
Molecular Classification and Therapeutic Targets in Ependymoma


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. History
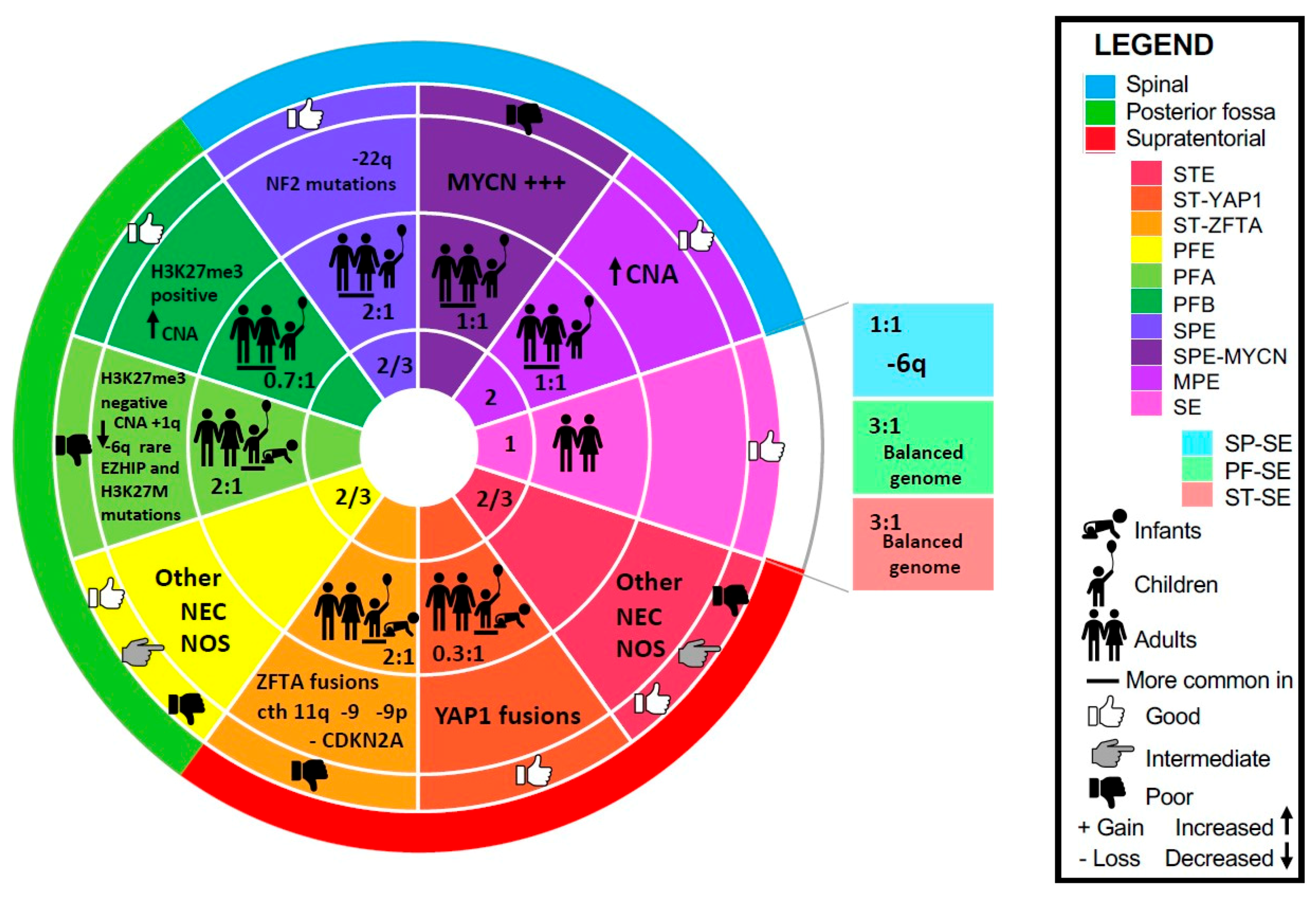
3. Subependymoma (SE)
3.1. Spinal Subependymoma (SP-SE)
3.2. Posterior Fossa Subependymoma (PF-SE)
3.3. Supratentorial Subependymoma (ST-SE)
4. Myxopapillary Ependymoma (MPE)
5. Spinal Ependymoma (SPE)
6. Spinal Ependymoma with MYCN Amplification (SPE-MYCN)
7. Posterior Fossa Ependymoma (PFE)
8. Posterior Fossa Ependymoma Group A (PFA)
9. Posterior Fossa Ependymoma Group B (PFB)
10. Supratentorial Ependymoma (STE)
11. Supratentorial Ependymoma with YAP1-Fusion (ST-YAP1)
12. Supratentorial Ependymoma with ZFTA-Fusion (ST-ZFTA)
13. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| IVthV | Fourth ventricle |
| 5AZA-DC | DNA methylase transferase inhibitor 5-aza-2′-deoxycytidine |
| AKT | Protein kinase B |
| ATRX/DAXX | Alpha-thalassemia, mental retardation, X-linked/death domain–associated protein complex |
| AURKA | Auro A-kinase |
| BET | Bromodomain and extraterminal domain |
| Brd4 | Bromodomain-containing protein 4 |
| c-KIT | Proto-oncogene encoding tyrosine-protein kinase KIT, CD117, or stem cell growth factor receptor |
| c-MYB | c-myeloblastosis |
| CAR T | Chimeric antigen receptor T cell |
| Cbp | CREB-binding protein |
| Cdc42 | Cell division cycle 42 |
| COX2 | Cyclooxygenase-2 |
| CNS | Central nervous system |
| CRL4 | Cullin Ring Ubiquitin Ligase 4 |
| CSF | Cerebrospinal fluid |
| CXCR4 | C-X-C chemokine receptor type 4 |
| EANO | The European Association of Neuro-Oncology |
| EED | Embryonic ectoderm development |
| EMA | Epithelial membrane antigen |
| Ep300 | Histone acetyltransferase p300 |
| EPHA7 | Ephrin type-A receptor 7 |
| ERK | Extracellular Signal-Regulated Kinase |
| ERM | Ezrin, radixin, moesin binding protein |
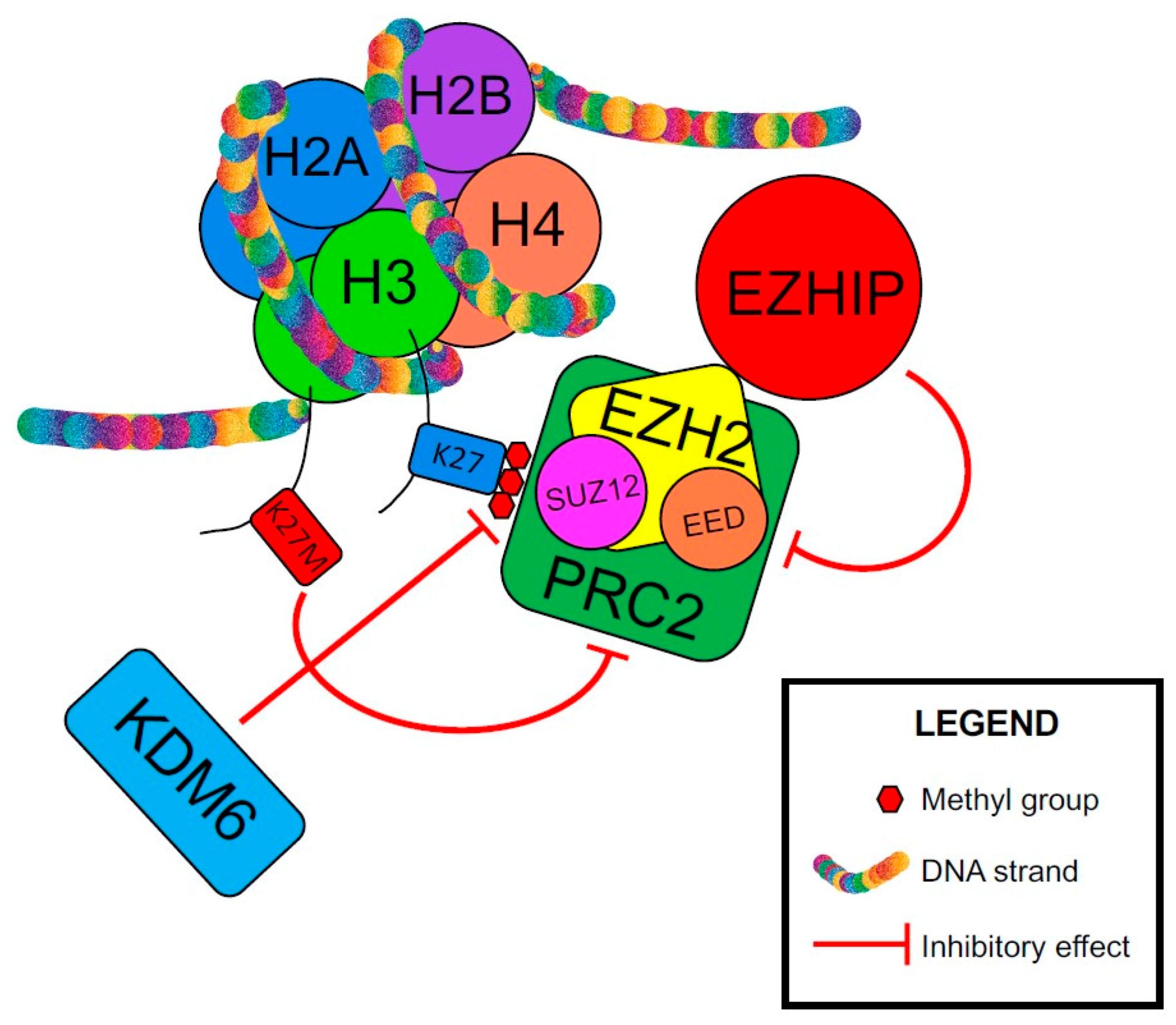
| EZHIP | EZH Inhibitory Protein or CXorf67 |
| EZH2 | Enhancer of zeste homolog 2 |
| FAK | Focal Adhesion Kinase |
| FAM 118B | Family with sequence similarity 118 member B |
| FGFR3 | Fibroblast growth factor receptor 3 |
| FOXJ1 | Forkhead box protein J1 |
| GFAP | Glial fibrillary acidic protein |
| GTR | Gross total resection |
| H3K27M | Histone H3 lysine 27 to methionine mutation |
| H3K27me3 | Trimethylated histone H3 at lysine 27 |
| HDAC | Histone deacetylase |
| HE | Hematoxylin and eosin |
| HIF-1a | Hypoxia inducible factor 1 alpha |
| HOXA13 | Homeobox A13 |
| HOXB13 | Homeobox B13 |
| HOXC10 | Homeobox C10 |
| HOXD10 | Homeobox D10 |
| hTERT | Human telomerase reverse transcriptase |
| IkBα | Nuclear factor of kappa light polypeptide gene enhancer in B cells alpha |
| IL-6 | Interleukin-6 |
| IT | Infratentorial |
| KDM6 | Lysine-specific demethylase 6 |
| KIT | Tyrosine protein kinase or stem cell growth factor receptor or CD117 |
| L1CAM | L1 Cell Adhesion Molecule |
| LATS1/2 | Large Tumor Suppressor Kinase 1 and 2 |
| LDOC | Leucine zipper downregulated in cancer 1 |
| MAML2 | Mastermind like transcription coactivator 2 |
| MAMLD1 | Mastermind like domain containing 1 |
| MDM2 | Mouse double minute 2 homolog or E3 ubiquitin-protein ligase Mdm2 |
| MEF | Myocyte enhancer factor |
| MPE | Spinal myxopapillary ependymoma |
| mTORC1 | Mammalian Target of Rapamycin Complex 1 |
| MYCN | Myelocytomatosis-N |
| NCOA1 | Nuclear receptor coactivator 1 |
| NCOA2 | Nuclear receptor coactivator 2 |
| NEFL | Neurofilament light chain |
| NEC | Not elsewhere classified |
| NF-κB | Nuclear factor-κB |
| NF2 | Neurofibromatosis type 2 |
| NHERF1/EBP50 | Na+/H+ exchanger regulatory factor/ezrin-radixin-moesin binding protein 50 |
| NOS | Not otherwise specified |
| OS | Overall survival |
| PAK | P21 activated kinase |
| PARP | Poly (ADP-ribose) polymerase |
| PDGFRA | Platelet derived growth factor alpha |
| PFA | Posterior fossa ependymoma Group A |
| PFB | Posterior fossa ependymoma Group B |
| PFE | Posterior fossa ependymoma |
| PF-SE | Posterior fossa subependymoma |
| PFS | Progression-free survival |
| PI3K | Phosphoinositide 3-kinase |
| PIKE-L | Phosphoinositide 3-Kinase Enhancer-brain specific isoform |
| PLAGL | PLAG1 like zinc finger 1 |
| PRC2 | Polycomb repressive complex 2 |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| RAC | Ras-related C3 botulinum toxin |
| RAF | Rapidly accelerated fibrosarcoma |
| Ras | Rat sarcoma virus |
| RELA | REL-associated protein |
| SAM | S-adenosyl methionine |
| SE | Subependymoma |
| SET | Su(var)3-9/enhancer-of-zeste/trithorax |
| Shh | Sonic hedgehog |
| SP | Spinal |
| SPE | Spinal ependymoma |
| SPE-MYCN | Spinal ependymoma with MYCN amplification |
| SP-SE | Spinal subependymoma |
| ST | Supratentorial |
| ST-RELA | Supratentorial ependymoma with RELA fusion |
| ST-SE | Supratentorial subependymoma |
| ST-YAP1 | Supratentorial ependymoma with YAP1 fusion |
| STAT | Signal transducer and activator of transcription |
| STE | Supratentorial ependymoma |
| STR | Subtotal resection |
| SUZ12 | Suppressor of zeste 12 homolog |
| TEAD | Transcriptional enhancer factor domain |
| TAZ | Gene encoding protein Tafazzin |
| VEGF | Vascular endothelial growth factor |
| WHO | World Health Organization |
| WP744 | 4′-O-benzylated doxorubicin analog |
| WP1066 | JAK2/STAT3 inhibitor |
| WP1193 | JAK2/STAT3 inhibitor |
| YAP1 | Yes1 associated transcriptional regulator or YAP or YAP65 |
| ZFTA | Zinc finger translocation-associated |
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Armstrong, T.S.; Gilbert, M.R. Biology and management of ependymomas. Neuro-Oncology 2016, 18, 902–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro-Oncology 2020, 22, IV1–IV96. [Google Scholar] [CrossRef] [PubMed]
- McGuire, C.S.; Sainani, K.L.; Fisher, P.G. Incidence patterns for ependymoma: A Surveillance, Epidemiology, and End Results study—Clinical article. J. Neurosurg. 2009, 110, 725–729. [Google Scholar] [CrossRef]
- Vera-Bolanos, E.; Aldape, K.; Yuan, Y.; Wu, J.; Wani, K.; Necesito-Reyes, M.J.; Colman, H.; Dhall, G.; Lieberman, F.S.; Metellus, P.; et al. Clinical course and progression-free survival of adult intracranial and spinal ependymoma patients. Neuro-Oncology 2015, 17, 440–447. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Mack, S.C.; Ramaswamy, V.; Smith, C.A.; Witt, H.; Smith, A.; Hansford, J.R.; von Hoff, K.; Wright, K.D.; Hwang, E.; et al. The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Acta Neuropathol. 2017, 133, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Reni, M.; Gatta, G.; Mazza, E.; Vecht, C. Ependymoma. Crit. Rev. Oncol. Hematol. 2007, 63, 81–89. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network. Central Nervous System Cancers. Available online: https://www.nccn.org/professionals/physician_gls/pdf/cns.pdf (accessed on 9 September 2021).
- Metellus, P.; Guyotat, J.; Chinot, O.; Durand, A.; Barrie, M.; Giorgi, R.; Jouvet, A.; Figarella-Branger, D. Adult intracranial WHO grade II ependymomas: Long-term outcome and prognostic factor analysis in a series of 114 patients. Neuro-Oncology 2010, 12, 976–984. [Google Scholar] [CrossRef]
- Rogers, L.; Pueschel, J.; Spetzler, R.; Shapiro, W.; Coons, S.; Thomas, T.; Speiser, B. Is gross-total resection sufficient treatment for posterior fossa ependymomas? J. Neurosurg. 2005, 102, 629–636. [Google Scholar] [CrossRef]
- Rudà, R.; Reifenberger, G.; Frappaz, D.; Pfister, S.M.; Laprie, A.; Santarius, T.; Roth, P.; Tonn, J.C.; Soffietti, R.; Weller, M.; et al. EANO guidelines for the diagnosis and treatment of ependymal tumors. Neuro-Oncology 2018, 20, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Aizer, A.A.; Ancukiewicz, M.; Nguyen, P.L.; MacDonald, S.M.; Yock, T.I.; Tarbell, N.J.; Shih, H.A.; Loeffler, J.S.; Oh, K.S. Natural history and role of radiation in patients with supratentorial and infratentorial WHO grade II ependymomas: Results from a population-based study. J. Neurooncol. 2013, 115, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Yuan, Y.; Wu, J.; Mendoza, T.; Vera, E.; Omuro, A.; Lieberman, F.; Robins, H.I.; Gerstner, E.R.; Wu, J.; et al. A phase II study of dose-dense temozolomide and lapatinib for recurrent low-grade and anaplastic supratentorial, infratentorial, and spinal cord ependymoma. Neuro-Oncology 2021, 23, 468–477. [Google Scholar] [CrossRef]
- Brandes, A.A.; Cavallo, G.; Reni, M.; Tosoni, A.; Nicolardi, L.; Scopece, L.; Franceschi, E.; Sotti, G.; Talacchi, A.; Turazzi, S.; et al. A multicenter retrospective study of chemotherapy for recurrent intracranial ependymal tumors in adults by the Gruppo Italiano Cooperative di Neuro-Oncologia. Cancer 2005, 104, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, M.C.; Johnston, S.K. Temozolomide for recurrent intracranial supratentorial platinum-refractory ependymoma. Cancer 2009, 115, 4775–4782. [Google Scholar] [CrossRef] [PubMed]
- Green, R.M.; Cloughesy, T.F.; Stupp, R.; Deangelis, L.M.; Woyshner, E.A.; Ney, D.E.; Lassman, A.B. Bevacizumab for recurrent ependymoma. Neurology 2009, 73, 1677–1680. [Google Scholar] [CrossRef] [Green Version]
- Rudà, R.; Bosa, C.; Magistrello, M.; Franchino, F.; Pellerino, A.; Fiano, V.; Trevisan, M.; Cassoni, P.; Soffietti, R. Temozolomide as salvage treatment for recurrent intracranial ependymomas of the adult: A retrospective study. Neuro-Oncology 2016, 18, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Gramatzki, D.; Roth, P.; Felsberg, J.; Hofer, S.; Rushing, E.J.; Hentschel, B.; Westphal, M.; Krex, D.; Simon, M.; Schnell, O.; et al. Chemotherapy for intracranial ependymoma in adults. BMC Cancer 2016, 16, 287. [Google Scholar] [CrossRef] [Green Version]
- Kieran, M.W.; Goumnerova, L.; Manley, P.; Chi, S.N.; Marcus, K.J.; Manzanera, A.G.; Polanco, M.L.S.; Guzik, B.W.; Aguilar-Cordova, E.; Diaz-Montero, C.M.; et al. Phase I study of gene-mediated cytotoxic immunotherapy with AdV-tk as adjuvant to surgery and radiation for pediatric malignant glioma and recurrent ependymoma. Neuro-Oncology 2019, 21, 537–546. [Google Scholar] [CrossRef]
- National Cancer Institute. Childhood Ependymoma Treatment (PDQ®)–Health Professional Version. Available online: https://www.cancer.gov/types/brain/hp/child-ependymoma-treatment-pdq (accessed on 30 November 2021).
- Duffner, P.K.; Horowitz, M.E.; Krischer, J.P.; Friedman, H.S.; Burger, P.C.; Cohen, M.E.; Sanford, R.A.; Mulhern, R.K.; James, H.E.; Freeman, C.R.; et al. Postoperative Chemotherapy and Delayed Radiation in Children Less Than Three Years of Age with Malignant Brain Tumors. N. Engl. J. Med. 1993, 328, 1725–1731. [Google Scholar] [CrossRef]
- Snider, C.A.; Yang, K.; Mack, S.C.; Suh, J.H.; Chao, S.T.; Merchant, T.E.; Murphy, E.S. Impact of radiation therapy and extent of resection for ependymoma in young children: A population-based study. Pediatr. Blood Cancer 2018, 65, e26880. [Google Scholar] [CrossRef]
- Grundy, R.G.; Wilne, S.A.; Weston, C.L.; Robinson, K.; Lashford, L.S.; Ironside, J.; Cox, T.; Chong, W.K.; Campbell, R.H.; Bailey, C.C.; et al. Primary postoperative chemotherapy without radiotherapy for intracranial ependymoma in children: The UKCCSG/SIOP prospective study. Lancet Oncol. 2007, 8, 696–705. [Google Scholar] [CrossRef]
- Strother, D.R.; Lafay-Cousin, L.; Boyett, J.M.; Burger, P.; Aronin, P.; Constine, L.; Duffner, P.; Kocak, M.; Kun, L.E.; Horowitz, M.E.; et al. Benefit from prolonged dose-intensive chemotherapy for infants with malignant brain tumors is restricted to patients with ependymoma: A report of the pediatric oncology group randomized controlled trial 9233/34. Neuro-Oncology 2014, 16, 457–465. [Google Scholar] [CrossRef] [Green Version]
- Merchant, T.E.; Bendel, A.E.; Sabin, N.D.; Burger, P.C.; Shaw, D.W.; Chang, E.; Wu, S.; Zhou, T.; Eisenstat, D.D.; Foreman, N.K.; et al. Conformal radiation therapy for pediatric ependymoma, chemotherapy for incompletely resected ependymoma, and observation for completely resected, supratentorial ependymoma. J. Clin. Oncol. 2019, 37, 974–983. [Google Scholar] [CrossRef] [Green Version]
- Massimino, M.; Solero, C.L.; Garrè, M.L.; Biassoni, V.; Cama, A.; Genitori, L.; Di Rocco, C.; Sardi, I.; Viscardi, E.; Modena, P.; et al. Second-look surgery for ependymoma: The Italian experience—Clinical article. J. Neurosurg. Pediatr. 2011, 8, 246–250. [Google Scholar] [CrossRef]
- Garvin, J.H.; Selch, M.T.; Holmes, E.; Berger, M.S.; Finlay, J.L.; Flannery, A.; Goldwein, J.W.; Packer, R.J.; Rorke-Adams, L.B.; Shiminski-Maher, T.; et al. Phase II study of pre-irradiation chemotherapy for childhood intracranial ependymoma. Children’s Cancer Group protocol 9942: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2012, 59, 1183–1189. [Google Scholar] [CrossRef]
- Zacharoulis, S.; Ashley, S.; Moreno, L.; Gentet, J.C.; Massimino, M.; Frappaz, D. Treatment and outcome of children with relapsed ependymoma: A multi-institutional retrospective analysis. Childs Nerv. Syst. 2010, 26, 905–911. [Google Scholar] [CrossRef]
- Jakacki, R.I.; Foley, M.A.; Horan, J.; Wang, J.; Kieran, M.W.; Bowers, D.C.; Bouffet, E.; Zacharoulis, S.; Gill, S.C. Single-agent erlotinib versus oral etoposide in patients with recurrent or refractory pediatric ependymoma: A randomized open-label study. J. Neurooncol. 2016, 129, 131–138. [Google Scholar] [CrossRef]
- Bouffet, E.; Capra, M.; Bartels, U. Salvage chemotherapy for metastatic and recurrent ependymoma of childhood. Childs Nerv. Syst. 2009, 25, 1293–1301. [Google Scholar] [CrossRef]
- Iqbal, M.S.; Lewis, J. An overview of the management of adult ependymomas with emphasis on relapsed disease. Clin. Oncol. 2013, 25, 726–733. [Google Scholar] [CrossRef]
- Donovan, L.K.; Delaidelli, A.; Joseph, S.K.; Bielamowicz, K.; Fousek, K.; Holgado, B.L.; Manno, A.; Srikanthan, D.; Gad, A.Z.; Van Ommeren, R.; et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med. 2020, 26, 720–731. [Google Scholar] [CrossRef]
- Ellison, D.W.; Kocak, M.; Figarella-Branger, D.; Felice, G.; Catherine, G.; Pietsch, T.; Frappaz, D.; Massimino, M.; Grill, J.; Boyett, J.M.; et al. Histopathological grading of pediatric ependymoma: Reproducibility and clinical relevance in European trial cohorts. J. Negat. Results Biomed. 2011, 10, 7. [Google Scholar] [CrossRef]
- Hübner, J.-M.; Kool, M.; Pfister, S.M.; Pajtler, K.W. Epidemiology, molecular classification and WHO grading of ependymoma. J. Neurosurg. Sci. 2018, 62, 46–50. [Google Scholar] [CrossRef]
- Tihan, T.; Zhou, T.; Holmes, E.; Burger, P.C.; Ozuysal, S.; Rushing, E.J. The prognostic value of histological grading of posterior fossa ependymomas in children: A Children’s Oncology Group study and a review of prognostic factors. Mod. Pathol. 2008, 21, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Xi, S.; Sai, K.; Hu, W.; Wang, F.; Chen, Y.; Wang, J.; Zeng, J.; Chen, Z. Clinical significance of the histological and molecular characteristics of ependymal tumors: A single institution case series from China. BMC Cancer 2019, 19, 717. [Google Scholar] [CrossRef] [Green Version]
- Witt, H.; Gramatzki, D.; Hentschel, B.; Pajtler, K.W.; Felsberg, J.; Schackert, G.; Löffler, M.; Capper, D.; Sahm, F.; Sill, M.; et al. DNA methylation-based classification of ependymomas in adulthood: Implications for diagnosis and treatment. Neuro-Oncology 2018, 20, 1616–1624. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, F.M.G.; Hübner, J.M.; Sharma, T.; Luu, B.; Sill, M.; Zapotocky, M.; Mack, S.C.; Witt, H.; Lin, T.; Shih, D.J.H.; et al. Heterogeneity within the PF-EPN-B ependymoma subgroup. Acta Neuropathol. 2018, 136, 227–237. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Hielscher, T.; Mack, S.C.; Lassaletta, A.; Lin, T.; Pajtler, K.W.; Jones, D.T.W.; Luu, B.; Cavalli, F.M.G.; Aldape, K.; et al. Therapeutic impact of cytoreductive surgery and irradiation of posterior fossa ependymoma in the molecular era: A retrospective multicohort analysis. J. Clin. Oncol. 2016, 34, 2468–2477. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Witt, H.; Sill, M.; Jones, D.T.W.; Hovestadt, V.; Kratochwil, F.; Wani, K.; Tatevossian, R.; Punchihewa, C.; Johann, P.; et al. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 2015, 27, 728–743. [Google Scholar] [CrossRef] [Green Version]
- Mack, S.C.; Witt, H.; Piro, R.M.; Gu, L.; Zuyderduyn, S.; Stütz, A.M.; Wang, X.; Gallo, M.; Garzia, L.; Zayne, K.; et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 2014, 506, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, L.M.; Donson, A.M.; Nakachi, I.; Griesinger, A.M.; Birks, D.K.; Amani, V.; Hemenway, M.S.; Liu, A.K.; Wang, M.; Hankinson, T.C.; et al. Molecular sub-group-specific immunophenotypic changes are associated with outcome in recurrent posterior fossa ependymoma. Acta Neuropathol. 2014, 127, 731–745. [Google Scholar] [CrossRef] [Green Version]
- Witt, H.; Mack, S.C.; Ryzhova, M.; Bender, S.; Sill, M.; Isserlin, R.; Benner, A.; Hielscher, T.; Milde, T.; Remke, M.; et al. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 2011, 20, 143–157. [Google Scholar] [CrossRef] [Green Version]
- Wani, K.; Armstrong, T.S.; Vera-Bolanos, E.; Raghunathan, A.; Ellison, D.; Gilbertson, R.; Vaillant, B.; Goldman, S.; Packer, R.J.; Fouladi, M.; et al. A prognostic gene expression signature in infratentorial ependymoma. Acta Neuropathol. 2012, 123, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Carter, M.; Nicholson, J.; Ross, F.; Crolla, J.; Allibone, R.; Balaji, V.; Perry, R.; Walker, D.; Gilbertson, R.; Ellison, D.W. Genetic abnormalities detected in ependymomas by comparative genomic hybridisation. Br. J. Cancer 2002, 86, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Puget, S.; Grill, J.; Valent, A.; Bieche, I.; Dantas-Barbosa, C.; Kauffmann, A.; Dessen, P.; Lacroix, L.; Geoerger, B.; Job, B.; et al. Candidate genes on chromosome 9q33-34 involved in the progression of childhood ependymomas. J. Clin. Oncol. 2009, 27, 1884–1892. [Google Scholar] [CrossRef]
- Korshunov, A.; Hielscher, T.; Ryzhova, M. Molecular Staging of Intracranial Ependymoma in Children and Adults Structure and Mechanism of Key Nonsense-Mediated mRNA Decay Factor Complexes View project EURAT: Ethical and legal aspects of genome sequencing View project. J. Clin. Oncol. 2010, 28, 3182–3190. [Google Scholar] [CrossRef]
- Modena, P.; Lualdi, E.; Facchinetti, F.; Veltman, J.; Reid, J.F.; Minardi, S.; Janssen, I.; Giangaspero, F.; Forni, M.; Finocchiaro, G.; et al. Identification of tumor-specific molecular signatures in intracranial ependymoma and association with clinical characteristics. J. Clin. Oncol. 2006, 24, 5223–5233. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Ellison, D.W.; Aldape, K.D.; Capper, D.; Fouladi, M.; Gilbert, M.R.; Gilbertson, R.J.; Hawkins, C.; Merchant, T.E.; Pajtler, K.; Venneti, S.; et al. cIMPACT-NOW update 7: Advancing the molecular classification of ependymal tumors. Brain Pathol. 2020, 30, 863–866. [Google Scholar] [CrossRef]
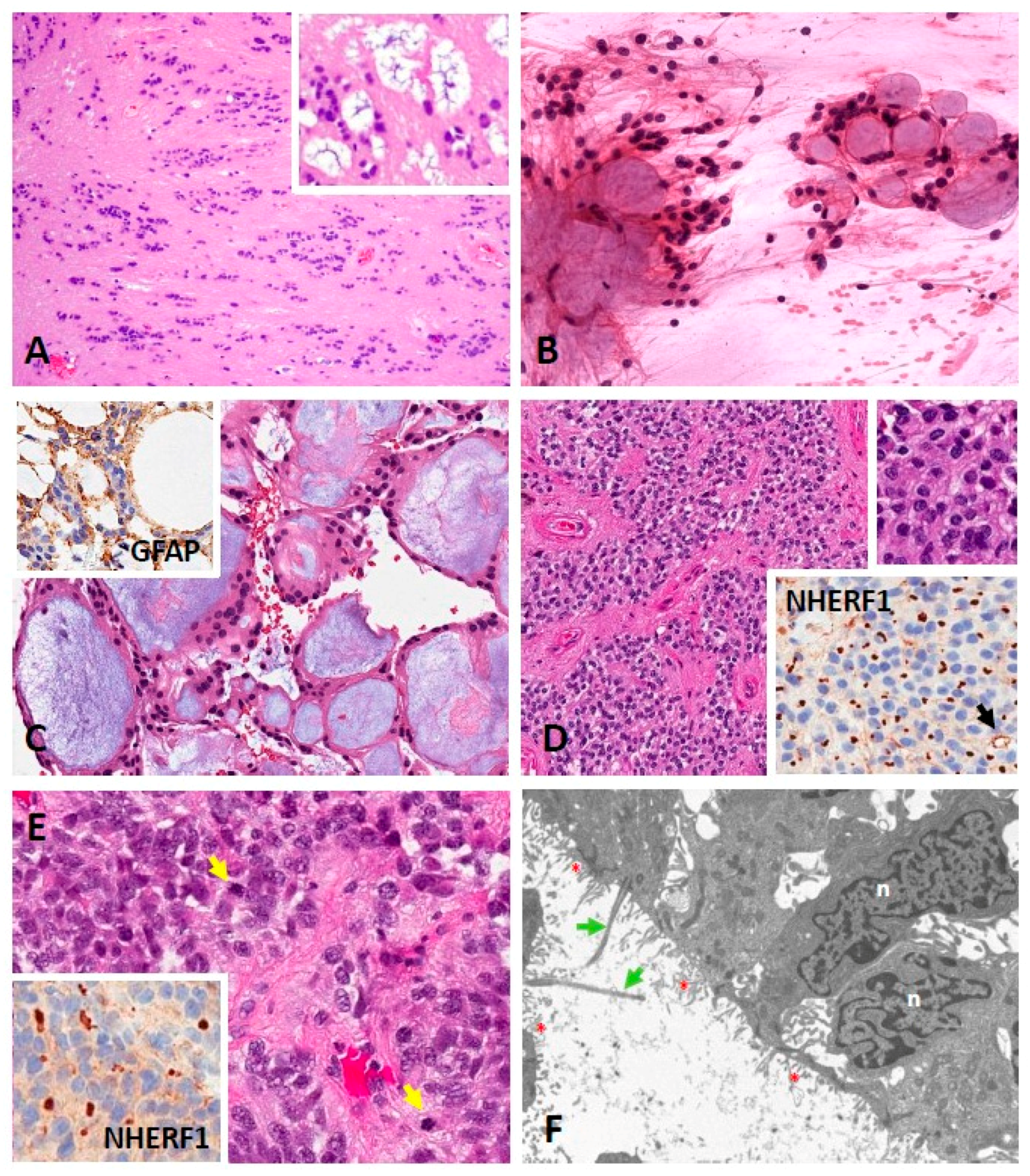
- Georgescu, M.M.; Yell, P.; Mobley, B.C.; Shang, P.; Georgescu, T.; Wang, S.H.J.; Canoll, P.; Hatanpaa, K.J.; White, C.L.; Raisanen, J.M. NHERF1/EBP50 is an organizer of polarity structures and a diagnostic marker in ependymoma. Acta Neuropathol. Commun. 2015, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, R.S.; Praver, M.; Zanazzi, G.J.; Englander, Z.K.; Sims, J.S.; Samanamud, J.L.; Ogden, A.T.; McCormick, P.C.; Feldstein, N.A.; McKhann, G.M.; et al. Subependymomas Are Low-Grade Heterogeneous Glial Neoplasms Defined by Subventricular Zone Lineage Markers. World Neurosurg. 2017, 107, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Shimada, S.; Ishizawa, K.; Horiguchi, H.; Shimada, T.; Hirose, T. Subependymoma of the spinal cord and review of the literature. Pathol. Int. 2003, 53, 169–173. [Google Scholar] [CrossRef]
- Krishnan, S.S.; Panigrahi, M.; Pendyala, S.; Rao, S.I.; Varma, D.R. Cervical Subependymoma: A rare case report with possible histogenesis. J. Neurosci. Rural Pract. 2012, 3, 366–369. [Google Scholar] [CrossRef]
- Soleiman, H.A.; Ironside, J.; Kealey, S.; Demetriades, A.K. Spinal subependymoma surgery: Do no harm. Little may be more! Neurosurg. Rev. 2020, 43, 1047–1053. [Google Scholar] [CrossRef]
- Wu, L.; Yang, T.; Deng, X.; Yang, C.; Zhao, L.; Fang, J.; Wang, G.; Yang, J.; Xu, Y. Surgical outcomes in spinal cord subependymomas: An institutional experience. J. Neurooncol. 2014, 116, 99–106. [Google Scholar] [CrossRef]
- Korshunov, A.; Neben, K.; Wrobel, G.; Tews, B.; Benner, A.; Hahn, M.; Golanov, A.; Lichter, P. Gene Expression Patterns in Ependymomas Correlate with Tumor Location, Grade, and Patient Age. Am. J. Pathol. 2003, 163, 1721–1727. [Google Scholar] [CrossRef] [Green Version]
- Monoranu, C.M.; Huang, B.; Zangen, I.L.; Rutkowski, S.; Vince, G.H.; Gerber, N.U.; Puppe, B.; Roggendorf, W. Correlation between 6q25.3 deletion status and survival in pediatric intracranial ependymomas. Cancer Genet. Cytogenet. 2008, 182, 18–26. [Google Scholar] [CrossRef]
- Rajaram, V.; Gutmann, D.H.; Prasad, S.K.; Mansur, D.B.; Perry, A. Alterations of protein 4.1 family members in ependymomas: A study of 84 cases. Mod. Pathol. 2005, 18, 991–997. [Google Scholar] [CrossRef]
- George, O.L.; Ness, S.A. Situational awareness: Regulation of the myb transcription factor in differentiation, the cell cycle and oncogenesis. Cancers 2014, 6, 2049–2071. [Google Scholar] [CrossRef]
- López-Nieva, P.; Vaquero, C.; Fernández-Navarro, P.; González-Sánchez, L.; Villa-Morales, M.; Santos, J.; Esteller, M.; Fernández-Piqueras, J. EPHA7, a new target gene for 6q deletion in T-cell lymphoblastic lymphomas. Carcinogenesis 2012, 33, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.S.; Doan, N.; Gelsomino, M.; Shabani, S. Intracranial Subependymoma: A SEER Analysis 2004–2013. World Neurosurg. 2017, 101, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Friede, R.L.; Pollak, A. The cytogenetic basis for classifying ependymomas. J. Neuropathol. Exp. Neurol. 1978, 37, 103–118. [Google Scholar] [CrossRef]
- Ragel, B.T.; Osborn, A.G.; Whang, K.; Townsend, J.J.; Jensen, R.L.; Couldwell, W.T. Subependymomas: An analysis of clinical and imaging features. Neurosurgery 2006, 58, 881–889. [Google Scholar] [CrossRef]
- Rushing, E.J.; Cooper, P.B.; Quezado, M.; Begnami, M.; Crespo, A.; Smirniotopoulos, J.G.; Ecklund, J.; Olsen, C.; Santi, M. Subependymoma revisited: Clinicopathological evaluation of 83 cases. J. Neuro-Oncol. 2007, 85, 297–305. [Google Scholar] [CrossRef]
- Leeper, H.; Felicella, M.M.; Walbert, T. Recent Advances in the Classification and Treatment of Ependymomas. Curr. Treat. Options Oncol. 2017, 18, 55. [Google Scholar] [CrossRef]
- Bi, Z.; Ren, X.; Zhang, J.; Jia, W. Clinical, radiological, and pathological features in 43 cases of intracranial subependymoma. J. Neurosurg. 2015, 122, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.Y.; Wei, J.; Haider, A.S.; Liebelt, B.D.; Ling, X.; Conrad, C.A.; Fuller, G.N.; Levine, N.B.; Priebe, W.; Sawaya, R.; et al. Therapeutic targets in subependymoma. J. Neuroimmunol. 2014, 277, 168–175. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A. Glivec (ST1571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discov. 2002, 1, 493–502. [Google Scholar] [CrossRef]
- Donson, A.; Werner, E.; Amani, V.; Griesinger, A.; Witt, D.; Nellan, A.; Vibhakar, R.; Hankinson, T.; Handler, M.; Dorris, K.; et al. Tyrosine kinase inhibitors axitinib, imatinib and pazopanib are selectively potent in ependymoma. Neuro-Oncology 2017, 19, iv17. [Google Scholar] [CrossRef]
- Newton, H.B.; Ray-Chaudhury, A. Overview of Brain Tumor Epidemiology and Histopathology. Handb. Brain Tumor Chemother. 2006, 3–20. [Google Scholar] [CrossRef]
- Kleihues, P.; Burger, P.C.; Scheithauer, B.W. Histological Typing of Tumours of the Central Nervous System; Springer: Berlin/Heidelberg, Germany, 1993. [Google Scholar] [CrossRef]
- Kweh, B.T.S.; Rosenfeld, J.V.; Hunn, M.; Tee, J.W. Tumor characteristics and surgical outcomes of intracranial subependymomas: A systematic review and meta-analysis. J. Neurosurg. 2021, 20, 1–13. [Google Scholar] [CrossRef]
- Jooma, R.; Torrens, M.J.; Bradshaw, J.; Brownell, B. Subependymomas of the fourth ventricle. Surgical treatment in 12 cases. J. Neurosurg. 1985, 62, 508–512. [Google Scholar] [CrossRef]
- Varma, A.; Giraldi, D.; Mills, S.; Brodbelt, A.R.; Jenkinson, M.D. Surgical management and long-term outcome of intracranial subependymoma. Acta Neurochir. 2018, 160, 1793–1799. [Google Scholar] [CrossRef] [Green Version]
- Scheithauer, B.W. Symptomatic subependymoma. Report of 21 cases with review of the literature. J. Neurosurg. 1978, 49, 689–696. [Google Scholar] [CrossRef]
- Hou, Z.; Wu, Z.; Zhang, J.; Zhang, L.; Tian, R.; Liu, B.; Wang, Z. Lateral ventricular subependymomas: An analysis of the clinical features of 27 adult cases at a single institute. Neurol. India 2012, 60, 379. [Google Scholar] [CrossRef]
- Limaiem, F.; Das, J.M. Myxopapillary Ependymoma. Available online: http://www.ncbi.nlm.nih.gov/pubmed/32644598 (accessed on 7 July 2021).
- Sonneland, P.R.L.; Scheithauer, B.W.; Onofrio, B.M. Myxopapillary ependymoma. A clinicopathologic and immunocytochemical study of 77 cases. Cancer 1985, 56, 883–893. [Google Scholar] [CrossRef]
- Mørk, S.J.; Løken, A.C. Ependymoma. A follow-up study of 101 cases. Cancer 1977, 40, 907–915. [Google Scholar] [CrossRef]
- Kraetzig, T.; McLaughlin, L.; Bilsky, M.H.; Laufer, I. Metastases of spinal myxopapillary ependymoma: Unique characteristics and clinical management. J. Neurosurg. Spine 2018, 28, 201–208. [Google Scholar] [CrossRef]
- Bagley, C.A.; Wilson, S.; Kothbauer, K.F.; Bookland, M.J.; Epstein, F.; Jallo, G.I. Long term outcomes following surgical resection of myxopapillary ependymomas. Neurosurg. Rev. 2009, 32, 321–334. [Google Scholar] [CrossRef]
- Feldman, W.B.; Clark, A.J.; Safaee, M.; Ames, C.P.; Parsa, A.T. Tumor control after surgery for spinal myxopapillary ependymomas: Distinct outcomes in adults versus children. J. Neurosurg. Spine 2013, 19, 471–476. [Google Scholar] [CrossRef]
- Liu, T.; Yang, C.; Deng, X.; Li, A.; Xin, Y.; Yang, J.; Xu, Y. Clinical characteristics and surgical outcomes of spinal myxopapillary ependymomas. Neurosurg. Rev. 2020, 43, 1351–1356. [Google Scholar] [CrossRef]
- Akyurek, S.; Chang, E.L.; Yu, T.K.; Little, D.; Allen, P.K.; McCutcheon, I.; Mahajan, A.; Maor, M.H.; Woo, S.Y. Spinal myxopapillary ependymoma outcomes in patients treated with surgery and radiotherapy at M.D. Anderson Cancer Center. J. Neurooncol. 2006, 80, 177–183. [Google Scholar] [CrossRef]
- Waldron, J.N.; Laperriere, N.J.; Jaakkimainen, L.; Simpson, W.J.; Payne, D.; Milosevic, M.; Wong, C.S. Spinal cord ependymomas: A retrospective analysis of 59 cases. Int. J. Radiat. Oncol. Biol. Phys. 1993, 27, 223–229. [Google Scholar] [CrossRef]
- Whitaker, S.J.; Bessell, E.M.; Ashley, S.E.; Bloom, H.J.G.; Bell, B.A.; Brada, M. Postoperative radiotherapy in the management of spinal cord ependymoma. J. Neurosurg. 1991, 74, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Barton, V.N.; Donson, A.M.; Kleinschmidt-Demasters, B.K.; Birks, D.K.; Handler, M.H.; Foreman, N.K. Unique molecular characteristics of pediatric myxopapillary ependymoma. Brain Pathol. 2010, 20, 560–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellik, D.M. Hox patterning of the vertebrate axial skeleton. Dev. Dyn. 2007, 236, 2454–2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alharbi, R.A.; Pandha, H.S.; Simpson, G.R.; Pettengell, R.; Poterlowicz, K.; Thompson, A.; Harrington, K.; El-Tanani, M.; Morgan, R. Inhibition of HOX/PBX dimer formation leads to necroptosis in acute myeloid leukemia cells. Oncotarget 2017, 8, 89566–89579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Hamada, J.I.; Murakawa, K.; Takada, M.; Tada, M.; Nogami, I.; Hayashi, N.; Nakamori, S.; Monden, M.; Miyamoto, M.; et al. Expression profiles of 39 HOX genes in normal human adult organs and anaplastic thyroid cancer cell lines by quantitative real-time RT-PCR system. Exp. Cell Res. 2004, 293, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, O.; Chapman, R.; Storer, L.C.; Luo, L.; Heath, P.R.; Resar, L.; Cohen, K.J.; Grundy, R.G.; Lourdusamy, A. Integrative molecular characterization of pediatric spinal ependymoma: The UK Children’s Cancer and Leukaemia Group study. Neuro-Oncol. Adv. 2021, 3, vdab043. [Google Scholar] [CrossRef]
- Toomey, D.P.; Murphy, J.F.; Conlon, K.C. COX-2, VEGF and tumour angiogenesis. Surgeon 2009, 7, 174–180. [Google Scholar] [CrossRef]
- Schonthal, A.H. Exploiting Cyclooxygenase-(in)Dependent Properties of COX-2 Inhibitors for Malignant Glioma Therapy. Anticancer Agents Med. Chem. 2012, 10, 450–461. [Google Scholar] [CrossRef]
- Axelsson, H.; Lönnroth, C.; Wang, W.; Svanberg, E.; Lundholm, K. Cyclooxygenase inhibition in early onset of tumor growth and related angiogenesis evaluated in EP1 and EP3 knockout tumor-bearing mice. Angiogenesis 2006, 8, 339–348. [Google Scholar] [CrossRef]
- Celano, E.; Salehani, A.; Malcolm, J.G.; Reinertsen, E.; Hadjipanayis, C.G. Spinal cord ependymoma: A review of the literature and case series of ten patients. J. Neurooncol. 2016, 128, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Salunke, P. Understanding Ependymoma Oncogenesis: An Update on Recent Molecular Advances and Current Perspectives. Mol. Neurobiol. 2017, 54, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Guyotat, J.; Metellus, P.; Giorgi, R.; Barrie, M.; Jouvet, A.; Fevre-Montange, M.; Chinot, O.; Durand, A.; Figarella-Branger, D. Infratentorial ependymomas: Prognostic factors and outcome analysis in a multi-center retrospective series of 106 adult patients. Acta Neurochir. 2009, 151, 947–960. [Google Scholar] [CrossRef] [PubMed]
- Gerszten, P.C.; Pollack, I.F.; Martínez, A.J.; Lo, K.H.; Janosky, J.; Albright, A.L. Intracranial ependymomas of childhood lack of correlation of histopathology and clinical outcome. Pathol. Res. Pract. 1996, 192, 515–522. [Google Scholar] [CrossRef]
- Schiffer, D.; Chiò, A.; Giordana, M.T.; Migheli, A.; Palma, L.; Pollo, B.; Soffietti, R.; Tribolo, A. Histologic prognostic factors in ependymoma. Childs Nerv. Syst. 1991, 7, 177–182. [Google Scholar] [CrossRef]
- Korshunov, A.; Golanov, A.; Sycheva, R.; Timirgaz, V. The Histologic Grade Is a Main Prognostic Factor for Patients with Intracranial Ependymomas Treated in the Microneurosurgical Era: An Analysis of 258 Patients. Cancer 2004, 100, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rivera, V.; Dono, A.; Abdelkhaleq, R.; Sheth, S.A.; Chen, P.R.; Chandra, A.; Ballester, L.Y.; Esquenazi, Y. Treatment trends and overall survival in patients with grade II/III ependymoma: The role of tumor grade and location. Clin. Neurol. Neurosurg. 2020, 199, 106282. [Google Scholar] [CrossRef]
- Tarapore, P.E.; Modera, P.; Naujokas, A.; Oh, M.C.; Amin, B.; Tihan, T.; Parsa, A.T.; Ames, C.P.; Chou, D.; Mummaneni, P.V.; et al. Pathology of spinal ependymomas: An institutional experience over 25 years in 134 patients. Neurosurgery 2013, 73, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Chung, C.K.; Kim, C.H.; Yoon, S.H.; Hyun, S.J.; Kim, K.J.; Kim, E.S.; Eoh, W.; Kim, H.J. Long-term outcomes of surgical resection with or without adjuvant radiation therapy for treatment of spinal ependymoma: A retrospective multicenter study by the Korea Spinal Oncology Research Group. Neuro-Oncology 2013, 15, 921–929. [Google Scholar] [CrossRef] [Green Version]
- Savoor, R.; Sita, T.L.; Dahdaleh, N.S.; Helenowski, I.; Kalapurakal, J.A.; Marymont, M.H.; Lukas, R.; Kruser, T.J.; Smith, Z.A.; Koski, T.; et al. Long-term outcomes of spinal ependymomas: An institutional experience of more than 60 cases. J. Neuro-Oncol. 2020, 151, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, A.M.; Fernández-Valle, C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene 2016, 35, 537–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coy, S.; Rashid, R.; Stemmer-Rachamimov, A.; Santagata, S. An update on the CNS manifestations of neurofibromatosis type 2. Acta Neuropathol. 2020, 139, 643–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abylkassov, R.; Xie, Y. Role of yes-associated protein in cancer: An update (Review). Oncol. Lett. 2016, 12, 2277–2282. [Google Scholar] [CrossRef] [Green Version]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, J.C.; Gan, H.K.; Blagden, S.P.; Plummer, R.; Arkenau, H.T.; Ranson, M.; Evans, T.R.J.; Zalcman, G.; Bahleda, R.; Hollebecque, A.; et al. A phase I, pharmacokinetic and pharmacodynamic study of GSK2256098, a focal adhesion kinase inhibitor, in patients with advanced solid tumors. Ann. Oncol. 2016, 27, 2268–2274. [Google Scholar] [CrossRef]
- Sato, T.; Sekido, Y. NF2/merlin inactivation and potential therapeutic targets in mesothelioma. Int. J. Mol. Sci. 2018, 19, 988. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Rossi, N.; Priddy, S.; Pierson, C.R.; Studebaker, A.W.; Johnson, R.A. EphB2 activation is required for ependymoma development as well as inhibits differentiation and promotes proliferation of the transformed cell. Sci. Rep. 2015, 5, 9248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 724–742. [Google Scholar]
- Pavon, L.F.; Sibov, T.T.; de Toledo, S.R.C.; de Oliveira, D.M.; Cabral, F.R.; de Souza, J.G.; Boufleur, P.; Marti, L.C.; Malheiros, J.M.; da Cruz, E.F.; et al. Establishment of primary cell culture and an intracranial xenograft model of pediatric ependymoma: A prospect for therapy development and understanding of tumor biology. Oncotarget 2018, 9, 21731. [Google Scholar] [CrossRef] [Green Version]
- Scheil, S.; Brüderlein, S.; Eicker, M.; Herms, J.; Herold-Mende, C.; Steiner, H.H.; Barth, T.F.E.; Möller, P. Low frequency of chromosomal imbalances in anaplastic ependymomas as detected by comparative genomic hybridization. Brain Pathol. 2001, 11, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, D.R.; Sill, M.; Okonechnikov, K.; Korshunov, A.; Yip, S.; Schutz, P.W.; Scheie, D.; Kruse, A.; Harter, P.N.; Kastelan, M.; et al. MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol. 2019, 138, 1075–1089. [Google Scholar] [CrossRef] [Green Version]
- Raffeld, M.; Abdullaev, Z.; Pack, S.D.; Xi, L.; Nagaraj, S.; Briceno, N.; Vera, E.; Pittaluga, S.; Lopes Abath Neto, O.; Quezado, M.; et al. High level MYCN amplification and distinct methylation signature define an aggressive subtype of spinal cord ependymoma. Acta Neuropathol. Commun. 2020, 8, 101. [Google Scholar] [CrossRef]
- Swanson, A.A.; Raghunathan, A.; Jenkins, R.B.; Messing-Jünger, M.; Pietsch, T.; Clarke, M.J.; Kaufmann, T.J.; Giannini, C. Spinal cord ependymomas with MYCN amplification show aggressive clinical behavior. J. Neuropathol. Exp. Neurol. 2019, 78, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Astolfi, A.; Vendemini, F.; Urbini, M.; Melchionda, F.; Masetti, R.; Franzoni, M.; Libri, V.; Serravalle, S.; Togni, M.; Paone, G.; et al. MYCN is a novel oncogenic target in pediatric T-cell Acute Lymphoblastic Leukemia. Oncotarget 2014, 5, 120–130. [Google Scholar] [CrossRef]
- Barone, G.; Anderson, J.; Pearson, A.D.J.; Petrie, K.; Chesler, L. New strategies in neuroblastoma: Therapeutic targeting of MYCN and ALK. Clin. Cancer Res. 2013, 19, 5814–5821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korshunov, A.; Schrimpf, D.; Ryzhova, M.; Sturm, D.; Chavez, L.; Hovestadt, V.; Sharma, T.; Habel, A.; Burford, A.; Jones, C.; et al. H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol. 2017, 134, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Phillips, J.W.; Smith, B.A.; Park, J.W.; Stoyanova, T.; McCaffrey, E.F.; Baertsch, R.; Sokolov, A.; Meyerowitz, J.G.; Mathis, C.; et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell 2016, 29, 536–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Pérez, M.V.; Henley, A.B.; Arsenian-Henriksson, M. The MYCN protein in health and disease. Genes 2017, 8, 113. [Google Scholar] [CrossRef]
- Stermann, A.; Huebener, N.; Seidel, D.; Fest, S.; Eschenburg, G.; Stauder, M.; Schramm, A.; Eggert, A.; Lode, H.N. Targeting of MYCN by means of DNA vaccination is effective against neuroblastoma in mice. Cancer Immunol. Immunother. 2015, 64, 1215–1227. [Google Scholar] [CrossRef]
- Mack, S.C.; Pajtler, K.W.; Chavez, L.; Okonechnikov, K.; Bertrand, K.C.; Wang, X.X.; Erkek, S.; Federation, A.; Song, A.; Lee, C.; et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 2018, 553, 101–105. [Google Scholar] [CrossRef]
- Panwalkar, P.; Clark, J.; Ramaswamy, V.; Hawes, D.; Yang, F.; Dunham, C.; Yip, S.; Hukin, J.; Sun, Y.; Schipper, M.J.; et al. Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol. 2017, 134, 705–714. [Google Scholar] [CrossRef]
- Bayliss, J.; Mukherjee, P.; Lu, C.; Jain, S.U.; Chung, C.; Martinez, D.; Sabari, B.; Margol, A.S.; Panwalkar, P.; Parolia, A.; et al. Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci. Transl. Med. 2016, 8, 366ra161. [Google Scholar] [CrossRef] [Green Version]
- Nambirajan, A.; Sharma, A.; Rajeshwari, M.; Boorgula, M.T.; Doddamani, R.; Garg, A.; Suri, V.; Sarkar, C.; Sharma, M.C. EZH2 inhibitory protein (EZHIP/Cxorf67) expression correlates strongly with H3K27me3 loss in posterior fossa ependymomas and is mutually exclusive with H3K27M mutations. Brain Tumor Pathol. 2021, 38, 30–40. [Google Scholar] [CrossRef]
- Tanrıkulu, B.; Danyeli, A.E.; Özek, M.M. Is H3K27me3 status really a strong prognostic indicator for pediatric posterior fossa ependymomas? A single surgeon, single center experience. Childs Nerv. Syst. 2020, 36, 941–949. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Yuh, E.L.; Barkovich, A.J.; Gupta, N. Imaging of ependymomas: MRI and CT. Childs Nerv. Syst. 2009, 25, 1203–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonezawa, U.; Karlowee, V.; Amatya, V.J.; Takayasu, T.; Takano, M.; Takeshima, Y.; Sugiyama, K.; Kurisu, K.; Yamasaki, F. Radiology Profile as a Potential Instrument to Differentiate Between Posterior Fossa Ependymoma (PF-EPN) Group A and B. World Neurosurg. 2020, 140, e320–e327. [Google Scholar] [CrossRef]
- Zhao, F.; Wu, T.; Wang, L.M.; Zhang, J.; Zhang, H.; Li, S.W.; Zhang, S.; Li, P.; Wang, B.; Luo, L.; et al. Survival and Prognostic Factors of Adult Intracranial Ependymoma: A Single-institutional Analysis of 236 Patients. Am. J. Surg. Pathol. 2021, 45, 979–987. [Google Scholar] [CrossRef]
- Baroni, L.V.; Sundaresan, L.; Heled, A.; Coltin, H.; Pajtler, K.W.; Lin, T.; Merchant, T.E.; McLendon, R.; Faria, C.; Buntine, M.; et al. Ultra high-risk PFA ependymoma is characterized by loss of chromosome 6q. Neuro-Oncology 2021, 23, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Pajtler, K.W.; Wen, J.; Sill, M.; Lin, T.; Orisme, W.; Tang, B.; Hübner, J.M.; Ramaswamy, V.; Jia, S.; Dalton, J.D.; et al. Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. 2018, 136, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Ryall, S.; Guzman, M.; Elbabaa, S.K.; Luu, B.; Mack, S.C.; Zapotocky, M.; Taylor, M.D.; Hawkins, C.; Ramaswamy, V. H3 K27M mutations are extremely rare in posterior fossa group A ependymoma. Childs Nerv. Syst. 2017, 33, 1047–1051. [Google Scholar] [CrossRef]
- De Sousa, G.R.; Lira, R.C.P.; de Almeida Magalhães, T.; da Silva, K.R.; Nagano, L.F.P.; Saggioro, F.P.; Baroni, M.; Marie, S.K.N.; Oba-Shinjo, S.M.; Brandelise, S.; et al. A coordinated approach for the assessment of molecular subgroups in pediatric ependymomas using low-cost methods. J. Mol. Med. 2021, 99, 1101–1113. [Google Scholar] [CrossRef]
- Jain, S.U.; Do, T.J.; Lund, P.J.; Rashoff, A.Q.; Diehl, K.L.; Cieslik, M.; Bajic, A.; Juretic, N.; Deshmukh, S.; Venneti, S.; et al. PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat. Commun. 2019, 10, 2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hübner, J.M.; Müller, T.; Papageorgiou, D.N.; Mauermann, M.; Krijgsveld, J.; Russell, R.B.; Ellison, D.W.; Pfister, S.M.; Pajtler, K.W.; Kool, M. EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro-Oncology 2019, 21, 878–889. [Google Scholar] [CrossRef]
- Krug, B.; Harutyunyan, A.S.; Deshmukh, S.; Jabado, N. Polycomb repressive complex 2 in the driver’s seat of childhood and young adult brain tumours. Trends Cell Biol. 2021, 31, 814–828. [Google Scholar] [CrossRef]
- Jain, S.U.; Rashoff, A.Q.; Krabbenhoft, S.D.; Hoelper, D.; Do, T.J.; Gibson, T.J.; Lundgren, S.M.; Bondra, E.R.; Deshmukh, S.; Harutyunyan, A.S.; et al. H3 K27M and EZHIP Impede H3K27-Methylation Spreading by Inhibiting Allosterically Stimulated PRC2. Mol. Cell 2020, 80, 726–735. [Google Scholar] [CrossRef]
- Dyer, M.A.; Qadeer, Z.A.; Valle-Garcia, D.; Bernstein, E. ATRX and DAXX: Mechanisms and mutations. Cold Spring Harb. Perspect. Med. 2017, 7, a026567. [Google Scholar] [CrossRef]
- Michealraj, K.A.; Kumar, S.A.; Kim, L.J.Y.; Cavalli, F.M.G.; Przelicki, D.; Wojcik, J.B.; Delaidelli, A.; Bajic, A.; Saulnier, O.; MacLeod, G.; et al. Metabolic Regulation of the Epigenome Drives Lethal Infantile Ependymoma. Cell 2020, 181, 1329–1345. [Google Scholar] [CrossRef]
- Griesinger, A.M.; Witt, D.A.; Grob, S.T.; Georgio Westover, S.R.; Donson, A.M.; Sanford, B.; Mulcahy Levy, J.M.; Wong, R.; Moreira, D.C.; Desisto, J.A.; et al. NF-κB upregulation through epigenetic silencing of LDOC1 drives tumor biology and specific immunophenotype in Group A ependymoma. Neuro-Oncology 2017, 19, 1350–1360. [Google Scholar] [CrossRef] [Green Version]
- Griesinger, A.M.; Josephson, R.J.; Donson, A.M.; Levy, J.M.M.; Amani, V.; Birks, D.K.; Hoffman, L.M.; Furtek, S.L.; Reigan, P.; Handler, M.H.; et al. Interleukin-6/STAT3 pathway signaling drives an inflammatory phenotype in group a ependymoma. Cancer Immunol. Res. 2015, 3, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Gojo, J.; Lötsch, D.; Spiegl-Kreinecker, S.; Pajtler, K.W.; Neumayer, K.; Korbel, P.; Araki, A.; Brandstetter, A.; Mohr, T.; Hovestadt, V.; et al. Telomerase activation in posterior fossa group A ependymomas is associated with dismal prognosis and chromosome 1q gain. Neuro-Oncology 2017, 19, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Meel, M.H.; Kaspers, G.J.L.; Hulleman, E. Preclinical therapeutic targets in diffuse midline glioma. Drug Resist. Updates 2019, 44, 15–25. [Google Scholar] [CrossRef]
- Lin, G.L.; Wilson, K.M.; Ceribelli, M.; Stanton, B.Z.; Woo, P.J.; Kreimer, S.; Qin, E.Y.; Zhang, X.; Lennon, J.; Nagaraja, S.; et al. Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Sandberg, D.I.; Yu, B.; Patel, R.; Hagan, J.; Miesner, E.; Sabin, J.; Smith, S.; Fletcher, S.; Shah, M.N.; Sirianni, R.W.; et al. Infusion of 5-Azacytidine (5-AZA) into the fourth ventricle or resection cavity in children with recurrent posterior Fossa Ependymoma: A pilot clinical trial. J. Neurooncol. 2019, 141, 449–457. [Google Scholar] [CrossRef]
- Han, J.; Yu, M.; Bai, Y.; Yu, J.; Jin, F.; Li, C.; Zeng, R.; Peng, J.; Li, A.; Song, X.; et al. Elevated CXorf67 Expression in PFA Ependymomas Suppresses DNA Repair and Sensitizes to PARP Inhibitors. Cancer Cell 2020, 38, 844–856. [Google Scholar] [CrossRef]
- Gojo, J.; Englinger, B.; Jiang, L.; Hübner, J.M.; Shaw, M.L.; Hack, O.A.; Madlener, S.; Kirchhofer, D.; Liu, I.; Pyrdol, J.; et al. Single-Cell RNA-Seq Reveals Cellular Hierarchies and Impaired Developmental Trajectories in Pediatric Ependymoma. Cancer Cell 2020, 38, 44–59. [Google Scholar] [CrossRef]
- Cruz, C.; Ribes, V.; Kutejova, E.; Cayuso, J.; Lawson, V.; Norris, D.; Stevens, J.; Davey, M.; Blight, K.; Bangs, F.; et al. Foxj1 regulates floor plate cilia architecture and modifies the response of cells to sonic hedgehog signalling. Development 2010, 137, 4271–4282. [Google Scholar] [CrossRef] [Green Version]
- Abedalthagafi, M.S.; Wu, M.P.; Merrill, P.H.; Du, Z.; Woo, T.; Sheu, S.H.; Hurwitz, S.; Ligon, K.L.; Santagata, S. Decreased FOXJ1 expression and its ciliogenesis programme in aggressive ependymoma and choroid plexus tumours. J. Pathol. 2016, 238, 584–597. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cai, X.; Xia, L.; Zhou, J.; Xin, J.; Liu, M.; Shang, X.; Liu, J.; Li, X.; Chen, Z.; et al. Decreased Expression of FOXJ1 is a Potential Prognostic Predictor for Progression and Poor Survival of Gastric Cancer. Ann. Surg. Oncol. 2015, 22, 685–692. [Google Scholar] [CrossRef]
- Zhu, P.; Piao, Y.; Dong, X.; Jin, Z. Forkhead box J1 expression is upregulated and correlated with prognosis in patients with clear cell renal cell carcinoma. Oncol. Lett. 2015, 10, 1487–1494. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Fan, J.; Wu, J. Forkhead box protein J1 (FOXJ1) is overexpressed in colorectal cancer and promotes nuclear translocation of β-catenin in SW620 cells. Med. Sci. Monit. 2017, 23, 856–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgescu, M.M.; Gagea, M.; Cote, G. NHERF1/EBP50 Suppresses Wnt-β-Catenin Pathway–Driven Intestinal Neoplasia. Neoplasia 2016, 18, 512–523. [Google Scholar] [CrossRef] [Green Version]
- Kreimann, E.L.; Morales, F.C.; De Orbeta-Cruz, J.; Takahashi, Y.; Adams, H.; Liu, T.J.; McCrea, P.D.; Georgescu, M.M. Cortical stabilization of β-catenin contributes to NHERF1/EBP50 tumor suppressor function. Oncogene 2007, 26, 5290–5299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dihlmann, S.; Von Knebel Doeberitz, M. Wnt/β-catenin-pathway as a molecular target for future anti-cancer therapeutics. Int. J. Cancer 2005, 113, 515–524. [Google Scholar] [CrossRef]
- Jang, G.B.; Hong, I.S.; Kim, R.J.; Lee, S.Y.; Park, S.J.; Lee, E.S.; Park, J.H.; Yun, C.H.; Chung, J.U.; Lee, K.J.; et al. Wnt/β-catenin small-molecule inhibitor CWP232228 preferentially inhibits the growth of breast cancer stem-like cells. Cancer Res. 2015, 75, 1691–1702. [Google Scholar] [CrossRef] [Green Version]
- Pak, S.; Park, S.; Kim, Y.; Park, J.H.; Park, C.H.; Lee, K.J.; Kim, C.S.; Ahn, H. The small molecule WNT/β-catenin inhibitor CWP232291 blocks the growth of castration-resistant prostate cancer by activating the endoplasmic reticulum stress pathway. J. Exp. Clin. Cancer Res. 2019, 38, 342. [Google Scholar] [CrossRef]
- Coluccia, A.; La Regina, G.; Naccarato, V.; Nalli, M.; Orlando, V.; Biagioni, S.; De Angelis, M.L.; Baiocchi, M.; Gautier, C.; Gianni, S.; et al. Drug Design and Synthesis of First in Class PDZ1 Targeting NHERF1 Inhibitors as Anticancer Agents. ACS Med. Chem. Lett. 2019, 10, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Lester, A.; McDonald, K.L. Intracranial ependymomas: Molecular insights and translation to treatment. Brain Pathol. 2020, 30, 3–12. [Google Scholar] [CrossRef]
- Andreiuolo, F.; Varlet, P.; Tauziède-Espariat, A.; Jünger, S.T.; Dörner, E.; Dreschmann, V.; Kuchelmeister, K.; Waha, A.; Haberler, C.; Slavc, I.; et al. Childhood supratentorial ependymomas with YAP1-MAMLD1 fusion: An entity with characteristic clinical, radiological, cytogenetic and histopathological features. Brain Pathol. 2019, 29, 205–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajtler, K.W.; Wei, Y.; Okonechnikov, K.; Silva, P.B.G.; Vouri, M.; Zhang, L.; Brabetz, S.; Sieber, L.; Gulley, M.; Mauermann, M.; et al. YAP1 subgroup supratentorial ependymoma requires TEAD and nuclear factor I-mediated transcriptional programmes for tumorigenesis. Nat. Commun. 2019, 10, 3914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristal Kaan, H.Y.; Chan, S.W.; Tan, S.K.J.; Guo, F.; Lim, C.J.; Hong, W.; Song, H. Crystal structure of TAZ-TEAD complex reveals a distinct interaction mode from that of YAP-TEAD complex. Sci. Rep. 2017, 7, 2035. [Google Scholar] [CrossRef]
- Eder, N.; Roncaroli, F.; Dolmart, M.C.; Horswell, S.; Andreiuolo, F.; Flynn, H.R.; Lopes, A.T.; Claxton, S.; Kilday, J.P.; Collinson, L.; et al. YAP1/TAZ drives ependymoma-like tumour formation in mice. Nat. Commun. 2020, 11, 2380. [Google Scholar] [CrossRef]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. β-catenin driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151. [Google Scholar] [CrossRef] [Green Version]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Szulzewsky, F.; Holland, E.C.; Vasioukhin, V. YAP1 and its fusion proteins in cancer initiation, progression and therapeutic resistance. Dev. Biol. 2021, 475, 205–221. [Google Scholar] [CrossRef]
- Pan, Z.; Tian, Y.; Cao, C.; Niu, G. The emerging role of YAP/TAZ in tumor immunity. Mol. Cancer Res. 2019, 17, 1777–1786. [Google Scholar] [CrossRef] [Green Version]
- Oku, Y.; Nishiya, N.; Shito, T.; Yamamoto, R.; Yamamoto, Y.; Oyama, C.; Uehara, Y. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio 2015, 5, 542–549. [Google Scholar] [CrossRef] [Green Version]
- Lindauer, M.; Hochhaus, A. Dasatinib. Recent Results Cancer Res. 2018, 212, 29–68. [Google Scholar]
- Miyamoto, S.; Kakutani, S.; Sato, Y.; Hanashi, A.; Kinoshita, Y.; Ishikawa, A. Drug review: Pazopanib. Jpn. J. Clin. Oncol. 2018, 48, 503–513. [Google Scholar] [CrossRef]
- Hayashi, K.; Nakazato, Y.; Morito, N.; Sagi, M.; Fujita, T.; Anzai, N.; Chida, M. Fluvastatin is effective against thymic carcinoma. Life Sci. 2020, 240, 117110. [Google Scholar] [CrossRef]
- Giraud, J.; Molina-Castro, S.; Seeneevassen, L.; Sifré, E.; Izotte, J.; Tiffon, C.; Staedel, C.; Boeuf, H.; Fernandez, S.; Barthelemy, P.; et al. Verteporfin targeting YAP1/TAZ-TEAD transcriptional activity inhibits the tumorigenic properties of gastric cancer stem cells. Int. J. Cancer 2020, 146, 2255–2267. [Google Scholar] [CrossRef]
- Sallam, Y.T.; Zhang, Q.; Pandey, S.K. Cortically based cystic supratentorial RELA fusion-positive ependymoma: A case report with unusual presentation and appearance and review of literature. Radiol. Case Rep. 2020, 15, 2495–2499. [Google Scholar] [CrossRef]
- Gessi, M.; Giagnacovo, M.; Modena, P.; Elefante, G.; Gianno, F.; Buttarelli, F.R.; Arcella, A.; Donofrio, V.; Diomedi Camassei, F.; Nozza, P.; et al. Role of immunohistochemistry in the identification of supratentorial C11ORF95-RELA fused ependymoma in routine neuropathology. Am. J. Surg. Pathol. 2019, 43, 56–63. [Google Scholar] [CrossRef]
- Tauziède-Espariat, A.; Siegfried, A.; Nicaise, Y.; Kergrohen, T.; Sievers, P.; Vasiljevic, A.; Roux, A.; Dezamis, E.; Benevello, C.; Machet, M.C.; et al. Supratentorial non-RELA, ZFTA-fused ependymomas: A comprehensive phenotype genotype correlation highlighting the number of zinc fingers in ZFTA-NCOA1/2 fusions. Acta Neuropathol. Commun. 2021, 9, 135. [Google Scholar] [CrossRef]
- Parker, M.; Mohankumar, K.M.; Punchihewa, C.; Weinlich, R.; Dalton, J.D.; Li, Y.; Lee, R.; Tatevossian, R.G.; Phoenix, T.N.; Thiruvenkatam, R.; et al. C11orf95-RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature 2014, 506, 451–455. [Google Scholar] [CrossRef] [Green Version]
- Zschernack, V.; Jünger, S.T.; Mynarek, M.; Rutkowski, S.; Garre, M.L.; Ebinger, M.; Neu, M.; Faber, J.; Erdlenbruch, B.; Claviez, A.; et al. Supratentorial ependymoma in childhood: More than just RELA or YAP. Acta Neuropathol. 2021, 141, 455–466. [Google Scholar] [CrossRef]
- Tomomasa, R.; Arai, Y.; Kawabata-Iwakawa, R.; Fukuoka, K.; Nakano, Y.; Hama, N.; Nakata, S.; Suzuki, N.; Ishi, Y.; Tanaka, S.; et al. Ependymoma-like tumor with mesenchymal differentiation harboring C11orf95-NCOA1/2 or -RELA fusion: A hitherto unclassified tumor related to ependymoma. Brain Pathol. 2021, 31, e12943. [Google Scholar] [CrossRef]
- Tamai, S.; Nakano, Y.; Kinoshita, M.; Sabit, H.; Nobusawa, S.; Arai, Y.; Hama, N.; Totoki, Y.; Shibata, T.; Ichimura, K.; et al. Ependymoma with C11orf95-MAML2 fusion: Presenting with granular cell and ganglion cell features. Brain Tumor Pathol. 2021, 38, 64–70. [Google Scholar] [CrossRef]
- Zheng, T.; Ghasemi, D.R.; Okonechnikov, K.; Korshunov, A.; Sill, M.; Maass, K.K.; Benites Goncalves da Silva, P.; Ryzhova, M.; Gojo, J.; Stichel, D.; et al. Cross-Species Genomics Reveals Oncogenic Dependencies in ZFTA/C11orf95 Fusion–Positive Supratentorial Ependymomas. Cancer Discov. 2021, 11, 2230–2247. [Google Scholar] [CrossRef]
- Kupp, R.; Ruff, L.; Terranova, S.; Nathan, E.; Ballereau, S.; Stark, R.; Sekhar Reddy Chilamakuri, C.; Hoffmann, N.; Wickham-Rahrmann, K.; Widdess, M.; et al. ZFTA Translocations Constitute Ependymoma Chromatin Remodeling and Transcription Factors. Cancer Discov. 2021, 11, 2216–2229. [Google Scholar] [CrossRef]
- Ozawa, T.; Arora, S.; Szulzewsky, F.; Juric-Sekhar, G.; Miyajima, Y.; Bolouri, H.; Yasui, Y.; Barber, J.; Kupp, R.; Dalton, J.; et al. A De Novo Mouse Model of C11orf95-RELA Fusion-Driven Ependymoma Identifies Driver Functions in Addition to NF-κB. Cell Rep. 2018, 23, 3787–3797. [Google Scholar] [CrossRef]
- Ozawa, T.; Kaneko, S.; Szulzewsky, F.; Qiao, Z.; Takadera, M.; Narita, Y.; Kondo, T.; Holland, E.C.; Hamamoto, R.; Ichimura, K. C11orf95-RELA fusion drives aberrant gene expression through the unique epigenetic regulation for ependymoma formation. Acta Neuropathol. Commun. 2021, 9, 36. [Google Scholar] [CrossRef]
- Didonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef]
- Arabzade, A.; Zhao, Y.; Varadharajan, S.; Chen, H.-C.; Jessa, S.; Rivas, B.; Stuckert, A.J.; Solis, M.; Kardian, A.; Tlais, D.; et al. ZFTA–RELA Dictates Oncogenic Transcriptional Programs to Drive Aggressive Supratentorial Ependymoma. Cancer Discov. 2021, 11, 2200–2215. [Google Scholar] [CrossRef]
- Lötsch, D.; Kirchhofer, D.; Englinger, B.; Jiang, L.; Okonechnikov, K.; Senfter, D.; Laemmerer, A.; Gabler, L.; Pirker, C.; Donson, A.M.; et al. Targeting fibroblast growth factor receptors to combat aggressive ependymoma. Acta Neuropathol. 2021, 142, 339–360. [Google Scholar] [CrossRef]
- Wang, L.; Han, S.; Yan, C.; Yang, Y.; Li, Z.; Yang, Z. The role of clinical factors and immunocheckpoint molecules in the prognosis of patients with supratentorial extraventricular ependymoma: A single-center retrospective study. J. Cancer Res. Clin. Oncol. 2021, 147, 1259–1270. [Google Scholar] [CrossRef]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Rausch, T.; Jones, D.T.W.; Zapatka, M.; Stütz, A.M.; Zichner, T.; Weischenfeldt, J.; Jäger, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larrew, T.; Saway, B.F.; Lowe, S.R.; Olar, A. Molecular Classification and Therapeutic Targets in Ependymoma. Cancers 2021, 13, 6218. https://doi.org/10.3390/cancers13246218
Larrew T, Saway BF, Lowe SR, Olar A. Molecular Classification and Therapeutic Targets in Ependymoma. Cancers. 2021; 13(24):6218. https://doi.org/10.3390/cancers13246218
Chicago/Turabian StyleLarrew, Thomas, Brian Fabian Saway, Stephen R. Lowe, and Adriana Olar. 2021. "Molecular Classification and Therapeutic Targets in Ependymoma" Cancers 13, no. 24: 6218. https://doi.org/10.3390/cancers13246218
APA StyleLarrew, T., Saway, B. F., Lowe, S. R., & Olar, A. (2021). Molecular Classification and Therapeutic Targets in Ependymoma. Cancers, 13(24), 6218. https://doi.org/10.3390/cancers13246218