECK | [58] | |
| miR-128-3p | Colorectal cancer | FHC cells | Parental cells transfection using Lipofectamine | Upregulation of E-cadherin and inhibition oxaliplatin-induced epithelial mesenchymal transition by downregulation of Bmi1, and decreasing oxaliplatin efflux via suppression of MRP5 | [59] | |
| miR-335-5p | Hepatocellular carcinoma | LX2 cells | Parental cells transfection using Lipofectamine | Inhibition of hepatocellular carcinoma cells proliferation and invasion through downregulation of 13 mRNA | [60] | |
| miR-379 | Breast cancer cells | MSCs | Lentiviral transfection of parental cells | Suppression of tumor growth via downregulate cyclooxygenase-2 | [61] | |
| miR-26a | HepG2 cells | 293T cells | Electroporation | Decreasing cell migration and proliferation via downregulation of CCNE2 and CDK6 | [62] | |
| miR-124a | Glioblastoma | MSCs | Lentiviral transfection of parental cells | Significant reduction in viability due to abnormal lipid accumulation through silencing FOXA2 | [63] | |
| miR-584 | Glioma | MSCs | Lentiviral transfection of parental cells | Inducing tumor cell apoptosis and reducing tumor cell invasion via enhancing caspase-3 and reducing matrix metalloproteinase-2 expression | [64] | |
| miR-122 | Hepatocellular carcinoma | adipose tissue-derived MSCs | Parental cells transfection using Lipofectamine | Increasing chemosensitivity through downregulation of CCNG1, ADAM10, and insulin-like growth factor 1 receptor | [65] | |
| let-7a | Breast cancer | HEK293 cells | Parental cells transfection using HiPerFect reagent | Suppressing tumor growth in vivo | [66] | |
| miR-146b | Glioma | MSCs | Parental cells transfection using electroporation | Reducing tumor size via suppressing EGFR and NF-κB | [49] |
| Targeting Molecule | Target to | Cancer Type | How to Add Targeting Molecule | References |
|---|---|---|---|---|
| Antibody | ||||
| anti-Her2-scFv | Her2 | Breast cancer | Binding of anti-Her2-scFv to C1C2 domain of lactadherin that can bind to phosphatidylserine | [104] |
| A33 antibody | A33 | Colorectal cancer | EVs isolated from A33 positive LIM1215 were coated with surface-carboxyl superparamagnetic iron oxide particles with A33 antibodies | [107] |
| somatostatin receptor-2 antibody | somatostatin receptor-2 | Neuroendocrine cancer | Coincubation of anti-SSTR Ab with 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine (DSPE)-PEG-N-hydroxysuccinimide and mPEG-DSPE-EV | [108] |
| EGFR targeting nanobody | EGFR | Lung cancer | Simple enzymatic method to bind peptides and nanobodies to EVs via covalent bonds using Sortase | [109] |
| Peptide or other molecules | ||||
| c(RGDyK) peptide | αVβ3 integrin | Glioblastoma | Coincubated with micelles formed by DSPE-PEG2000-c(RGDyK) | [110] |
| Folate | Folate receptor | Breast cancer | Coincubated with folate conjugated with DSPE-PEG2000 | [103] |
| Folate | Folate receptor | Lung cancer | Covalently conjugation using standard stable amide chemistry | [39] |
| RGERPPR peptide(RGE peptide) | Neuropilin-1 | Glioma | The alkyne group was conjugated with phosphatidylethanolamine on the exosome surface, and the RGE peptide with an azide group was conjugated with the alkyne group by a triazole linkages. | [111] |
| iRGD peptide | αv integrin | Breast cancer | Parental cells were transfected with the vector expressing iRGD-Lamp2b fusion protein | [112] |
| GE11 | EGFR | Breast cancer | Parental cells were transfected with the plasmid containing platelet-derived growth factor receptor transmembrane domain fused with GE-11 | [66] |
| Interleukin-3 (IL3) | IL3-R | Chronic myeloid leukemia | Parental cells were transfected with the plasmid containing Lamp2b gene fused with the IL3 gene fragment | [54] |
| T7 | Transferrin receptor | Glioblastoma | Parental cells were transfected with the plasmid containing Lamp2b gene fused with a T7 | [56] |
| Aptamer | ||||
| AS1411 aptamer | nucleolin | Breast cancer | Extrusion of dendritic cells labeled with Aptamer conjugated with PEGylated cholesterol | [113] |
| PSMA aptamer | PSMA | Prostate cancer | Conjugation of aptamer with pRNA-3WJ fused with cholesterol. | [55] |
| MUC1 aptamer | MUC-1 | Colorectal cancer | Utilizing amine groups on the surface of EVs to bind via amide bonds | [114] |
| scgc8 aptamer | Protein tyrosine kinase 7 | T-cell leukemia | Diacyllipid conjugated aptamer decorated onto EVs through hydrophobic interaction between the diacyllipid tail and the phospholipid bilayer of EVs. | [115] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuriyama, N.; Yoshioka, Y.; Kikuchi, S.; Okamura, A.; Azuma, N.; Ochiya, T. Challenges for the Development of Extracellular Vesicle-Based Nucleic Acid Medicines. Cancers 2021, 13, 6137. https://doi.org/10.3390/cancers13236137
Kuriyama N, Yoshioka Y, Kikuchi S, Okamura A, Azuma N, Ochiya T. Challenges for the Development of Extracellular Vesicle-Based Nucleic Acid Medicines. Cancers. 2021; 13(23):6137. https://doi.org/10.3390/cancers13236137
Chicago/Turabian StyleKuriyama, Naoya, Yusuke Yoshioka, Shinsuke Kikuchi, Akihiko Okamura, Nobuyoshi Azuma, and Takahiro Ochiya. 2021. "Challenges for the Development of Extracellular Vesicle-Based Nucleic Acid Medicines" Cancers 13, no. 23: 6137. https://doi.org/10.3390/cancers13236137
APA StyleKuriyama, N., Yoshioka, Y., Kikuchi, S., Okamura, A., Azuma, N., & Ochiya, T. (2021). Challenges for the Development of Extracellular Vesicle-Based Nucleic Acid Medicines. Cancers, 13(23), 6137. https://doi.org/10.3390/cancers13236137