
Methylation Markers in Cutaneous Melanoma: Unravelling the Potential Utility of Their Tracking by Liquid Biopsy

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
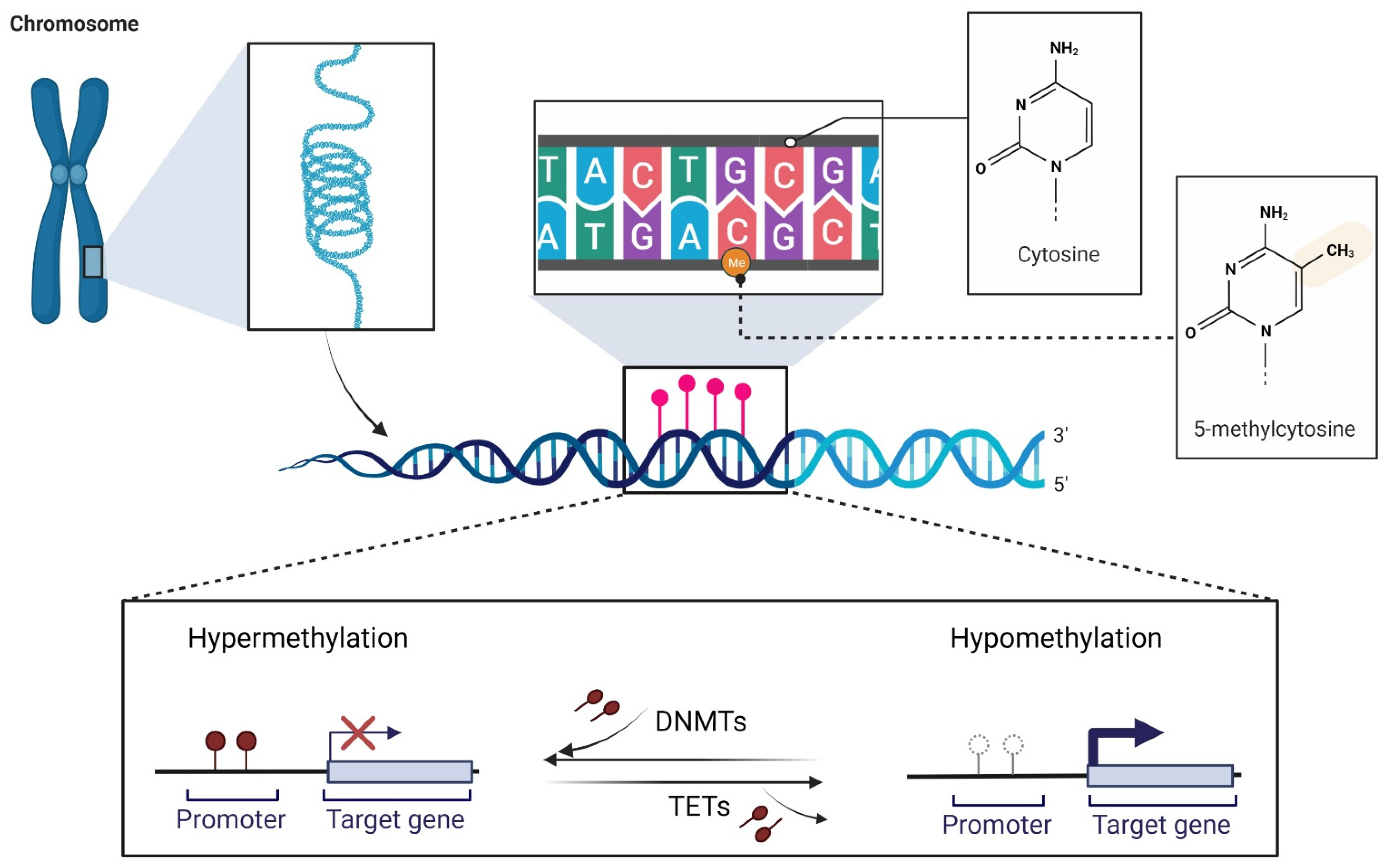
2. DNA Methylation and Melanoma
2.1. DNA Methylation at a Glance
2.2. DNA Methylation in Melanoma Development
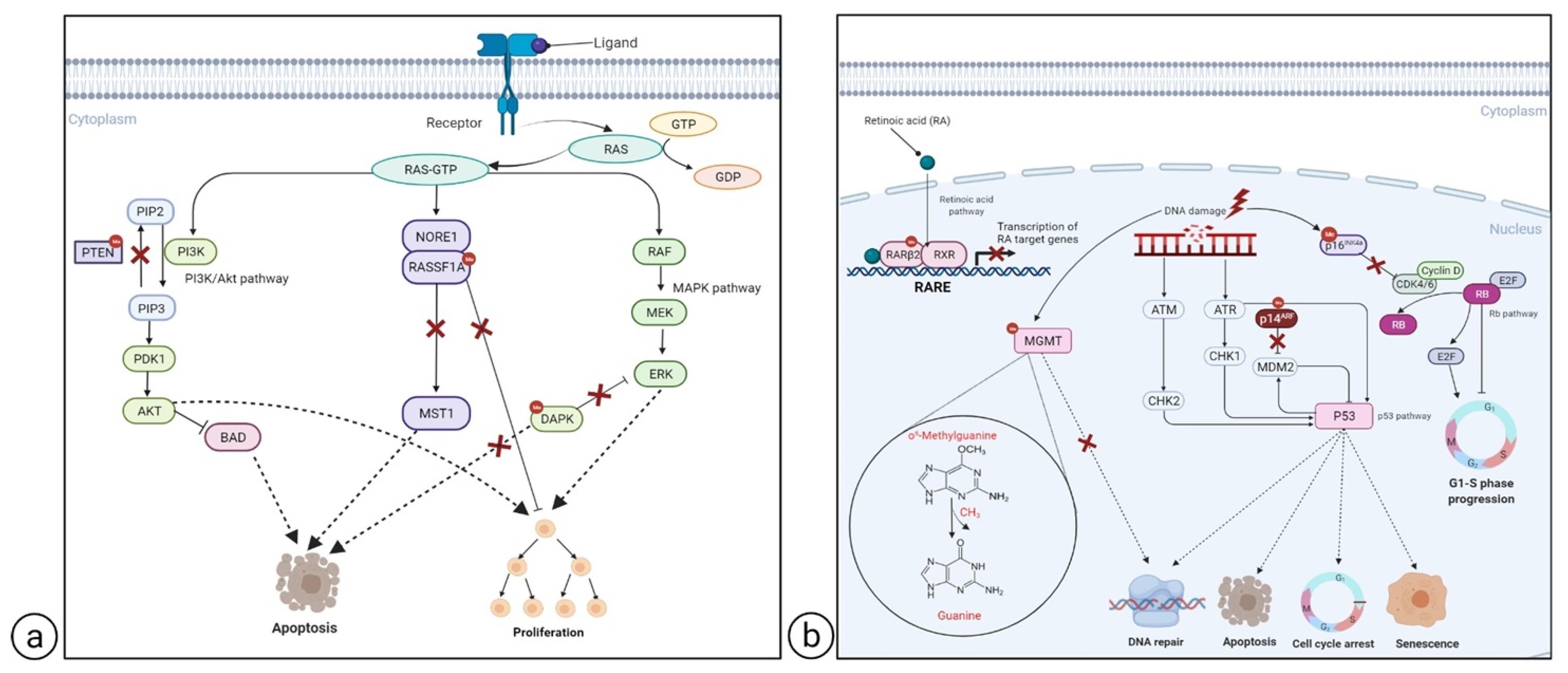
2.2.1. Hypermethylated Genes
RASSF1A
DAPK
PTEN
CDKN2A
MGMT
RARβ2
CpG Methylator Phenotype (CIMP) in Melanoma
2.2.2. Hypomethylated Genes
TBC1D16
PDGFD, ZEB1, and THRB
TKTL1
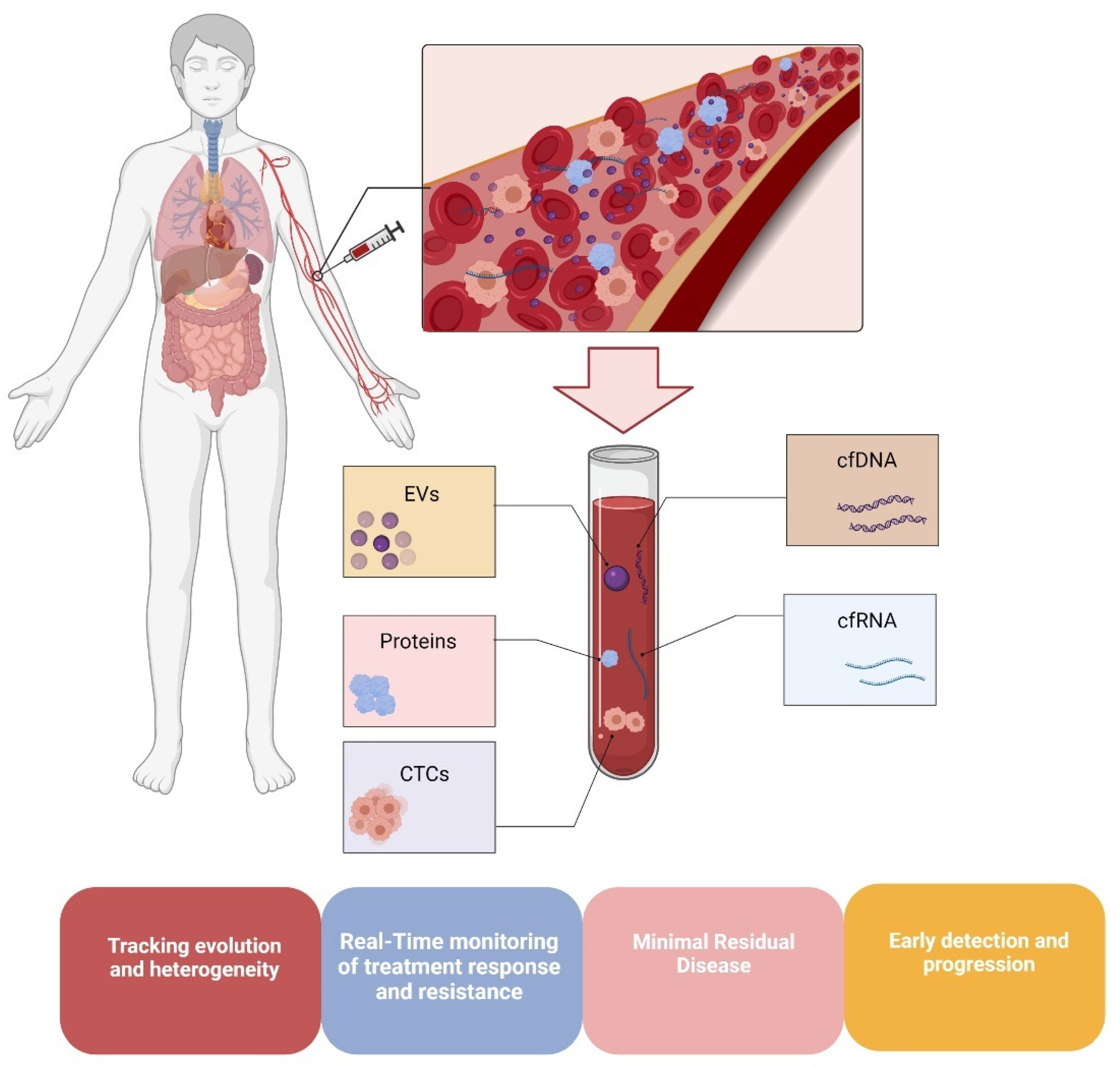
3. DNA Methylation in Melanoma Liquid Biopsies
3.1. DNA Methylation in Circulating Melanoma Cells
3.2. Methylation in Circulating Melanoma DNA
3.3. Methylation in Melanoma Extracellular Vesicle-Derived DNA (evDNA)
4. Circulating Methylated DNA Biomarkers for Tracking Response to Therapy and Resistance Onset
5. Methylation Markers from Bench to Bedside
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karimkhani, C.; Green, A.; Nijsten, T.; Weinstock, M.; Dellavalle, R.; Naghavi, M.; Fitzmaurice, C. The global burden of melanoma: Results from the Global Burden of Disease Study 2015. Br. J. Dermatol. 2017, 177, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Sileni, V.C.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage IIIBRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Sileni, V.C.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, Z.; Ribas, A. Combination therapy with BRAF and MEK inhibitors for melanoma: Latest evidence and place in therapy. Ther. Adv. Med Oncol. 2016, 8, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; De Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozar, I.; Margue, C.; Rothengatter, S.; Haan, C.; Kreis, S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim. Biophys. Acta BBA Rev. 2019, 1871, 313–322. [Google Scholar] [CrossRef]
- Gorges, K.; Wiltfang, L.; Gorges, T.M.; Sartori, A.; Hildebrandt, L.; Keller, L.; Volkmer, B.; Peine, S.; Babayan, A.; Moll, I.; et al. Intra-Patient Heterogeneity of Circulating Tumor Cells and Circulating Tumor DNA in Blood of Melanoma Patients. Cancers 2019, 11, 1685. [Google Scholar] [CrossRef] [Green Version]
- Frederick, D.T.; Piris, A.; Cogdill, A.; Cooper, Z.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF Inhibition Is Associated with Enhanced Melanoma Antigen Expression and a More Favorable Tumor Microenvironment in Patients with Metastatic Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; Sileni, V.C.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Wilmott, J.S.; Long, G.V.; Howle, J.R.; Haydu, L.E.; Sharma, R.N.; Thompson, J.F.; Kefford, R.F.; Hersey, P.; Scolyer, R.A. Selective BRAF Inhibitors Induce Marked T-cell Infiltration into Human Metastatic Melanoma. Clin. Cancer Res. 2011, 18, 1386–1394. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Serratì, S.; De Summa, S.; Pilato, B.; Petriella, D.; Lacalamita, R.; Tommasi, S.; Pinto, R. Next-generation sequencing: Advances and applications in cancer diagnosis. OncoTargets Ther. 2016, 9, 7355–7365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.; Kim, M.; Kim, M.J.; Kim, H.; Ock, C.-Y.; Keam, B.; Kim, T.M.; Kim, D.-W.; Kim, J.-I.; Heo, D.S. Clinical application of next-generation sequencing-based panel to BRAF wild-type advanced melanoma identifies key oncogenic alterations and therapeutic strategies. Mol. Cancer Ther. 2019, 19, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Pleasance, E.D.; Cheetham, R.K.; Stephens, P.J.; McBride, D.J.; Humphray, S.J.; Greenman, C.D.; Varela, I.; Lin, M.-L.; Ordóñez, G.R.; Bignell, G.R.; et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2009, 463, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Papadodima, O.; Kontogianni, G.; Piroti, G.; Maglogiannis, I.; Chatziioannou, A. Contralateral axillary metastasis: Is surgical treatment the best option? J. Transl. Genet. Genom. 2019, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Scatena, C.; Murtas, D.; Tomei, S. Cutaneous Melanoma Classification: The Importance of High-Throughput Genomic Technologies. Front. Oncol. 2021, 11, 1313. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The Epigenomics of Cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCabe, M.T.; Brandes, J.; Vertino, P.M. Cancer DNA Methylation: Molecular Mechanisms and Clinical Implications. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 3927–3937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, P.; Liu, D.; Dong, J.; Xing, M. The BRAFV600E Causes Widespread Alterations in Gene Methylation in the Genome of Melanoma Cells. Cell Cycle Georget. Tex 2012, 11, 286–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothhammer, T.; Bosserhoff, A.-K. Epigenetic events in malignant melanoma. Pigment. Cell Res. 2007, 20, 92–111. [Google Scholar] [CrossRef] [PubMed]
- Ewarton, K.; Esamimi, G. Methylation of cell-free circulating DNA in the diagnosis of cancer. Front. Mol. Biosci. 2015, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Waterland, R.A. Epigenetic mechanisms and gastrointestinal development. J. Pediatr. 2006, 149, S137–S142. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Y. Role of Epigenetic Regulation in Plasticity of Tumor Immune Microenvironment. Front. Immunol. 2021, 12, 640369. [Google Scholar] [CrossRef]
- Ortiz-Barahona, V.; Joshi, R.S.; Esteller, M. Use of DNA methylation profiling in translational oncology. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Esteller, M. 2-DNA Methylation and Cancer. In Advances in Genetics; Herceg, Z., Ushijima, T., Eds.; Epigenetics and Cancer, Part A; Academic Press: Cambridge, MA, USA, 2010; Volume 70, pp. 27–56. [Google Scholar]
- Witte, T.; Plass, C.; Gerhauser, C. Pan-cancer patterns of DNA methylation. Genome Med. 2014, 6, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [Green Version]
- Karpf, A.R.; Matsui, S.-I. Genetic Disruption of Cytosine DNA Methyltransferase Enzymes Induces Chromosomal Instability in Human Cancer Cells. Cancer Res. 2005, 65, 8635–8639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheaffer, K.L.; Elliott, E.N.; Kaestner, K.H. DNA Hypomethylation Contributes to Genomic Instability and Intestinal Cancer Initiation. Cancer Prev. Res. 2016, 9, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Holm, T.M.; Jackson-Grusby, L.; Brambrink, T.; Yamada, Y.; Rideout, W.M.; Jaenisch, R. Global loss of imprinting leads to widespread tumorigenesis in adult mice. Cancer Cell 2005, 8, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Iskow, R.; Yang, L.; Gokcumen, O.; Haseley, P.; Luquette, L.J.; Lohr, J.G.; Harris, C.C.; Ding, L.; Wilson, R.K.; et al. Landscape of Somatic Retrotransposition in Human Cancers. Science 2012, 337, 967–971. [Google Scholar] [CrossRef] [Green Version]
- Tubio, J.M.; Li, Y.; Ju, Y.S.; Martincorena, I.; Cooke, S.L.; Tojo, M.; Gundem, G.; Pipinikas, C.P.; Zamora, J.; Raine, K.; et al. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science 2014, 345, 1251343. [Google Scholar] [CrossRef] [Green Version]
- Micevic, G.; Theodosakis, N.; Bosenberg, M. Aberrant DNA methylation in melanoma: Biomarker and therapeutic opportunities. Clin. Epigenet. 2017, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinke, C.; Mo, Y.; Yu, Y.; Amiri, K.; Sosman, J.; Greally, J.; Verma, A. Aberrant DNA methylation in malignant melanoma. Melanoma Res. 2010, 20, 253–265. [Google Scholar] [CrossRef]
- Dobre, E.-G.; Constantin, C.; Costache, M.; Neagu, M. Interrogating Epigenome toward Personalized Approach in Cutaneous Melanoma. J. Pers. Med. 2021, 11, 901. [Google Scholar] [CrossRef] [PubMed]
- Howell, J.P.M.; Liu, S.; Ren, S.; Behlen, C.; Fodstad, O.; Riker, A.I. Epigenetics in Human Melanoma. Cancer Control 2009, 16, 200–218. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-Castañeda, L.D.; Nova, J.A.; Tovar-Parra, J.D. Frequency of mutations in BRAF, NRAS, and KIT in different populations and histological subtypes of melanoma: A systemic review. Melanoma Res. 2020, 30, 62–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Jia, P.; Hutchinson, K.E.; Dahlman, K.B.; Johnson, D.; Sosman, J.; Pao, W.; Zhao, Z. A Meta-analysis of Somatic Mutations from Next Generation Sequencing of 241 Melanomas: A Road Map for the Study of Genes with Potential Clinical Relevance. Mol. Cancer Ther. 2014, 13, 1918–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanemura, A.; Terando, A.M.; Sim, M.-S.; Van Hoesel, A.Q.; De Maat, M.F.; Morton, D.L.; Hoon, D.S. CpG Island Methylator Phenotype Predicts Progression of Malignant Melanoma. Clin. Cancer Res. 2009, 15, 1801–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef] [Green Version]
- Spugnardi, M.; Tommasi, S.; Dammann, R.; Pfeifer, G.P.; Hoon, D.S.B. Epigenetic inactivation of RAS association domain family protein 1 (RASSF1A) in malignant cutaneous melanoma. Cancer Res. 2003, 63, 1639–1643. [Google Scholar] [PubMed]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Baylin, S.B.; Herman, J.G. DNA hypermethylation in tumorigenesis: Epigenetics joins genetics. Trends Genet. 2000, 16, 168–174. [Google Scholar] [CrossRef]
- Esteller, M.; Corn, P.G.; Baylin, S.B.; Herman, J.G. A gene hypermethylation profile of human cancer. Cancer Res. 2001, 61, 3225–3229. [Google Scholar] [PubMed]
- Koroknai, V.; Szász, I.; Hernandez-Vargas, H.; Fernandez-Jimenez, N.; Cuenin, C.; Herceg, Z.; Vízkeleti, L.; Ádány, R.; Ecsedi, S.; Balázs, M. DNA hypermethylation is associated with invasive phenotype of malignant melanoma. Exp. Dermatol. 2020, 29, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Wu, H.; Zhang, H.; Lian, C.G.; Lu, Q. DNA methylation/hydroxymethylation in melanoma. Oncotarget 2017, 8, 78163–78173. [Google Scholar] [CrossRef] [Green Version]
- Bustos, B.D.U.; Estal, R.M.; Simó, G.P.; Farinos, J.S.; Marco, C.P.; Mira, M.N.; de Miquel, V.A.; Sánchez, R.B.; Marco, V.S.; Ros, M.L.; et al. Aberrant DNA methylation is associated with aggressive clinicopathological features and poor survival in cutaneous melanoma. Br. J. Dermatol. 2017, 179, 394–404. [Google Scholar] [CrossRef]
- Christmann, M.; Pick, M.; Lage, H.; Schadendorf, D.; Kaina, B. Acquired resistance of melanoma cells to the antineoplastic agent fotemustine is caused by reactivation of the DNA repair gene MGMT. Int. J. Cancer 2001, 92, 123–129. [Google Scholar] [CrossRef]
- Fujimoto, A.; Morita, R.; Hatta, N.; Takehara, K.; Takata, M. p16INK4a inactivation is not frequent in uncultured sporadic primary cutaneous melanoma. Oncogene 1999, 18, 2527–2532. [Google Scholar] [CrossRef] [Green Version]
- Dammann, R.H.; Richter, A.M.; Jiménez, A.P.; Woods, M.; Küster, M.; Witharana, C. Impact of Natural Compounds on DNA Methylation Levels of the Tumor Suppressor Gene RASSF1A in Cancer. Int. J. Mol. Sci. 2017, 18, 2160. [Google Scholar] [CrossRef] [Green Version]
- Marzese, D.M.; Scolyer, R.A.; Roqué, M.; Vargas-Roig, L.M.; Huynh, J.L.; Wilmott, J.S.; Murali, R.; Buckland, M.E.; Barkhoudarian, G.; Thompson, J.F.; et al. DNA methylation and gene deletion analysis of brain metastases in melanoma patients identifies mutually exclusive molecular alterations. Neuro-Oncology 2014, 16, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Worm, J.; Christensen, C.; Grønbæk, K.; Tulchinsky, E.; Guldberg, P. Genetic and epigenetic alterations of the APC gene in malignant melanoma. Oncogene 2004, 23, 5215–5226. [Google Scholar] [CrossRef] [Green Version]
- Carmona, F.J.; Villanueva, A.; Vidal, A.; Muñoz, C.; Puertas, S.; Penin, R.M.; Gomà, M.; Lujambio, A.; Piulats, J.M.; Mesía, R.; et al. Epigenetic disruption of cadherin-11 in human cancer metastasis. J. Pathol. 2012, 228, 230–240. [Google Scholar] [CrossRef] [Green Version]
- Kaehler, K.C.; Politz, O.; Henderson, D.; Ulbrich, H.-F.; Hauschild, A.; Mund, C.; Egberts, F. Novel DNA methylation markers with potential prognostic relevance in advanced malignant melanoma identified using COBRA assays. Melanoma Res. 2015, 25, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Hurk, K.V.D.; Moerkerk, P.T.; Goeman, J.; Beck, S.; Gruis, N.; Oord, J.J.V.D.; Winnepenninckx, V.J.; van Engeland, M.; van Doorn, R. Promoter CpG Island Hypermethylation in Dysplastic Nevus and Melanoma: CLDN11 as an Epigenetic Biomarker for Malignancy. J. Investig. Dermatol. 2014, 134, 2957–2966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuphal, S.; Martyn, A.C.; Pedley, J.; Crowther, L.M.; Bonazzi, V.F.; Parsons, P.G.; Bosserhoff, A.K.; Hayward, N.K.; Boyle, G.M. H-Cadherin expression reduces invasion of malignant melanoma. Pigment Cell Melanoma Res. 2009, 22, 296–306. [Google Scholar] [CrossRef]
- Frequent intra-tumoural heterogeneity of promoter hypermethylation in malignant melanoma. Histol. Histopathol 2007, 22, 1005–1015.
- Jonsson, A.; Tuominen, R.; Grafström, E.; Hansson, J.; Egyhazi, S. High Frequency of p16INK4A Promoter Methylation in NRAS-Mutated Cutaneous Melanoma. J. Investig. Dermatol. 2010, 130, 2809–2817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedberg, D.E.; Rigas, S.H.; Russak, J.; Gai, W.; Kaplow, M.; Osman, I.; Turner, F.; Randerson-Moor, J.A.; Houghton, A.; Busam, K.; et al. Frequent p16-Independent Inactivation of p14ARF in Human Melanoma. J. Natl. Cancer Inst. 2008, 100, 784–795. [Google Scholar] [CrossRef] [Green Version]
- Sherr, C.J. The Pezcoller lecture: Cancer cell cycles revisited. Cancer Res. 2000, 60, 3689–3695. [Google Scholar]
- Pomerantz, J.; Schreiber-Agus, N.; Liégeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.-W.; et al. The Ink4a Tumor Suppressor Gene Product, p19Arf, Interacts with MDM2 and Neutralizes MDM2’s Inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.; Lee, H.-W.; Chin, L.; Cordon-Cardo, C.; Beach, D.; DePinho, R. Role of the INK4a Locus in Tumor Suppression and Cell Mortality. Cell 1996, 85, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, G.I.; Edwards, C.D.; Ewen, M.E.; Rollins, B.J. p16 INK4A Participates in a G 1 Arrest Checkpoint in Response to DNA Damage. Mol. Cell. Biol. 1998, 18, 378–387. [Google Scholar] [CrossRef] [Green Version]
- Marini, A.; Mirmohammadsadegh, A.; Nambiar, S.; Gustrau, A.; Ruzicka, T.; Hengge, U.R. Epigenetic Inactivation of Tumor Suppressor Genes in Serum of Patients with Cutaneous Melanoma. J. Investig. Dermatol. 2006, 126, 422–431. [Google Scholar] [CrossRef]
- Venza, M.; Visalli, M.; Biondo, C.; Lentini, M.; Catalano, T.; Teti, D.; Venza, I. Epigenetic regulation of p14ARF and p16INK4A expression in cutaneous and uveal melanoma. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2015, 1849, 247–256. [Google Scholar] [CrossRef]
- Walesch, S.K.; Richter, A.M.; Helmbold, P.; Dammann, R.H. Claudin11 Promoter Hypermethylation Is Frequent in Malignant Melanoma of the Skin, but Uncommon in Nevus Cell Nevi. Cancers 2015, 7, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Smit, M.A.; Oord, J.J.V.D.; Goeman, J.J.; Verdegaal, E.M.E.; Van Der Burg, S.H.; Stas, M.; Beck, S.; Gruis, N.A.; Tensen, C.; et al. Genome-wide promoter methylation analysis identifies epigenetic silencing ofMAPK13in primary cutaneous melanoma. Pigment Cell Melanoma Res. 2013, 26, 542–554. [Google Scholar] [CrossRef]
- Hoon, D.S.B.; Spugnardi, M.; Kuo, C.; Huang, S.K.; Morton, D.L.; Taback, B. Profiling epigenetic inactivation of tumor suppressor genes in tumors and plasma from cutaneous melanoma patients. Oncogene 2004, 23, 4014–4022. [Google Scholar] [CrossRef] [Green Version]
- Merhavi, E.; Cohen, Y.; Avraham, B.C.R.; Frenkel, S.; Chowers, I.; Pe’Er, J.; Goldenberg-Cohen, N. Promoter Methylation Status of Multiple Genes in Uveal Melanoma. Investig. Opthalmol. Vis. Sci. 2007, 48, 4403–4406. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.; O’Day, S.J.; Umetani, N.; Martinez, S.R.; Kitago, M.; Koyanagi, K.; Kuo, C.; Takeshima, T.-L.; Milford, R.; Wang, H.-J.; et al. Predictive Utility of Circulating Methylated DNA in Serum of Melanoma Patients Receiving Biochemotherapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 9351–9358. [Google Scholar] [CrossRef] [PubMed]
- Olvedy, M.; Tisserand, J.C.; Luciani, F.; Boeckx, B.; Wouters, J.; Lopez, S.; Rambow, F.; Aibar, S.; Thienpont, B.; Barra, J.; et al. Comparative Oncogenomics Identifies Tyrosine Kinase FES as a Tumor Suppressor in Melanoma. Available online: https://www.jci.org/articles/view/91291/pdf (accessed on 19 August 2021).
- Wouters, J.; Vizoso, M.; Martinez-Cardus, A.; Carmona, F.J.; Govaere, O.; Laguna, T.; Joseph, J.; Dynoodt, P.; Aura, C.; Foth, M.; et al. Comprehensive DNA methylation study identifies novel progression-related and prognostic markers for cutaneous melanoma. BMC Med. 2017, 15, 101. [Google Scholar] [CrossRef]
- Diefenbach, R.J.; Lee, J.H.; Chandler, D.; Wang, Y.; Pflueger, C.; Long, G.V.; Scolyer, R.A.; Carlino, M.S.; Menzies, A.M.; Kefford, R.F.; et al. Hypermethylation of Circulating Free DNA in Cutaneous Melanoma. Appl. Sci. 2019, 9, 5074. [Google Scholar] [CrossRef] [Green Version]
- Hassel, J.C.; Sucker, A.; Edler, L.; Kurzen, H.; Moll, I.; Stresemann, C.; Spieth, K.; Mauch, C.; Rass, K. MGMT gene promoter methylation correlates with tolerance of temozolomide treatment in melanoma but not with clinical outcome. Br. J. Cancer 2010, 103, 820–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuominen, R.; Jewell, R.; Oord, J.J.V.D.; Wolter, P.; Stierner, U.; Lindholm, C.; Johansson, C.H.; Lindén, D.; Johansson, H.; Stolt, M.F.; et al. MGMT promoter methylation is associated with temozolomide response and prolonged progression-free survival in disseminated cutaneous melanoma. Int. J. Cancer 2014, 136, 2844–2853. [Google Scholar] [CrossRef] [Green Version]
- Guadagni, S.; Fiorentini, G.; Clementi, M.; Palumbo, G.; Masedu, F.; Deraco, M.; De Manzoni, G.; Chiominto, A.; Valenti, M.; Pellegrini, C. MGMT methylation correlates with melphalan pelvic perfusion survival in stage III melanoma patients: A pilot study. Melanoma Res. 2017, 27, 439–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tellez, C.S.; Shen, L.; Estecio, M.; Jelinek, J.; Gershenwald, J.E.; Issa, J.-P. CpG island methylation profiling in human melanoma cell lines. Melanoma Res. 2009, 19, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Czyz, M. MITF in melanoma: Mechanisms behind its expression and activity. Cell. Mol. Life Sci. 2015, 72, 1249–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Araújo, É.S.S.; Pramio, D.T.; Kashiwabara, A.Y.; Pennacchi, P.C.; Maria-Engler, S.S.; Achatz, M.I.; Campos, A.H.J.F.M.; Duprat, J.P.; Rosenberg, C.; Carraro, D.M.; et al. DNA Methylation Levels of Melanoma Risk Genes Are Associated with Clinical Characteristics of Melanoma Patients. BioMed Res. Int. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Micevic, G.; Thakral, D.; McGeary, M.; Bosenberg, M.W. PD-L1 methylation regulates PD-L1 expression and is associated with melanoma survival. Pigment Cell Melanoma Res. 2018, 32, 435–440. [Google Scholar] [CrossRef]
- Stahl, J.M.; Cheung, M.; Sharma, A.; Trivedi, N.R.; Shanmugam, S.; Robertson, G.P. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res. 2003, 63, 2881–2890. [Google Scholar]
- Mirmohammadsadegh, A.; Marini, A.; Nambiar, S.; Hassan, M.; Tannapfel, A.; Ruzicka, T.; Hengge, U.R. Epigenetic Silencing of the PTEN Gene in Melanoma. Cancer Res. 2006, 66, 6546–6552. [Google Scholar] [CrossRef] [Green Version]
- Lahtz, C.; Stranzenbach, R.; Fiedler, E.; Helmbold, P.; Dammann, R.H. Methylation of PTEN as a Prognostic Factor in Malignant Melanoma of the Skin. J. Investig. Dermatol. 2010, 130, 620–622. [Google Scholar] [CrossRef] [Green Version]
- Roh, M.R.; Gupta, S.; Park, K.-H.; Chung, K.Y.; Lauss, M.; Flaherty, K.T.; Jönsson, G.; Rha, S.Y.; Tsao, H. Promoter Methylation of PTEN Is a Significant Prognostic Factor in Melanoma Survival. J. Investig. Dermatol. 2016, 136, 1002–1011. [Google Scholar] [CrossRef]
- Furuta, J.; Umebayashi, Y.; Miyamoto, K.; Kikuchi, K.; Otsuka, F.; Sugimura, T.; Ushijima, T. Promoter methylation profiling of 30 genes in human malignant melanoma. Cancer Sci. 2004, 95, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ren, S.; Howell, P.; Fodstad, O.; Riker, A.I. Identification of novel epigenetically modified genes in human melanoma via promoter methylation gene profiling. Pigment. Cell Melanoma Res. 2008, 21, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Salvianti, F.; Orlando, C.; Massi, D.; de Giorgi, V.; Grazzini, M.; Pazzagli, M.; Pinzani, P. Tumor-Related Methylated Cell-Free DNA and Circulating Tumor Cells in Melanoma. Front Mol. Biosci. 2016, 2, 76. [Google Scholar] [CrossRef] [Green Version]
- Reifenberger, J.; Knobbe, C.B.; Sterzinger, A.A.; Blaschke, B.; Schulte, K.W.; Ruzicka, T.; Reifenberger, G. Frequent alterations of Ras signaling pathway genes in sporadic malignant melanomas. Int. J. Cancer 2004, 109, 377–384. [Google Scholar] [CrossRef]
- Maat, W.; Van Der Velden, P.A.; Out-Luiting, C.; Plug, M.; Dirks-Mulder, A.; Jager, M.J.; Gruis, N.A. Epigenetic Inactivation of RASSF1a in Uveal Melanoma. Investig. Ophthalmol. Vis. Sci. 2007, 48, 486–490. [Google Scholar] [CrossRef] [Green Version]
- Donninger, H.; Vos, M.D.; Clark, G.J. The RASSF1A tumor suppressor. J. Cell Sci. 2007, 120, 3163–3172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Gutiérrez, L.; McKenna, S.; Kolch, W.; Matallanas, D. RASSF1A Tumour Suppressor: Target the Network for Effective Cancer Therapy. Cancers 2020, 12, 229. [Google Scholar] [CrossRef] [Green Version]
- Yi, M.; Yang, J.; Chen, X.; Li, J.; Li, X.; Wang, L.; Tan, Y.; Xiong, W.; Zhou, M.; McCarthy, J.B.; et al. RASSF1A suppresses melanoma development by modulating apoptosis and cell-cycle progression. J. Cell. Physiol. 2011, 226, 2360–2369. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, R.; Bialik, S.; Kimchi, A. The DAPK family: A structure—function analysis. Apoptosis 2014, 19, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Bialik, S.; Kimchi, A. The Death-Associated Protein Kinases: Structure, Function, and Beyond. Annu. Rev. Biochem. 2006, 75, 189–210. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Ravanan, P.; Talwar, P. Death Associated Protein Kinase 1 (DAPK1): A Regulator of Apoptosis and Autophagy. Front. Mol. Neurosci. 2016, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Inbal, B.; Bialik, S.; Sabanay, I.; Shani, G.; Kimchi, A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J. Cell Biol. 2002, 157, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Zalckvar, E.; Berissi, H.; Eisenstein, M.; Kimchi, A. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 2009, 5, 720–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozuacik, D.; Kimchi, A. DAPk protein family and cancer. Autophagy 2006, 2, 74–79. [Google Scholar] [CrossRef] [Green Version]
- Kissil, J.L.; Feinstein, E.; Cohen, O.; Jones, P.A.; Tsai, Y.C.; Knowles, M.A.; Eydmann, M.E.; Kimchi, A. DAP-kinase loss of expression in various carcinoma and B-cell lymphoma cell lines: Possible implications for role as tumor suppressor gene. Oncogene 1997, 15, 403–407. [Google Scholar] [CrossRef] [Green Version]
- Raveh, T.; Droguett, G.; Horwitz, M.S.; DePinho, R.; Kimchi, A. DAP kinase activates a p19ARF/p53-mediated apoptotic checkpoint to suppress oncogenic transformation. Nat. Cell Biol. 2000, 3, 1–7. [Google Scholar] [CrossRef]
- Inbal, B.; Cohen, O.; Polak-Charcon, S.; Kopolovic, J.; Vadai, E.; Eisenbach, L.R.; Kimchi, A. DAP kinase links the control of apoptosis to metastasis. Nature 1997, 390, 180–184. [Google Scholar] [CrossRef]
- Kuo, J.-C.; Wang, W.-J.; Yao, C.-C.; Wu, P.-R.; Chen, R.-H. The tumor suppressor DAPK inhibits cell motility by blocking the integrin-mediated polarity pathway. J. Cell Biol. 2006, 172, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Simpson, L.; Parsons, R. PTEN: Life as a Tumor Suppressor. Exp. Cell Res. 2001, 264, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiong, Y.; Yarbrough, W.G. ARF Promotes MDM2 Degradation and Stabilizes p53: ARF-INK4a Locus Deletion Impairs Both the Rb and p53 Tumor Suppression Pathways. Cell 1998, 92, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.G. Hypermethylation of tumor suppressor genes in cancer. Semin. Cancer Biol. 1999, 9, 359–367. [Google Scholar] [CrossRef]
- Tano, K.; Shiota, S.; Collier, J.; Foote, R.S.; Mitra, S. Isolation and structural characterization of a cDNA clone encoding the human DNA repair protein for O6-alkylguanine. Proc. Natl. Acad. Sci. USA 1990, 87, 686–690. [Google Scholar] [CrossRef] [Green Version]
- Hegi, M.E.; Hamou, M.-F.; De Tribolet, N.; Kros, J.M.; Mariani, L.; Mirimanoff, R.O.; Stupp, R. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Olsson, M.; Lindahl, T. Repair of alkylated DNA in Escherichia coli. Methyl group transfer from O6-methylguanine to a protein cysteine residue. J. Biol. Chem. 1980, 255, 10569–10571. [Google Scholar] [CrossRef]
- Costello, J.F.; Futscher, B.W.; Tano, K.; Graunke, D.M.; Pieper, R.O. Graded methylation in the promoter and body of the O6-methylguanine DNA methyltransferase (MGMT) gene correlates with MGMT expression in human glioma cells. J. Biol. Chem. 1994, 269, 17228–17237. [Google Scholar] [CrossRef]
- Kato, Y. The Role and Epigenetic Modification of the Retinoic Acid Receptor. In Handbook of Nutrition, Diet, and Epigenetics; Patel, V., Preedy, V., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 1–10. [Google Scholar]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.-P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Alvarez, A.; Ortiz, C.; Muñoz-Couselo, E. Current Perspectives and Novel Strategies of NRAS-Mutant Melanoma. OncoTargets Ther. 2021, 14, 3709–3719. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.-G.; Xiong, W.; Wu, X.; Yang, L.; Pfeifer, G.P. The DNA methylation landscape of human melanoma. Genomics 2015, 106, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Gama-Sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.D.; Kim, J. Global DNA Hypomethylation in Peripheral Blood Leukocytes as a Biomarker for Cancer Risk: A Meta-Analysis. PLoS ONE 2012, 7, e34615. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Flanagan, J. Is There a Link Between Genome-Wide Hypomethylation in Blood and Cancer Risk? Cancer Prev. Res. 2012, 5, 1345–1357. [Google Scholar] [CrossRef] [Green Version]
- Ecsedi, S.I.; Hernandez-Vargas, H.; Lima, S.C.; Herceg, Z.; Adany, R.; Balazs, M. Transposable hypomethylation is associated with metastatic capacity of primary melanomas. Int. J. Clin. Exp. Pathol. 2013, 6, 2943–2948. [Google Scholar]
- Estécio, M.R.; Gallegos, J.; Vallot, C.; Castoro, R.J.; Chung, W.; Maegawa, S.; Oki, Y.; Kondo, Y.; Jelinek, J.; Shen, L.; et al. Genome architecture marked by retrotransposons modulates predisposition to DNA methylation in cancer. Genome Res. 2010, 20, 1369–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poage, G.M.; Houseman, E.A.; Christensen, B.C.; Butler, R.A.; Avissar-Whiting, M.; McClean, M.; Waterboer, T.; Pawlita, M.; Marsit, C.; Kelsey, K.T. Global Hypomethylation Identifies Loci Targeted for Hypermethylation in Head and Neck Cancer. Clin. Cancer Res. 2011, 17, 3579–3589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshimoto, S.; Kuo, C.T.; Chong, K.K.; Takeshima, T.-L.; Takei, Y.; Li, M.W.; Huang, S.K.; Sim, M.-S.; Morton, D.L.; Hoon, D.S. AIM1 and LINE-1 Epigenetic Aberrations in Tumor and Serum Relate to Melanoma Progression and Disease Outcome. J. Investig. Dermatol. 2012, 132, 1689–1697. [Google Scholar] [CrossRef] [Green Version]
- Venza, M.; Visalli, M.; Catalano, T.; Beninati, C.; Teti, D.; Venza, I. DSS1 promoter hypomethylation and overexpression predict poor prognosis in melanoma and squamous cell carcinoma patients. Hum. Pathol. 2017, 60, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Pramio, D.T.; Pennacchi, P.C.; Maria-Engler, S.S.; Campos, A.H.J.F.M.; Duprat, J.P.; Carraro, D.M.; Krepischi, A.C.V. LINE-1 hypomethylation and mutational status in cutaneous melanomas. J. Investig. Med. 2016, 64, 899–904. [Google Scholar] [CrossRef]
- De Smet, C.; De Backer, O.; Faraoni, I.; Lurquin, C.; Brasseur, F.; Boon, T. The activation of human gene MAGE-1 in tumor cells is correlated with genome-wide demethylation. Proc. Natl. Acad. Sci. USA 1996, 93, 7149–7153. [Google Scholar] [CrossRef] [Green Version]
- Denk, A.E.; Bettstetter, M.; Wild, P.J.; Hoek, K.; Bataille, F.; Dietmaier, W.; Bosserhoff, A. Loss of maspin expression contributes to a more invasive potential in malignant melanoma. Pigment Cell Res. 2007, 20, 112–119. [Google Scholar] [CrossRef]
- Wada, K.; Maesawa, C.; Akasaka, T.; Masuda, T. Aberrant Expression of the Maspin Gene Associated with Epigenetic Modification in Melanoma Cells. J. Investig. Dermatol. 2004, 122, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Jia, P.; Hutchinson, K.E.; Johnson, D.B.; Sosman, J.A.; Zhao, Z. Clinically relevant genes and regulatory pathways associated with NRASQ61 mutations in melanoma through an integrative genomics approach. Oncotarget 2014, 6, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, F.; Zarbl, R.; Niebel, D.; Sirokay, J.; Fröhlich, A.; Posch, C.; Holderried, T.A.W.; Brossart, P.; Saavedra, G.; Kuster, P.; et al. Prognostic and predictive value of PD-L2 DNA methylation and mRNA expression in melanoma. Clin. Epigenet. 2020, 12, 1–12. [Google Scholar] [CrossRef]
- Vizoso, M.; Ferreira, H.; Lopez-Serra, P.; Carmona, F.J.; Cardus, A.M.; Girotti, M.R.; Villanueva, A.; Guil, S.; Moutinho, C.; Liz, J.; et al. Epigenetic activation of a cryptic TBC1D16 transcript enhances melanoma progression by targeting EGFR. Nat. Med. 2015, 21, 741–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayachandran, A.; Lo, P.-H.; Chueh, A.C.; Prithviraj, P.; Molania, R.; Davalos-Salas, M.; Anaka, M.; Walkiewicz, M.; Cebon, J.; Behren, A. Transketolase-like 1 ectopic expression is associated with DNA hypomethylation and induces the Warburg effect in melanoma cells. BMC Cancer 2016, 16, 134. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Kong, D.; Li, Y.; Sarkar, F.H. PDGF-D Signaling: A Novel Target in Cancer Therapy. Curr. Drug Targets 2009, 10, 38–41. [Google Scholar] [CrossRef]
- Wels, C.; Joshi, S.; Koefinger, P.; Bergler, H.; Schaider, H. Transcriptional Activation of ZEB1 by Slug Leads to Cooperative Regulation of the Epithelial–Mesenchymal Transition-Like Phenotype in Melanoma. J. Investig. Dermatol. 2011, 131, 1877–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiya, Y.; Puzianowska-Kuznicka, M.; McPHIE, P.; Nauman, J.; Cheng, S.-Y.; Nauman, A. Expression of mutant thyroid hormone nuclear receptors is associated with human renal clear cell carcinoma. Carcinogenesis 2002, 23, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamińska, P.; Buszka, K.; Zabel, M.; Nowicki, M.; Alix-Panabières, C.; Budna-Tukan, J. Liquid Biopsy in Melanoma: Significance in Diagnostics, Prediction and Treatment Monitoring. Int. J. Mol. Sci. 2021, 22, 9714. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Alix-Panabières, C. Circulating tumour cells in cancer patients: Challenges and perspectives. Trends Mol. Med. 2010, 16, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Pantel, K. Liquid Biopsy: From Discovery to Clinical Application. Cancer Discov. 2021, 11, 858–873. [Google Scholar] [CrossRef]
- Cabel, L.; Proudhon, C.; Gortais, H.; Loirat, D.; Coussy, F.; Pierga, J.-Y.; Bidard, F.-C. Circulating tumor cells: Clinical validity and utility. Int. J. Clin. Oncol. 2017, 22, 421–430. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Schwarzenbach, H.; Pantel, K. Circulating Tumor Cells and Circulating Tumor DNA. Annu. Rev. Med. 2012, 63, 199–215. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Marsavela, G.; Aya-Bonilla, C.A.; Warkiani, M.E.; Gray, E.S.; Ziman, M. Melanoma circulating tumor cells: Benefits and challenges required for clinical application. Cancer Lett. 2018, 424, 1–8. [Google Scholar] [CrossRef]
- Rapanotti, M.C.; Campione, E.; Spallone, G.; Orlandi, A.; Bernardini, S.; Bianchi, L. Minimal residual disease in melanoma: Circulating melanoma cells and predictive role of MCAM/MUC18/MelCAM/CD146. Cell Death Discov. 2017, 3, 17005. [Google Scholar] [CrossRef] [Green Version]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9, 2415. [Google Scholar] [CrossRef] [Green Version]
- Gray, E.S.; Witkowski, T.; Pereira, M.; Calapre, L.; Herron, K.; Irwin, D.; Chapman, B.; Khattak, M.A.; Raleigh, J.; Hatzimihalis, A.; et al. Genomic Analysis of Circulating Tumor DNA Using a Melanoma-Specific UltraSEEK Oncogene Panel. J. Mol. Diagn. JMD 2019, 21, 418–426. [Google Scholar] [CrossRef]
- Cheng, F.; Su, L.; Qian, C. Circulating tumor DNA: A promising biomarker in the liquid biopsy of cancer. Oncotarget 2016, 7, 48832–48841. [Google Scholar] [CrossRef] [Green Version]
- Cree, I.A.; Uttley, L.; Woods, H.B.; Kikuchi, H.; Reiman, A.; Harnan, S.; Whiteman, B.L.; Philips, S.T.; Messenger, M. The Evidence Base for Circulating Tumour DNA Blood-Based Biomarkers for the Early Detection of Cancer: A Systematic Mapping Review. BMC Cancer 2017, 17, 697. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.S.; Rizos, H.; Reid, A.L.; Boyd, S.C.; Pereira, M.R.; Lo, J.; Tembe, V.; Freeman, J.; Lee, J.H.; Scolyer, R.A.; et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015, 6, 42008–42018. [Google Scholar] [CrossRef] [Green Version]
- Santiago-Walker, A.; Gagnon, R.; Mazumdar, J.; Casey, M.; Long, G.; Schadendorf, D.; Flaherty, K.T.; Kefford, R.; Hauschild, A.; Hwu, P.; et al. Correlation of BRAF Mutation Status in Circulating-Free DNA and Tumor and Association with Clinical Outcome across Four BRAFi and MEKi Clinical Trials. Clin. Cancer Res. 2016, 22, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Wang, Z.; Liu, Z.; Liang, G.; Gu, W.; Ge, Q. Technical progress in circulating tumor DNA analysis using next generation sequencing. Mol. Cell. Probes 2020, 49, 101480. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Lin, J.; Li, J.; Huang, B.; Liu, J.; Chen, X.; Chen, X.-M.; Xu, Y.-M.; Huang, L.-F.; Wang, X.-Z. Exosomes: Novel Biomarkers for Clinical Diagnosis. Sci. World J. 2015, 2015, 1–8. [Google Scholar] [CrossRef]
- Alegre, E.; Zubiri, L.; Perez-Gracia, J.L.; González-Cao, M.; Soria, L.; Martín-Algarra, S.; González, A. Circulating melanoma exosomes as diagnostic and prognosis biomarkers. Clin. Chim. Acta Int. J. Clin. Chem. 2016, 454, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Moran, B.; Silva, R.; Perry, A.; Gallagher, W.M. Epigenetics of malignant melanoma. Semin. Cancer Biol. 2018, 51, 80–88. [Google Scholar] [CrossRef]
- Diefenbach, R.J.; Lee, J.H.; Rizos, H. Methylated circulating tumor DNA as a biomarker in cutaneous melanoma. Melanoma Manag. 2020, 7, MMT46. [Google Scholar] [CrossRef] [PubMed]
- Constâncio, V.; Nunes, S.P.; Henrique, R.; Jerónimo, C. DNA Methylation-Based Testing in Liquid Biopsies as Detection and Prognostic Biomarkers for the Four Major Cancer Types. Cells 2020, 9, 624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating Tumor Cells, Disease Progression, and Survival in Metastatic Breast Cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [Green Version]
- De Bono, J.S.; Scher, H.I.; Montgomery, R.B.; Parker, C.; Miller, M.C.; Tissing, H.; Doyle, G.V.; Terstappen, L.; Pienta, K.; Raghavan, D. Circulating Tumor Cells Predict Survival Benefit from Treatment in Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 6302–6309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.J.; Punt, C.J.A.; Iannotti, N.; Saidman, B.H.; Sabbath, K.D.; Gabrail, N.Y.; Picus, J.; Morse, M.; Mitchell, E.; Miller, M.C.; et al. Relationship of Circulating Tumor Cells to Tumor Response, Progression-Free Survival, and Overall Survival in Patients with Metastatic Colorectal Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 3213–3221. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, K.; Mori, T.; O’Day, S.J.; Martinez, S.R.; Wang, H.-J.; Hoon, D.S. Association of Circulating Tumor Cells with Serum Tumor-Related Methylated DNA in Peripheral Blood of Melanoma Patients. Cancer Res. 2006, 66, 6111–6117. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Wu, X.; Zheng, J.; Dong, D. DNA methylome profiling of circulating tumor cells in lung cancer at single base-pair resolution. Oncogene 2021, 40, 1884–1895. [Google Scholar] [CrossRef]
- Chen, H.; Su, Z.; Li, R.; Zhang, N.; Guo, H.; Bai, F. Single-cell DNA methylome analysis of circulating tumor cells. Chin. J. Cancer Res. 2021, 33, 391–404. [Google Scholar] [CrossRef]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112.e14. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Toung, J.; Jassowicz, A.; Vijayaraghavan, R.; Kang, H.; Zhang, R.; Kruglyak, K.; Huang, H.; Hinoue, T.; Shen, H.; et al. Targeted methylation sequencing of plasma cell-free DNA for cancer detection and classification. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1445–1453. [Google Scholar] [CrossRef]
- Klein, E.; Richards, D.; Cohn, A.; Tummala, M.; Lapham, R.; Cosgrove, D.; Chung, G.; Clement, J.; Gao, J.; Hunkapiller, N.; et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann. Oncol. 2021, 32, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Catoni, C.; Di Paolo, V.; Rossi, E.; Quintieri, L.; Zamarchi, R. Cell-Secreted Vesicles: Novel Opportunities in Cancer Diagnosis, Monitoring and Treatment. Diagnostics 2021, 11, 1118. [Google Scholar] [CrossRef]
- Ibn Sina, A.A.; Lin, T.-Y.; Vaidyanathan, R.; Wang, Z.; Dey, S.; Wang, J.; Behren, A.; Wuethrich, A.; Carrascosa, L.G.; Trau, M. Methylation dependent gold adsorption behaviour identifies cancer derived extracellular vesicular DNA. Nanoscale Horiz. 2020, 5, 1317–1323. [Google Scholar] [CrossRef]
- Sadovska, L.; Santos, C.B.; Kalniņa, Z.; Linē, A. Biodistribution, Uptake and Effects Caused by Cancer-Derived Extracellular Vesicles. J. Circ. Biomark. 2015, 4, 4. [Google Scholar] [CrossRef]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, K.C.; Bond, D.T.; McKee, M.; Skog, J.; Paunescu, T.; Da Silva, N.; Brown, D.; Russo, L.M. Nucleic acids within urinary exosomes/microvesicles are potential biomarkers for renal disease. Kidney Int. 2010, 78, 191–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Romero, N.; Madurga, R.; Rackov, G.; Aliana, I.P.; Núñez-Torres, R.; Asensi-Puig, A.; Carrión-Navarro, J.; Esteban-Rubio, S.; Peinado, H.; Gonzalez-Neira, A.; et al. Polyethylene glycol improves current methods for circulating extracellular vesicle-derived DNA isolation. J. Transl. Med. 2019, 17, 75. [Google Scholar] [CrossRef] [PubMed]
- Kamyabi, N.; Abbasgholizadeh, R.; Maitra, A.; Ardekani, A.; Biswal, S.L.; Grande-Allen, K.J. Isolation and mutational assessment of pancreatic cancer extracellular vesicles using a microfluidic platform. Biomed. Microdevices 2020, 22, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zavridou, M.; Strati, A.; Bournakis, E.; Smilkou, S.; Tserpeli, V.; Lianidou, E. Prognostic Significance of Gene Expression and DNA Methylation Markers in Circulating Tumor Cells and Paired Plasma Derived Exosomes in Metastatic Castration Resistant Prostate Cancer. Cancers 2021, 13, 780. [Google Scholar] [CrossRef]
- Barıs, I.C.; Hacıoglu, S.; Turk, N.S.; Cetın, G.O.; Zencır, S.; Bagcı, G.; Caner, V. Expression and DNA methylation profiles of EZH2-target genes in plasma exosomes and matched primary tumor tissues of the patients with diffuse large B-cell lymphoma. Clin. Transl. Oncol. 2021, 23, 1152–1166. [Google Scholar] [CrossRef]
- Maire, C.L.; Fuh, M.M.; Kaulich, K.; Fita, K.D.; Stevic, I.; Heiland, D.H.; A Welsh, J.; Jones, J.C.; Görgens, A.; Ricklefs, T.; et al. Genome-wide methylation profiling of glioblastoma cell-derived extracellular vesicle DNA allows tumor classification. Neuro-Oncology 2021, 23, 1087–1099. [Google Scholar] [CrossRef] [PubMed]
- Koul, S.; McKiernan, J.M.; Narayan, G.; Houldsworth, J.; Bacik, J.; Dobrzynski, D.L.; Assaad, A.M.; Mansukhani, M.; Reuter, V.E.; Bosl, G.J.; et al. Role of promoter hypermethylation in Cisplatin treatment response of male germ cell tumors. Mol. Cancer 2004, 3, 16. [Google Scholar] [CrossRef] [Green Version]
- Fiegl, H.; Millinger, S.; Mueller-Holzner, E.; Marth, C.; Ensinger, C.; Berger, A.; Klocker, H.; Goebel, G.; Widschwendter, M. Circulating Tumor-Specific DNA: A Marker for Monitoring Efficacy of Adjuvant Therapy in Cancer Patients. Cancer Res. 2005, 65, 1141–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vos, L.; Jung, M.; Koerber, R.-M.; Bawden, E.G.; Holderried, T.A.; Dietrich, J.; Bootz, F.; Brossart, P.; Kristiansen, G.; Dietrich, D. Treatment Response Monitoring in Patients with Advanced Malignancies Using Cell-Free SHOX2 and SEPT9 DNA Methylation in Blood. J. Mol. Diagn. JMD 2020, 22, 920–933. [Google Scholar] [CrossRef]
- Bouchereau, S.; Chaplain, L.; Fort, M.; Beauchet, A.; Sidibé, T.; Chapalain, M.; Gonzalez-Lara, L.; Longvert, C.; Blom, A.; Saiag, P.; et al. Impact of prior treatment with immune checkpoint inhibitors on dacarbazine efficacy in metastatic melanoma. Br. J. Cancer 2021, 125, 948–954. [Google Scholar] [CrossRef] [PubMed]
- Makita, K.; Hara, H.; Sano, E.; Okamoto, Y.; Ochiai, Y.; Harada, T.; Ueda, T.; Nakayama, T.; Aizawa, S.; Yoshino, A. Interferon-β sensitizes human malignant melanoma cells to temozolomide-induced apoptosis and autophagy. Int. J. Oncol. 2019, 54, 1864–1874. [Google Scholar] [CrossRef] [PubMed]
- Zuhur, S.S.; Tanik, C.; Karaman, Ö.; Velet, S.; Çil, E.; Öztürk, F.Y.; Özkayalar, H.; Müslüman, A.M.; Altuntaş, Y. MGMT immunoexpression in growth hormone-secreting pituitary adenomas and its correlation with Ki-67 labeling index and cytokeratin distribution pattern. Endocrine 2011, 40, 222–227. [Google Scholar] [CrossRef]
- Kaina, B.; Fritz, G.; Mitra, S.; Coquerelle, T.; Bernd, K. Transfection and expression of human O6-methylguanine-DNA methyltransferase (MGMT) cDNA in Chinese hamster cells: The role of MGMT in protection against the genotoxic effects of alkylating agents. Carcinogenesis 1991, 12, 1857–1867. [Google Scholar] [CrossRef]
- Kaina, B.; Ziouta, A.; Ochs, K.; Coquerelle, T. Chromosomal instability, reproductive cell death and apoptosis induced by O6-methylguanine in Mex−, Mex+ and methylation-tolerant mismatch repair compromised cells: Facts and models. Mutat. Res. Mol. Mech. Mutagen. 1997, 381, 227–241. [Google Scholar] [CrossRef]
- Jian-Min, C.; Yang-Pei, Z.; Wang, C.; Sun, Y.; Fujimoto, J.; Ikenaga, M. O6-Methylguanine-DNA methyltransferase activity in human tumors. Carcinogenesis 1992, 13, 1503–1507. [Google Scholar] [CrossRef]
- Naumann, S.C.; Roos, W.; Jöst, E.; Belohlavek, C.; Lennerz, V.; Schmidt, C.; Christmann, M.; Kaina, B. Temozolomide- and fotemustine-induced apoptosis in human malignant melanoma cells: Response related to MGMT, MMR, DSBs, and P53. Br. J. Cancer 2009, 100, 322–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palanca-Ballester, C.; Rodriguez-Casanova, A.; Torres, S.; Calabuig-Fariñas, S.; Exposito, F.; Serrano, D.; Redin, E.; Valencia, K.; Jantus-Lewintre, E.; Diaz-Lagares, A.; et al. Cancer Epigenetic Biomarkers in Liquid Biopsy for High Incidence Malignancies. Cancers 2021, 13, 3016. [Google Scholar] [CrossRef] [PubMed]
- Collinson, P. Evidence and Cost Effectiveness Requirements for Recommending New Biomarkers. EJIFCC 2015, 26, 183–189. [Google Scholar]
- Schneegans, S.; Lück, L.; Besler, K.; Bluhm, L.; Stadler, J.-C.; Staub, J.; Greinert, R.; Volkmer, B.; Kubista, M.; Gebhardt, C.; et al. Pre-analytical factors affecting the establishment of a single tube assay for multiparameter liquid biopsy detection in melanoma patients. Mol. Oncol. 2020, 14, 1001–1015. [Google Scholar] [CrossRef]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, R.W.Y.; Jiang, P.; Peng, X.; Tam, L.-S.; Liao, G.J.W.; Li, E.K.M.; Wong, P.C.H.; Sun, H.; Chan, K.C.A.; Chiu, R.W.K.; et al. Plasma DNA aberrations in systemic lupus erythematosus revealed by genomic and methylomic sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, E5302–E5311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, T.; Zeybel, M.; Day, C.P.; Dipper, C.; Masson, S.; McPherson, S.; Henderson, E.; Tiniakos, D.; White, S.; French, J.; et al. Plasma DNA methylation: A potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut 2017, 66, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Syed, F.; Tersey, S.A.; Turatsinze, J.-V.; Felton, J.L.; Kang, N.J.; Nelson, J.B.; Sims, E.K.; Defrance, M.; Bizet, M.; Fuks, F.; et al. Circulating unmethylated CHTOP and INS DNA fragments provide evidence of possible islet cell death in youth with obesity and diabetes. Clin. Epigenet. 2020, 12, 116. [Google Scholar] [CrossRef]
- Lianidou, E. Detection and relevance of epigenetic markers on ctDNA: Recent advances and future outlook. Mol. Oncol. 2021, 15, 1683–1700. [Google Scholar] [CrossRef]
- Eisenstein, A.; Gonzalez, E.C.; Raghunathan, R.; Xu, X.; Wu, M.; McLean, E.O.; McGee, J.; Ryu, B.; Alani, R.M. Emerging Biomarkers in Cutaneous Melanoma. Mol. Diagn. Ther. 2018, 22, 203–218. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | Relevance to Melanoma | Ref. |
|---|---|---|---|
| APC | Adenomatous Polyposis Coli | Decreased expression increases the proliferation potential of melanoma cells. Found in brain metastases in melanoma. | [59,60] |
| CDH11 | Cadherin 11 | The hypermethylated CDH11 facilitates the scattering of tumor cells by loosening contacts between them. Increased proliferation following its inactivation may encourage further establishment at secondary sites, contributing to melanoma progression. | [61,62,63] |
| CDH13 | Cadherin 13 | The loss of CDH13 is involved in the development of malignant melanoma. Found in brain metastases in melanoma. | [55,59,64] |
| CDKN2A | Cyclin-Dependent Kinase Inhibitor 2A | The hypermethylated p16INK4A promoter has been predominantly observed in NRAS-mutated metastatic melanomas. | [41,55,65,66,67,68,69,70,71,72,73] |
| CLDN11 | Claudin 11 | Its methylation level is a potential tool to help discriminate between malignant melanoma and nevus cell nevi. | [74,75] |
| DAPK | Death-Associated Protein Kinase | Detected in patients with both cutaneous and uveal melanoma: its epigenetic silencing is a common mechanism for tumor formation. | [65,76,77] |
| ESR1 | Estrogen Receptor 1 | The detection of methylated ESR1 in tissues or sera correlates with tumor progression and is, therefore, of prognostic importance in melanoma patients. In addition, it may identify a population of patients with poor response to systemic therapy, for whom alternative treatment management should be considered. | [55,59,78] |
| FES | FES Proto-Oncogene, Tyrosine Kinase | Its downregulation correlates with poor OS. FES loss drives tumor progression of BRAF V600E-induced murine melanoma. | [79] |
| MAPK13 | Mitogen-Activated Protein Kinase 13 | Its epigenetic silencing contributes to melanoma progression: restoration of its expression in melanoma cells with MAPK13 promoter methylation reduces these cells’ proliferative capacity. | [63,75] |
| MEOX2 | Mesenchyme Homeobox 2 | This gene’s degree of DNA methylation can predict the prognosis of melanoma patients. Its methylation is associated with melanoma progression and/or poor survival. | [80,81] |
| MGMT | O6-Methylguanine-DNA Methyltransferase | Its epigenetic silencing was associated with a better response to DTIC/TMZ therapy and longer PFS in patients with stage IV melanoma and patients with stage III melanoma treated with melphalan locoregional chemotherapy. | [65,72,76,78,82,83,84,85] |
| MITF | Melanocyte Inducing Transcription Factor | The MITF gene body was found to be hypermethylated in primary tumors compared to metastases. | [86,87] |
| OLIG3 | Oligodendrocyte Transcription Factor 3 | This gene’s degree of DNA methylation can predict the prognosis of melanoma patients. Its methylation is associated with melanoma progression and/or poor survival. | [80,81] |
| OVOL1 | Ovo Like Transcriptional Repressor 1 | Patients with high OVOL1 expression in the primary tumor had a significantly better prognosis than those with low expression. | [80] |
| PD-L1 | Programmed Cell Death 1 Ligand 1 | Decreased PD-L1 expression correlates with a shorter patient OS. | [88] |
| PON3 | Paraoxonase 3 | This gene’s degree of DNA methylation can predict the prognosis of melanoma patients. Its hypermethylation is significantly elevated in patients with metastatic melanoma. | [80,81] |
| PTEN | Phosphatase And Tensin Homolog | Reduced OS and DFS in stage III/IV patients. Found in brain metastases in melanoma. | [55,59,89,90,91,92] |
| RARβ2 | Retinoic Acid Receptor Beta 2 | Correlated with Breslow thickness of the primary tumor: its silencing may be a key epigenetic factor in melanocyte transformation and progression of the primary lesion. Found in brain metastases in melanoma. | [55,59,76,78,85,93,94] |
| RASSF1A | Ras-Association Domain Family Member 1 | Detected in patients with cutaneous and uveal melanoma. It can predict the response of patients with stage IV melanoma to biochemotherapy. Found in brain metastases in melanoma. | [47,49,55,59,65,76,77,78,93,95,96,97] |
| SOCS1/2 | Suppressor of Cytokine Signaling 1/2 | Frequently found hypermethylated in the serum of melanoma patients or melanoma cell lines. | [72,94] |
| Gene/Sequence Symbol | Gene/Sequence Name | Relevance to Melanoma | Ref. |
|---|---|---|---|
| DSS1 | Deleted in Split-Hand/Split-Foot 1 | Its increased expression has been associated with the presence of metastases, ulceration, and reduced OS and DFS, so that it may be used as a biomarker of poor prognosis in melanoma patients. | [131] |
| LINE-1 | Long Interspersed Nuclear Element-1 | Hypomethylation increase is correlated with advanced stages and a worse prognosis. | [85,130,132] |
| MAGE-1/2/3/4 | Melanoma-Associated Antigen 1/2/3/4 | Frequently hypomethylated in melanoma cell lines. | [93,133] |
| Maspin | Maspin | It behaves like a TSG in breast and prostate cancer, but its role in melanoma is controversial. | [85,134,135] |
| PDGFD, THRB, ZEB1 | Platelet-Derived Growth Factor D, Thyroid Hormone Receptor Beta, Zinc Finger E-Box Binding Homeobox 1 | Its higher expression in NRASQ61-mutated melanomas has been associated with patients’ survival time. It could be a potential candidate for drug development for NRAS-mutant melanomas. | [136] |
| PD-L2 | Programmed Cell Death 1 Ligand 2 | Predictor of longer PFS in patients referred for anti-PD-1 immunotherapy. | [137] |
| TBC1D16 | TBC1 Domain Family Member 16 | Associated with increased clinical response to BRAF inhibitors in patients harboring the BRAF V600E missense mutation. | [80,138] |
| TKTL1 | Transketolase Like 1 | It increases the metastatic potential of melanoma cells by contributing to the enhancement of the ’Warburg effect.” | [139] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aleotti, V.; Catoni, C.; Poggiana, C.; Rosato, A.; Facchinetti, A.; Scaini, M.C. Methylation Markers in Cutaneous Melanoma: Unravelling the Potential Utility of Their Tracking by Liquid Biopsy. Cancers 2021, 13, 6217. https://doi.org/10.3390/cancers13246217
Aleotti V, Catoni C, Poggiana C, Rosato A, Facchinetti A, Scaini MC. Methylation Markers in Cutaneous Melanoma: Unravelling the Potential Utility of Their Tracking by Liquid Biopsy. Cancers. 2021; 13(24):6217. https://doi.org/10.3390/cancers13246217
Chicago/Turabian StyleAleotti, Valentina, Cristina Catoni, Cristina Poggiana, Antonio Rosato, Antonella Facchinetti, and Maria Chiara Scaini. 2021. "Methylation Markers in Cutaneous Melanoma: Unravelling the Potential Utility of Their Tracking by Liquid Biopsy" Cancers 13, no. 24: 6217. https://doi.org/10.3390/cancers13246217
APA StyleAleotti, V., Catoni, C., Poggiana, C., Rosato, A., Facchinetti, A., & Scaini, M. C. (2021). Methylation Markers in Cutaneous Melanoma: Unravelling the Potential Utility of Their Tracking by Liquid Biopsy. Cancers, 13(24), 6217. https://doi.org/10.3390/cancers13246217