
Cancer Treatment-Induced Accelerated Aging in Cancer Survivors: Biology and Assessment


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
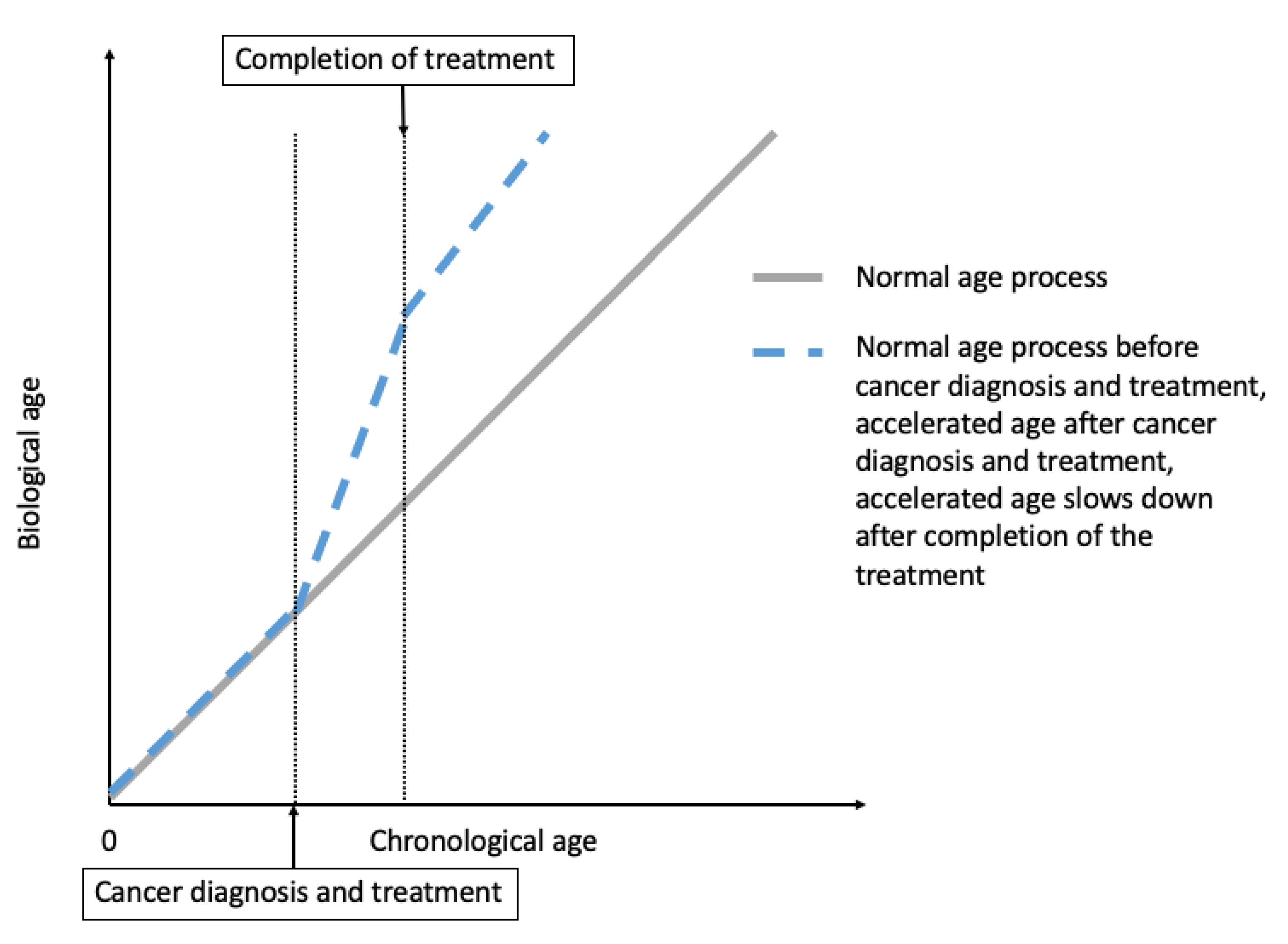
2. Cancer Treatment-Induced Accelerated Aging in Cancer Survivors
2.1. Epidemiological Evidence for Treatment-Induced Accelerated Aging
2.1.1. Increased Frailty in Cancer Survivors
2.1.2. Increased Risk of Comorbidities and Premature Mortality in Cancer Survivors
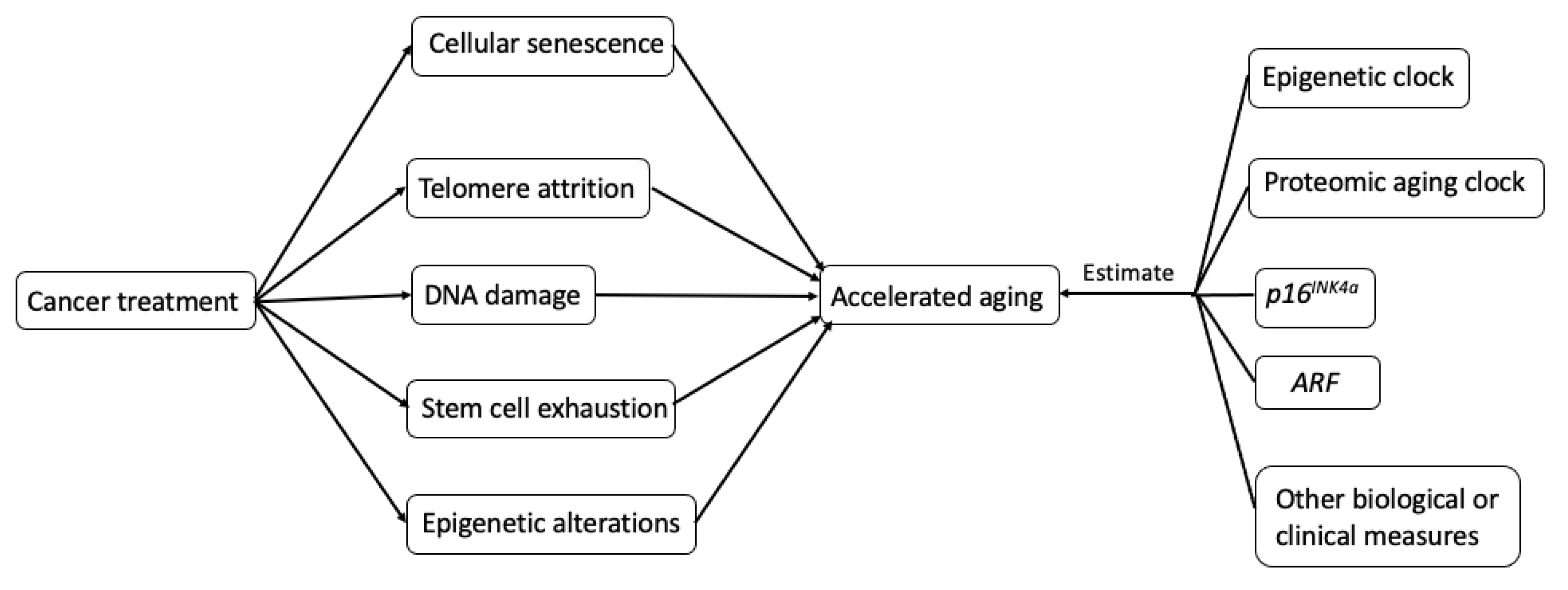
2.2. Biological Mechanisms Underlying Treatment-Induced Accelerated Aging in Cancer Survivors
2.2.1. Cellular Senescence and SASP
Cellular Senescence and Aging
SASP and Tumorigenesis
2.2.2. Telomere Attrition
2.2.3. Stem Cell Exhaustion
2.2.4. DNA Damage
2.2.5. Epigenetic Alterations
3. Measurements of Accelerated Aging in Cancer Survivors
3.1. Aging Clocks
3.1.1. Epigenetic Clock
3.1.2. Proteomic Aging Clock
3.2. Biomarkers of Cellular Senescence
3.2.1. p16INK4a
3.2.2. ARF (An Alternate Reading Frame Protein Product of the CDKN2A Locus)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sebastiani, P.; Thyagarajan, B.; Sun, F.; Schupf, N.; Newman, A.B.; Montano, M.; Perls, T.T. Biomarker signatures of aging. Aging Cell 2017, 16, 329–338. [Google Scholar] [CrossRef]
- Soto-Perez-De-Celis, E.; Li, D.; Yuan, Y.; Lau, Y.M.; Hurria, A. Functional versus chronological age: Geriatric assessments to guide decision making in older patients with cancer. Lancet Oncol. 2018, 19, e305–e316. [Google Scholar] [CrossRef]
- Olsen, J.H.; Möller, T.; Anderson, H.; Langmark, F.; Sankila, R.; Tryggvadóttír, L.; Winther, J.F.; Rechnitzer, C.; Jonmundsson, G.; Christensen, J.; et al. Lifelong Cancer Incidence in 47 697 Patients Treated for Childhood Cancer in the Nordic Countries. J. Natl. Cancer Inst. 2009, 101, 806–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reulen, R.C.; Frobisher, C.; Winter, D.L.; Kelly, J.; Lancashire, E.R.; Stiller, C.A.; Pritchard-Jones, K.; Jenkinson, H.C.; Hawkins, M.M.; British Childhood Cancer Survivor Study Steering Group. Long-term Risks of Subsequent Primary Neoplasms Among Survivors of Childhood Cancer. JAMA 2011, 305, 2311–2319. [Google Scholar] [CrossRef] [Green Version]
- Mohty, B.; Mohty, M. Long-term complications and side effects after allogeneic hematopoietic stem cell transplantation: An update. Blood Cancer J. 2011, 1, e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cupit-Link, M.C.; Kirkland, J.L.; Ness, K.K.; Armstrong, G.T.; Tchkonia, T.; Lebrasseur, N.K.; Armenian, S.H.; Ruddy, K.J.; Hashmi, S.K. Biology of premature ageing in survivors of cancer. ESMO Open 2017, 2, e000250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, A.; Sadda, J.; LaBarge, M.A.; Hurria, A. How cancer therapeutics cause accelerated aging: Insights from the hallmarks of aging. J. Geriatr. Oncol. 2020, 11, 191–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guida, J.L.; A Ahles, T.; Belsky, D.W.; Campisi, J.; Cohen, H.J.; DeGregori, J.; Fuldner, R.; Ferrucci, L.; Gallicchio, L.; Gavrilov, L.A.; et al. Measuring Aging and Identifying Aging Phenotypes in Cancer Survivors. J. Natl. Cancer Inst. 2019, 111, 1245–1254. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, M.C.; Mulrooney, D.A.; Steinberger, J.; Lee, J.; Baker, K.S.; Ness, K.K. Deficits in Physical Function Among Young Childhood Cancer Survivors. J. Clin. Oncol. 2013, 31, 2799–2805. [Google Scholar] [CrossRef] [Green Version]
- Lintermans, A.; Van Calster, B.; Van Hoydonck, M.; Pans, S.; Verhaeghe, J.; Westhovens, R.; Henry, N.L.; Wildiers, H.; Paridaens, R.; Dieudonné, A.S.; et al. Aromatase inhibitor-induced loss of grip strength is body mass index dependent: Hypothesis-generating findings for its pathogenesis. Ann. Oncol. 2011, 22, 1763–1769. [Google Scholar] [CrossRef] [PubMed]
- Lintermans, A.; Van Asten, K.; Wildiers, H.; Laenen, A.; Paridaens, R.; Weltens, C.; Verhaeghe, J.; Vanderschueren, D.; Smeets, A.; Van Limbergen, E.; et al. A prospective assessment of musculoskeletal toxicity and loss of grip strength in breast cancer patients receiving adjuvant aromatase inhibitors and tamoxifen, and relation with BMI. Breast Cancer Res. Treat. 2014, 146, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Uziel, O.; Lahav, M.; Shargian, L.; Beery, E.; Pasvolsky, O.; Rozovski, U.; Raanani, P.; Yeshurun, M. Premature ageing following allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2020, 55, 1438–1446. [Google Scholar] [CrossRef] [PubMed]
- Arora, M.; Sun, C.-L.; Ness, K.K.; Teh, J.B.; Wu, J.; Francisco, L.; Armenian, S.H.; Schad, A.; Namdar, G.; Bosworth, A.; et al. Physiologic Frailty in Nonelderly Hematopoietic Cell Transplantation Patients. JAMA Oncol. 2016, 2, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.L.; Whitton, J.; Leisenring, W.; Mertens, A.C.; Hammond, S.; Stovall, M.; Donaldson, S.S.; Meadows, A.T.; Robison, L.L.; Neglia, J.P. Subsequent Neoplasms in 5-Year Survivors of Childhood Cancer: The Childhood Cancer Survivor Study. J. Natl. Cancer Inst. 2010, 102, 1083–1095. [Google Scholar] [CrossRef]
- Bhakta, N.; Liu, Q.; Yeo, F.; Baassiri, M.; Ehrhardt, M.J.; Srivastava, D.K.; Metzger, M.L.; Krasin, M.J.; Ness, K.K.; Hudson, M.M.; et al. Cumulative burden of cardiovascular morbidity in paediatric, adolescent, and young adult survivors of Hodgkin’s lymphoma: An analysis from the St Jude Lifetime Cohort Study. Lancet Oncol. 2016, 17, 1325–1334. [Google Scholar] [CrossRef] [Green Version]
- Armenian, S.H.; Xu, L.; Ky, B.; Sun, C.; Farol, L.T.; Pal, S.K.; Douglas, P.S.; Bhatia, S.; Chao, C. Cardiovascular Disease Among Survivors of Adult-Onset Cancer: A Community-Based Retrospective Cohort Study. J. Clin. Oncol. 2016, 34, 1122–1130. [Google Scholar] [CrossRef]
- Strongman, H.; Gadd, S.; Matthews, A.; E Mansfield, K.; Stanway, S.; Lyon, A.R.; Dos-Santos-Silva, I.; Smeeth, L.; Bhaskaran, K. Medium and long-term risks of specific cardiovascular diseases in survivors of 20 adult cancers: A population-based cohort study using multiple linked UK electronic health records databases. Lancet 2019, 394, 1041–1054. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.B.; Chen, Y.; Yasui, Y.; Pui, C.-H.; Hunger, S.P.; Silverman, L.B.; Ness, K.K.; Green, D.M.; Howell, R.M.; Leisenring, W.M.; et al. Reduced Morbidity and Mortality in Survivors of Childhood Acute Lymphoblastic Leukemia: A Report From the Childhood Cancer Survivor Study. J. Clin. Oncol. 2020, 38, 3418–3429. [Google Scholar] [CrossRef]
- Yeh, J.M.; Nekhlyudov, L.; Goldie, S.J.; Mertens, A.C.; Diller, L. A Model-Based Estimate of Cumulative Excess Mortality in Survivors of Childhood Cancer. Ann. Intern. Med. 2010, 152, 409. [Google Scholar] [CrossRef]
- Pantziarka, P. Li Fraumeni syndrome, cancer and senescence: A new hypothesis. Cancer Cell Int. 2013, 13, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birkeland, A.C. Postoperative Clinical Radiosensitivity in Patients With Fanconi Anemia and Head and Neck Squamous Cell Carcinoma. Arch. Otolaryngol. Head Neck Surg. 2011, 137, 930–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J.; Di Fagagna, F.D. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef] [PubMed]
- I Evan, G.; Di Fagagna, F.D. Cellular senescence: Hot or what? Curr. Opin. Genet. Dev. 2009, 19, 25–31. [Google Scholar] [CrossRef]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.N.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; Van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lye, J.J.; Latorre, E.; Lee, B.P.; Bandinelli, S.; Holley, J.E.; Gutowski, N.J.; Ferrucci, L.; Harries, L.W. Astrocyte senescence may drive alterations in GFAPα, CDKN2A p14ARF, and TAU3 transcript expression and contribute to cognitive decline. GeroScience 2019, 41, 561–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diniz, B.S.; Reynolds, C.F.; Sibille, E.; Lin, C.-W.; Tseng, G.; Lotrich, F.; Aizenstein, H.J.; Butters, M.A. Enhanced Molecular Aging in Late-Life Depression: The Senescent-Associated Secretory Phenotype. Am. J. Geriatr. Psychiatry 2017, 25, 64–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.-Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; E Darnell, J. Stat3 as an Oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef]
- Kokkinos, M.I.; Wafai, R.; Wong, M.K.; Newgreen, D.F.; Thompson, E.W.; Waltham, M. Vimentin and Epithelial-Mesenchymal Transition in Human Breast Cancer—Observations in vitro and in vivo. Cells Tissues Organs 2007, 185, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.-W.; Liu, L.-J.; Huang, J. Interleukin-6-induced epithelial-mesenchymal transition through signal transducer and activator of transcription 3 in human cervical carcinoma. Int. J. Oncol. 2014, 45, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Kauser, K.; Campisi, J.; Beauséjour, C.M. Secretion of Vascular Endothelial Growth Factor by Primary Human Fibroblasts at Senescence. J. Biol. Chem. 2006, 281, 29568–29574. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, E.H.; Greider, C.W.; Szostak, J.W. Telomeres and telomerase: The path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 2006, 12, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A. Telomere length, stem cells and aging. Nat. Chem. Biol. 2007, 3, 640–649. [Google Scholar] [CrossRef]
- Carrero, J.J.; Stenvinkel, P.; Fellström, B.; Qureshi, A.R.; Lamb, K.; Heimbürger, O.; Bárány, P.; Radhakrishnan, K.; Lindholm, B.; Soveri, I.; et al. Telomere attrition is associated with inflammation, low fetuin-A levels and high mortality in prevalent haemodialysis patients. J. Intern. Med. 2007, 263, 302–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.G.; Zhang, R.P.; Wang, X.W.; Xie, H. Effects of cisplatin on telomerase activity and telomere length in BEL-7404 human hepatoma cells. Cell Res. 2002, 12, 55–62. [Google Scholar] [CrossRef]
- Hao, L.-Y.; Armanios, M.; Strong, M.A.; Karim, B.; Feldser, D.M.; Huso, D.; Greider, C.W. Short Telomeres, even in the Presence of Telomerase, Limit Tissue Renewal Capacity. Cell 2005, 123, 1121–1131. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-J.; Nam, C.-E.; Cho, S.-H.; Park, K.-S.; Chung, I.-J.; Kim, H.-J. Telomere length shortening in non-Hodgkin’s lymphoma patients undergoing chemotherapy. Ann. Hematol. 2003, 82, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, M.; Ozkaynak, M.F.; Drullinsky, P.; Sandoval, C.; Tugal, O.; Jayabose, S.; Moore, M. Telomerase activity and telomere length in pediatric patients with malignancies undergoing chemotherapy. Leuk 1998, 12, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Hattangadi, S.M.; Wong, P.; Zhang, L.; Flygare, J.; Lodish, H.F. From stem cell to red cell: Regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood 2011, 118, 6258–6268. [Google Scholar] [CrossRef] [Green Version]
- Kondo, M. Lymphoid and myeloid lineage commitment in multipotent hematopoietic progenitors. Immunol. Rev. 2010, 238, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Kollman, C.; Howe, C.W.S.; Anasetti, C.; Antin, J.H.; Davies, S.M.; Filipovich, A.H.; Hegland, J.; Kamani, N.; Kernan, N.A.; King, R.; et al. Donor characteristics as risk factors in recipients after transplantation of bone marrow from unrelated donors: The effect of donor age. Blood 2001, 98, 2043–2051. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Jamieson, C.H.M.; Weissman, I.L. Stems Cells and the Pathways to Aging and Cancer. Cell 2008, 132, 681–696. [Google Scholar] [CrossRef] [Green Version]
- Flach, J.; Bakker, S.T.; Mohrin, M.; Conroy, P.C.; Pietras, E.M.; Reynaud, D.; Alvarez, S.; Diolaiti, M.E.; Ugarte, F.; Forsberg, E.C.; et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nat. Cell Biol. 2014, 512, 198–202. [Google Scholar] [CrossRef] [Green Version]
- Cupit-Link, M.C.; Arora, M.; Wood, W.A.; Hashmi, S.K. Relationship between Aging and Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2018, 24, 1965–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, C.A.; Krishnan, K. Bone Marrow Failure; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Beauséjour, C. Bone marrow-derived cells: The influence of aging and cellular senescence. Handbook Exp. Pharmacol. 2007, 180, 67–88. [Google Scholar]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. From The Cover: Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [Green Version]
- MacCormick, R.E. Possible acceleration of aging by adjuvant chemotherapy: A cause of early onset frailty? Med. Hypotheses 2006, 67, 212–215. [Google Scholar] [CrossRef]
- Siddiqui, M.S.; François, M.; Fenech, M.F.; Leifert, W.R. Persistent γH2AX: A promising molecular marker of DNA damage and aging. Mutat. Res. Rev. Mutat. Res. 2015, 766, 1–19. [Google Scholar] [CrossRef]
- Heylmann, D.; Kaina, B. The γH2AX DNA damage assay from a drop of blood. Sci. Rep. 2016, 6, 22682. [Google Scholar] [CrossRef] [Green Version]
- Levine, E.G.; Bloomfield, C.D. Leukemias and myelodysplastic syndromes secondary to drug, radiation, and environmental exposure. Semin. Oncol. 1992, 19, 47–84. [Google Scholar]
- Davies, S.M. Therapy-related leukemia associated with alkylating agents. Med. Pediatr. Oncol. 2001, 36, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Takahashi, A.; Ono, K.; Ohnishi, T. DNA Damage Induced by Alkylating Agents and Repair Pathways. J. Nucleic Acids 2010, 2010, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoval, C.; Pui, C.H.; Bowman, L.C.; Heaton, D.; A Hurwitz, C.; Raimondi, S.C.; Behm, F.G.; Head, D.R. Secondary acute myeloid leukemia in children previously treated with alkylating agents, intercalating topoisomerase II inhibitors, and irradiation. J. Clin. Oncol. 1993, 11, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Pierre, R.V.; Bayrd, E.D. Multiple Myeloma and Acute Myelomonocytic Leukemia. New Engl. J. Med. 1970, 283, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, S.J. DNA damage by drugs and radiation: What is important and how is it measured? Eur. J. Cancer 1992, 28, 273–276. [Google Scholar] [CrossRef]
- Johnson, A.A.; Akman, K.; Calimport, S.R.; Wuttke, D.; Stolzing, A.; De Magalhães, J.P. The Role of DNA Methylation in Aging, Rejuvenation, and Age-Related Disease. Rejuvenation Res. 2012, 15, 483–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyce, J. Drug-induced DNA hypermethylation and drug resistance in human tumors. Cancer Res. 1989, 49, 5829–5836. [Google Scholar]
- Kirkland, J.L.; Tchkonia, T. Clinical strategies and animal models for developing senolytic agents. Exp. Gerontol. 2015, 68, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Hurria, A.; Jones, L.; Muss, H.B. Cancer Treatment as an Accelerated Aging Process: Assessment, Biomarkers, and Interventions. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e516–e522. [Google Scholar] [CrossRef]
- Buendía-Roldan, I.; Fernández-Plata, R.; Valdes-Bartolo, A.; Mejia, M.; Jaramillo, L.E.; Martínez-Briseño, D.; Santiago-Ruiz, A.; Tapia-Aguilar, H.; Gómez-Zamora, B.; Pardo, A.; et al. Determination of the phenotypic age in residents of Mexico City: Effect of accelerated ageing on lung function and structure. ERJ Open Res. 2020, 6. [Google Scholar]
- Korte, S.M.; Koolhaas, J.M.; Wingfield, J.C.; McEwen, B.S. The Darwinian concept of stress: Benefits of allostasis and costs of allostatic load and the trade-offs in health and disease. Neurosci. Biobehav. Rev. 2005, 29, 3–38. [Google Scholar] [CrossRef] [PubMed]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, K.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, S.C. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef]
- Galkin, F.; Mamoshina, P.; Kochetov, K. DeepMAge: A Methylation Aging Clock Developed with Deep Learning. Aging Dis. 2020. [Google Scholar] [CrossRef]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jöckel, K.H.; Erbel, R.; Mühleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef] [Green Version]
- Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Gori, D.; Giuliani, C.; Mari, D.; Di Blasio, A.M.; Gentilini, D.; Vitale, G.; Collino, S.; et al. Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell 2012, 11, 1132–1134. [Google Scholar] [CrossRef] [Green Version]
- Levine, M.E.; Hosgood, H.D.; Chen, B.; Absher, D.; Assimes, T.; Horvath, S.C. DNA methylation age of blood predicts future onset of lung cancer in the women’s health initiative. Aging 2015, 7, 690–700. [Google Scholar] [CrossRef] [Green Version]
- Chung, M.; Ruan, M.; Zhao, N.; Koestler, D.C.; De Vivo, I.; Kelsey, K.T.; Michaud, D.S. DNA methylation ageing clocks and pancreatic cancer risk: Pooled analysis of three prospective nested case-control studies. Epigenetics 2021, 1–11. [Google Scholar] [CrossRef]
- Kresovich, J.K.; Xu, Z.; O’Brien, K.M.; Weinberg, C.R.; Sandler, D.P.; Taylor, J.A. Methylation-Based Biological Age and Breast Cancer Risk. J. Natl. Cancer Inst. 2019, 111, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Durso, D.F.; Bacalini, M.G.; Sala, C.; Pirazzini, C.; Marasco, E.; Bonafé, M.; Valle, I.F.D.; Gentilini, D.; Castellani, G.; Faria, A.M.C.; et al. Acceleration of leukocytes’ epigenetic age as an early tumor and sex-specific marker of breast and colorectal cancer. Oncotarget 2017, 8, 23237–23245. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Joyce, B.T.; Colicino, E.; Liu, L.; Zhang, W.; Dai, Q.; Shrubsole, M.J.; Kibbe, W.A.; Gao, T.; Zhang, Z.; et al. Blood Epigenetic Age may Predict Cancer Incidence and Mortality. EBioMedicine 2016, 5, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Maden, S.K.; Luebeck, G.E.; Li, C.I.; Newcomb, P.A.; Ulrich, C.M.; Joo, J.-H.E.; Buchanan, D.D.; Milne, R.L.; Southey, M.C.; et al. Dysfunctional epigenetic aging of the normal colon and colorectal cancer risk. Clin. Epigenetics 2020, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sehl, M.E.; Carroll, J.E.; Horvath, S.C.; Bower, J.E. The acute effects of adjuvant radiation and chemotherapy on peripheral blood epigenetic age in early stage breast cancer patients. npj Breast Cancer 2020, 6, 23. [Google Scholar] [CrossRef]
- Tanaka, T.; Biancotto, A.; Moaddel, R.; Moore, A.Z.; Gonzalez-Freire, M.; Aon, M.A.; Candia, J.; Zhang, P.; Cheung, F.; Fantoni, G.; et al. Plasma proteomic signature of age in healthy humans. Aging Cell 2018, 17, e12799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaghlool, S.B.; Kühnel, B.; Elhadad, M.A.; Kader, S.; Halama, A.; Thareja, G.; Engelke, R.; Sarwath, H.; Al-Dous, E.K.; Mohamoud, Y.A.; et al. Epigenetics meets proteomics in an epigenome-wide association study with circulating blood plasma protein traits. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.B.; Maranville, J.C.; Peters, J.E.; Stacey, D.; Staley, J.R.; Blackshaw, J.; Burgess, S.; Jiang, T.; Paige, E.; Surendran, P.; et al. Genomic atlas of the human plasma proteome. Nat. Cell Biol. 2018, 558, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.A.; Shokhirev, M.N.; Wyss-Coray, T.; Lehallier, B. Systematic review and analysis of human proteomics aging studies unveils a novel proteomic aging clock and identifies key processes that change with age. Aging Res. Rev. 2020, 60, 101070. [Google Scholar] [CrossRef] [PubMed]
- Sathyan, S.; Ayers, E.; Gao, T.; Weiss, E.F.; Milman, S.; Verghese, J.; Barzilai, N. Plasma proteomic profile of age, health span, and all-cause mortality in older adults. Aging Cell 2020, 19, e13250. [Google Scholar] [CrossRef] [PubMed]
- Lehallier, B.; Gate, D.; Schaum, N.; Nanasi, T.; Lee, S.E.; Yousef, H.; Losada, P.M.; Berdnik, D.; Keller, A.; Verghese, J.; et al. Undulating changes in human plasma proteome profiles across the lifespan. Nat. Med. 2019, 25, 1843–1850. [Google Scholar] [CrossRef]
- Solovev, I.A.; Shaposhnikov, M.V.; Moskalev, A. An Overview of the Molecular and Cellular Biomarkers of Aging. In Healthy Ageing and Longevity; Springer Nature: Berlin/Heidelberg, Germany, 2019; pp. 67–78. [Google Scholar]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Halvorsen, T.L.; Beattie, G.M.; Lopez, A.D.; Hayek, A.; Levine, F. Accelerated telomere shortening and senescence in human pancreatic islet cells stimulated to divide in vitro. J. Endocrinol. 2000, 166, 103–109. [Google Scholar] [CrossRef]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 13742–13747. [Google Scholar] [CrossRef] [Green Version]
- Kamijo, T.; Zindy, F.; Roussel, M.F.; Quelle, D.E.; Downing, J.R.; Ashmun, R.A.; Grosveld, G.; Sherr, C.J. Tumor Suppression at the Mouse INK4a Locus Mediated by the Alternative Reading Frame Product p19 ARF. Cell 1997, 91, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Kiyono, T.; Foster, S.A.; Koop, J.I.; McDougall, J.K.; Galloway, D.A.; Klingelhutz, A.J. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nat. Cell Biol. 1998, 396, 84–88. [Google Scholar] [CrossRef]
- Park, I.-K.; Qian, D.; Kiel, M.; Becker, M.W.; Pihalja, M.; Weissman, I.L.; Morrison, S.J.; Clarke, M.F. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nat. Cell Biol. 2003, 423, 302–305. [Google Scholar] [CrossRef]
- Janzen, V.; Forkert, R.; Fleming, H.E.; Saito, Y.; Waring, M.T.; Dombkowski, D.M.; Cheng, T.; Depinho, R.A.; Sharpless, N.E.; Scadden, D.T. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nat. Cell Biol. 2006, 443, 421–426. [Google Scholar] [CrossRef]
- Molofsky, A.V.; Pardal, R.; Iwashita, T.; Park, I.-K.; Clarke, M.F.; Morrison, S.J. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nat. Cell Biol. 2003, 425, 962–967. [Google Scholar] [CrossRef] [Green Version]
- Lessard, J.; Sauvageau, G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nat. Cell Biol. 2003, 423, 255–260. [Google Scholar] [CrossRef]
- Liu, Y.; Sanoff, H.K.; Cho, H.; Burd, C.E.; Torrice, C.; Ibrahim, J.G.; Thomas, N.E.; Sharpless, N.E. Expression ofp16INK4ain peripheral blood T-cells is a biomarker of human aging. Aging Cell 2009, 8, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef]
- Edwards, M.G.; Anderson, R.M.; Yuan, M.; Kendziorski, C.; Weindruch, R.; Prolla, T.A. Gene expression profiling of aging reveals activation of a p53-mediated transcriptional program. BMC Genom. 2007, 8, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbig, U.; Ferreira, M.; Condel, L.; Carey, D.; Sedivy, J.M. Cellular Senescence in Aging Primates. Science 2006, 311, 1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melk, A.; Schmidt, B.M.; Takeuchi, O.; Sawitzki, B.; Rayner, D.C.; Halloran, P.F. Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int. 2004, 65, 510–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Signer, R.A.; Montecino-Rodriguez, E.; Witte, O.N.; Dorshkind, K. Aging and cancer resistance in lymphoid progenitors are linked processes conferred by p16Ink4a and Arf. Genes Dev. 2008, 22, 3115–3120. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, G.P.; O Stemmer-Rachamimov, A.; Shaw, J.; E Roy, J.; Koh, J.; Louis, D.N. Immunohistochemical survey of p16INK4A expression in normal human adult and infant tissues. Lab. Investig. 1999, 79, 1137–1143. [Google Scholar]
- Zindy, F.; E Quelle, D.; Roussel, M.F.; Sherr, C.J. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 1997, 15, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Sanoff, H.K.; Deal, A.M.; Krishnamurthy, J.; Torrice, C.; Dillon, P.; Sorrentino, J.; Ibrahim, J.G.; Jolly, T.A.; Williams, G.; Carey, L.A.; et al. Effect of Cytotoxic Chemotherapy on Markers of Molecular Age in Patients With Breast Cancer. J. Natl. Cancer Inst. 2014, 106, dju057. [Google Scholar] [CrossRef] [Green Version]
- Bourlon, M.T.; Velazquez, H.E.; Hinojosa, J.; Orozco, L.; Rios-Corzo, R.; Lima, G.; Llorente, L.; Hernandez-Ramirez, D.F.; Valentin-Cortez, F.J.; Medina-Rangel, I.; et al. Immunosenescence profile and expression of the aging biomarker (p16INK4a) in testicular cancer survivors treated with chemotherapy. BMC Cancer 2020, 20, 1–7. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Vital signs: Colorectal cancer screening, incidence, and mortality—United States, 2002–2010. In Morbidity and Mortality Weekly Report; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2011; pp. 884–890. [Google Scholar]
- Shen, J.; Song, R.; Fuemmeler, B.F.; McGuire, K.P.; Chow, W.-H.; Zhao, H. Biological Aging Marker p16INK4a in T Cells and Breast Cancer Risk. Cancers 2020, 12, 3122. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.; Han, S.Y.; Song, J. Regulatory Network of ARF in Cancer Development. Mol. Cells 2018, 41, 381–389. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Prizment, A.; Thyagarajan, B.; Blaes, A. Cancer Treatment-Induced Accelerated Aging in Cancer Survivors: Biology and Assessment. Cancers 2021, 13, 427. https://doi.org/10.3390/cancers13030427
Wang S, Prizment A, Thyagarajan B, Blaes A. Cancer Treatment-Induced Accelerated Aging in Cancer Survivors: Biology and Assessment. Cancers. 2021; 13(3):427. https://doi.org/10.3390/cancers13030427
Chicago/Turabian StyleWang, Shuo, Anna Prizment, Bharat Thyagarajan, and Anne Blaes. 2021. "Cancer Treatment-Induced Accelerated Aging in Cancer Survivors: Biology and Assessment" Cancers 13, no. 3: 427. https://doi.org/10.3390/cancers13030427
APA StyleWang, S., Prizment, A., Thyagarajan, B., & Blaes, A. (2021). Cancer Treatment-Induced Accelerated Aging in Cancer Survivors: Biology and Assessment. Cancers, 13(3), 427. https://doi.org/10.3390/cancers13030427