
Retrospective Validation of a 168-Gene Expression Signature for Glioma Classification on a Single Molecule Counting Platform

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
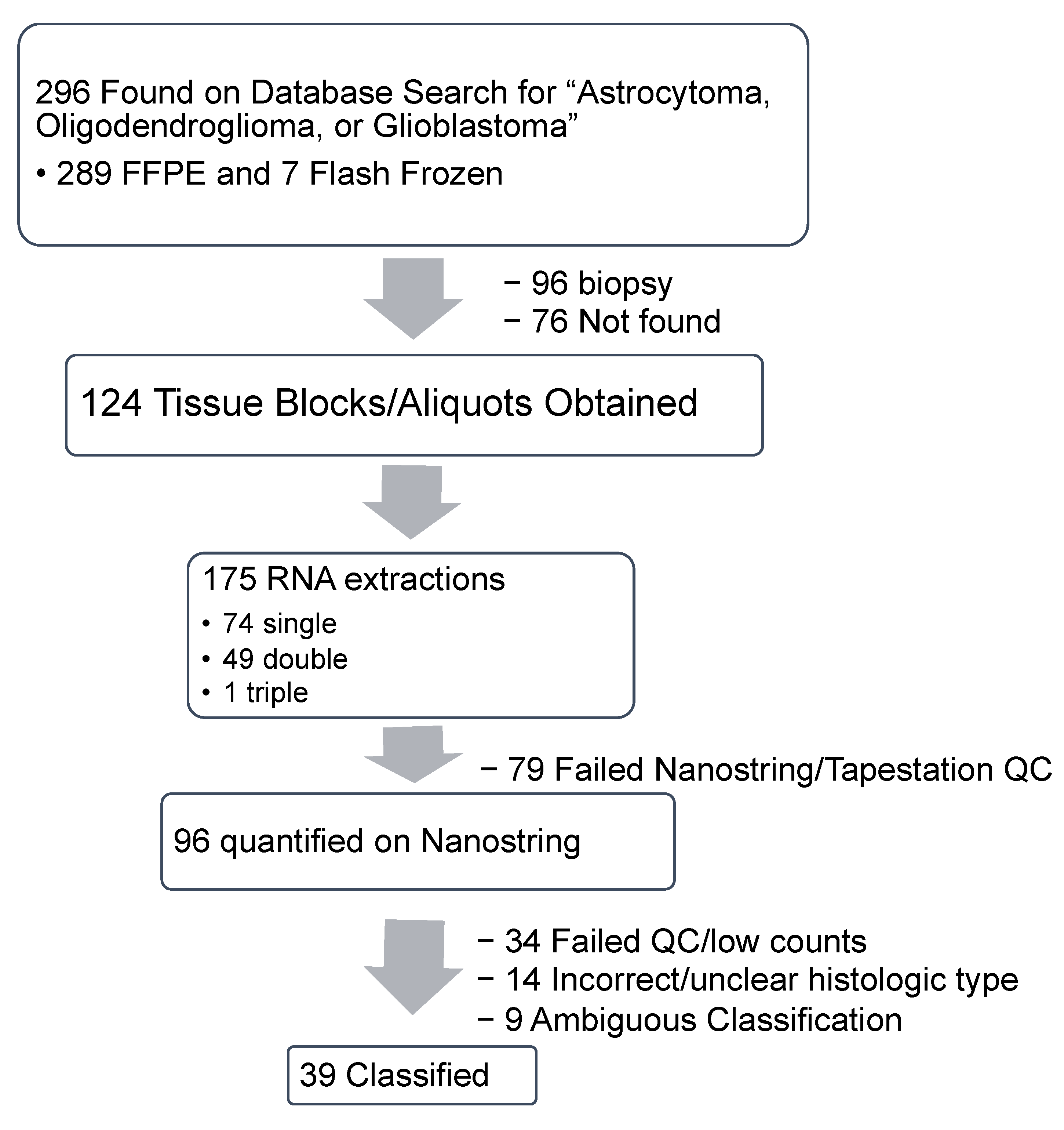
2.1. Specimen Acquisition, RNA Isolation and Quantification
2.1.1. Specimen Acquisition
2.1.2. Demographics
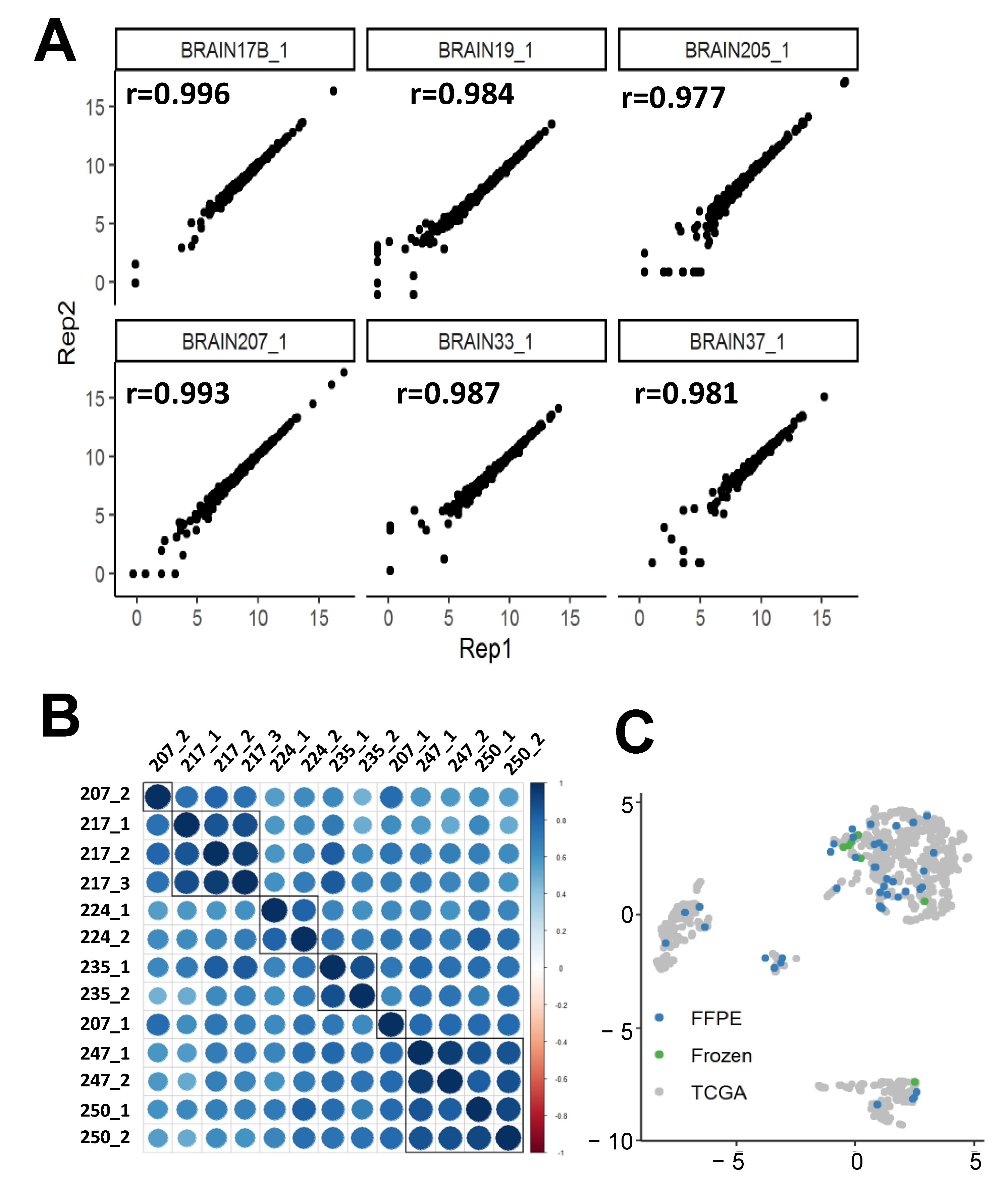
2.1.3. nCounter replicability
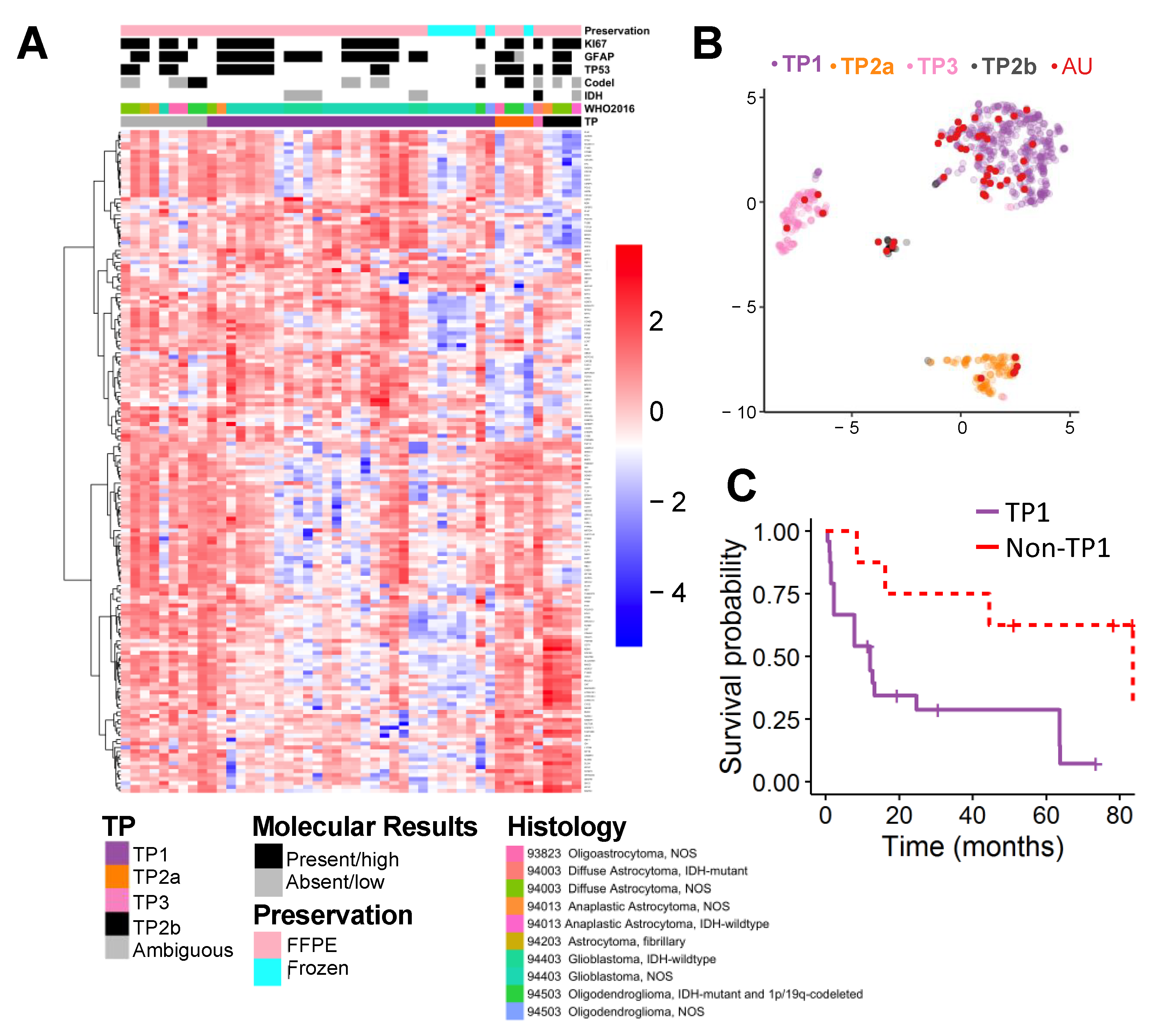
2.2. Supervised Classification
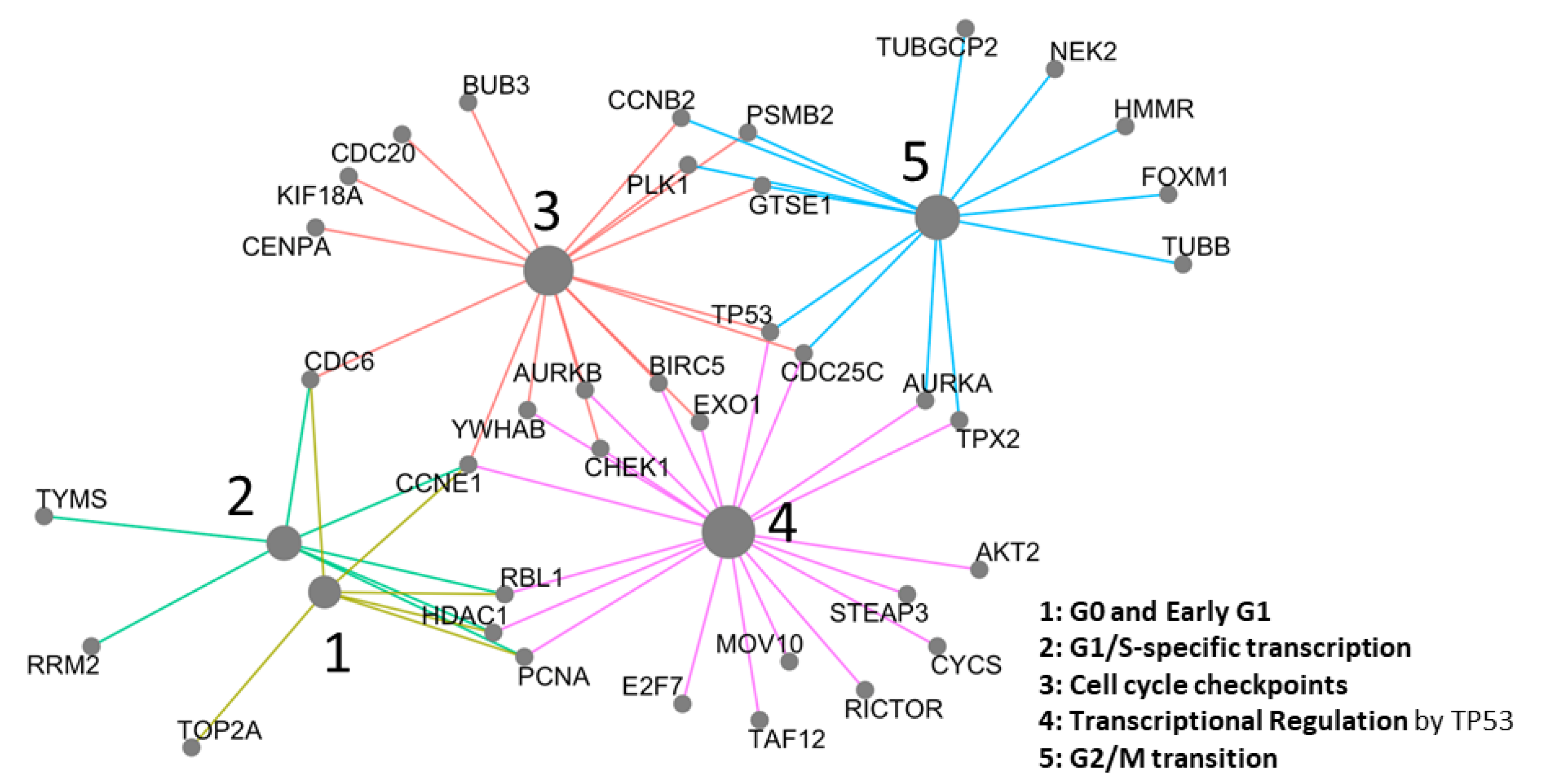
2.3. Network Analysis
3. Discussion
4. Materials and Methods
4.1. Augusta University Sample Collection and Processing
4.1.1. Study Participants
4.1.2. Specimen Characteristics and Assay Methods
4.1.3. Pipeline for FFPE Tissue RNA Extraction
4.2. Statistical Methods
4.2.1. Gene Expression Quantification
4.2.2. TCGA Dataset
4.2.3. Ensemble Transcriptomic Classification (ETC) Algorithm
4.2.4. Survival Analysis
4.2.5. Network Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- van den Bent, M.J. Practice changing mature results of RTOG study 9802: Another positive PCV trial makes adjuvant chemotherapy part of standard of care in low-grade glioma. Neuro-Oncology 2014, 16, 1570–1574. [Google Scholar] [CrossRef] [Green Version]
- van den Bent, M.J.; Brandes, A.A.; Taphoorn, M.J.; Kros, J.M.; Kouwenhoven, M.C.; Delattre, J.-Y.; Bernsen, H.J.; Frenay, M.; Tijssen, C.C.; Grisold, W. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: Long-term follow-up of EORTC brain tumor group study 26951. J. Clin. Oncol. 2012, 31, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, D.R.; Gaspar, L.E.; Cairncross, J.G. Successful chemotherapy for newly diagnosed aggressive oligodendroglioma. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1990, 27, 573–574. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K. WHO Classification of Tumours of the Central Nervous System, Revised 4th ed.; World Health Organization: Geneva, Switzerland, 2016; Volume 1, p. 408. [Google Scholar]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [CrossRef] [PubMed] [Green Version]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef] [Green Version]
- Ellison, D.W. Multiple Molecular Data Sets and the Classification of Adult Diffuse Gliomas. N. Engl. J. Med. 2015, 372, 2555–2557. [Google Scholar] [CrossRef]
- van den Bent, M.J. Interobserver variation of the histopathological diagnosis in clinical trials on glioma: A clinician’s perspective. Acta Neuropathol. 2010, 120, 297–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, P.M.H.; Tran, L.K.H.; Nechtman, J.; dos Santos, B.; Purohit, S.; Satter, K.B.; Dun, B.; Kolhe, R.; Sharma, S.; Bollag, R.; et al. Comparative analysis of transcriptomic profile, histology, and IDH mutation for classification of gliomas. Sci. Rep. 2020, 10, e20651. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Gusev, Y.; Bhuvaneshwar, K.; Song, L.; Zenklusen, J.-C.; Fine, H.; Madhavan, S. The REMBRANDT study, a large collection of genomic data from brain cancer patients. Sci. Data 2018, 5, e180158. [Google Scholar] [CrossRef]
- Geiss, G.K.; Bumgarner, R.E.; Birditt, B.; Dahl, T.; Dowidar, N.; Dunaway, D.L.; Fell, H.P.; Ferree, S.; George, R.D.; Grogan, T.; et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. 2008, 26, 317–325. [Google Scholar] [CrossRef]
- Laenkholm, A.V.; Jensen, M.B.; Eriksen, J.O.; Rasmussen, B.B.; Knoop, A.S.; Buckingham, W.; Ferree, S.; Schaper, C.; Nielsen, T.O.; Haffner, T.; et al. PAM50 Risk of Recurrence Score Predicts 10-Year Distant Recurrence in a Comprehensive Danish Cohort of Postmenopausal Women Allocated to 5 Years of Endocrine Therapy for Hormone Receptor-Positive Early Breast Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 735–740. [Google Scholar] [CrossRef]
- Ng, S.W.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016, 540, 433–437. [Google Scholar] [CrossRef]
- Reis, P.P.; Waldron, L.; Goswami, R.S.; Xu, W.; Xuan, Y.; Perez-Ordonez, B.; Gullane, P.; Irish, J.; Jurisica, I.; Kamel-Reid, S. mRNA transcript quantification in archival samples using multiplexed, color-coded probes. BMC Biotechnol. 2011, 11, e46. [Google Scholar] [CrossRef] [Green Version]
- Pesenacker, A.M.; Chen, V.; Gillies, J.; Speake, C.; Marwaha, A.K.; Sun, A.; Chow, S.; Tan, R.; Elliott, T.; Dutz, J.P.; et al. Treg gene signatures predict and measure type 1 diabetes trajectory. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Wallden, B.; Storhoff, J.; Nielsen, T.; Dowidar, N.; Schaper, C.; Ferree, S.; Liu, S.; Leung, S.; Geiss, G.; Snider, J.; et al. Development and verification of the PAM50-based Prosigna breast cancer gene signature assay. BMC Med. Genom. 2015, 8, e54. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro Stucklin, A.S.; Ryall, S.; Fukuoka, K.; Zapotocky, M.; Lassaletta, A.; Li, C.; Bridge, T.; Kim, B.; Arnoldo, A.; Kowalski, P.E.; et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat. Commun. 2019, 10, e4343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryall, S.; Arnoldo, A.; Krishnatry, R.; Mistry, M.; Khor, K.; Sheth, J.; Ling, C.; Leung, S.; Zapotocky, M.; Guerreiro Stucklin, A.; et al. Multiplex Detection of Pediatric Low-Grade Glioma Signature Fusion Transcripts and Duplications Using the NanoString nCounter System. J. Neuropathol. Exp. Neurol. 2017, 76, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Willemsma, K.; Yip, W.; LeVasseur, N.; Dobosz, K.; Illmann, C.; Baxter, S.; Lohrisch, C.; Simmons, C.E. Impact of Recurrence Score on type and duration of chemotherapy in breast cancer. Curr. Oncol. 2020, 27, e86–e92. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Tang, G.; Brauer, H.A.; Goerlitz, D.S.; Lucas, P.C.; Robidoux, A.; Harris, B.T.; Bandos, H.; Ren, Y.; Geyer, C.E., Jr.; et al. NSABP B-41, a Randomized Neoadjuvant Trial: Genes and Signatures Associated with Pathologic Complete Response. Clin. Cancer Res. 2020, 26, 4233–4241. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Shih, D.J.H.; Remke, M.; Cho, Y.-J.; Kool, M.; Hawkins, C.; Eberhart, C.G.; Dubuc, A.; Guettouche, T.; Cardentey, Y.; et al. Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol. 2012, 123, 615–626. [Google Scholar] [CrossRef] [Green Version]
- Pescarmona, R.; Belot, A.; Villard, M.; Besson, L.; Lopez, J.; Mosnier, I.; Mathieu, A.-L.; Lombard, C.; Garnier, L.; Frachette, C.; et al. Comparison of RT-qPCR and Nanostring in the measurement of blood interferon response for the diagnosis of type I interferonopathies. Cytokine 2019, 113, 446–452. [Google Scholar] [CrossRef]
- Kashofer, K.; Viertler, C.; Pichler, M.; Zatloukal, K. Quality Control of RNA Preservation and Extraction from Paraffin-Embedded Tissue: Implications for RT-PCR and Microarray Analysis. PLoS ONE 2013, 8, e70714. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Mooney, J.H.; Ilyas, A.; Chagoya, G.; Estevez-Ordonez, D.; Ibrahim, A.; Nakano, I. Molecular and cellular intratumoral heterogeneity in primary glioblastoma: Clinical and translational implications. J. Neurosurg. 2019, 133, 655–663. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Castro-Gamero, A.M.; Pezuk, J.A.; Brassesco, M.S.; Tone, L.G. G2/M inhibitors as pharmacotherapeutic opportunities for glioblastoma: The old, the new, and the future. Cancer Biol. Med. 2018, 15, 354–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsyth, P.; Cairncross, G.; Stewart, D.; Goodyear, M.; Wainman, N.; Eisenhauer, E. Phase II trial of docetaxel in patients with recurrent malignant glioma: A study of the National Cancer Institute of Canada Clinical Trials Group. Investig. New Drugs 1996, 14, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Sanson, M.; Napolitano, M.; Yaya, R.; Keime-Guibert, F.; Broët, P.; Hoang-Xuan, K.; Delattre, J.Y. Second line chemotherapy with docetaxel in patients with recurrent malignant glioma: A phase II study. J. Neuro-Oncol. 2000, 50, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Fogh, S.; Machtay, M.; Werner-Wasik, M.; Curran, W.J., Jr.; Bonanni, R.; Axelrod, R.; Andrews, D.; Dicker, A.P. Phase I trial using patupilone (epothilone B) and concurrent radiotherapy for central nervous system malignancies. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Peereboom, D.M.; Supko, J.G.; Carson, K.A.; Batchelor, T.; Phuphanich, S.; Lesser, G.; Mikkelsen, T.; Fisher, J.; Desideri, S.; He, X.; et al. A phase I/II trial and pharmacokinetic study of ixabepilone in adult patients with recurrent high-grade gliomas. J. Neuro-Oncol. 2010, 100, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Tosoni, A.; Bromberg, J.E.C.; Hau, P.; Campone, M.; Gijtenbeek, J.; Frenay, M.; Breimer, L.; Wiesinger, H.; Allgeier, A.; et al. Sagopilone (ZK-EPO, ZK 219477) for recurrent glioblastoma. A phase II multicenter trial by the European Organisation for Research and Treatment of Cancer (EORTC) Brain Tumor Group. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2011, 22, 2144–2149. [Google Scholar] [CrossRef]
- Pezuk, J.A.; Brassesco, M.S.; Morales, A.G.; de Oliveira, J.C.; de Paula Queiroz, R.G.; Machado, H.R.; Carlotti, C.G., Jr.; Neder, L.; Scrideli, C.A.; Tone, L.G. Polo-like kinase 1 inhibition causes decreased proliferation by cell cycle arrest, leading to cell death in glioblastoma. Cancer Gene 2013, 20, 499–506. [Google Scholar] [CrossRef] [Green Version]
- Danovi, D.; Folarin, A.; Gogolok, S.; Ender, C.; Elbatsh, A.M.O.; Engström, P.G.; Stricker, S.H.; Gagrica, S.; Georgian, A.; Yu, D.; et al. A high-content small molecule screen identifies sensitivity of glioblastoma stem cells to inhibition of polo-like kinase 1. PLoS ONE 2013, 8, e77053. [Google Scholar] [CrossRef]
- Raub, T.J.; Wishart, G.N.; Kulanthaivel, P.; Staton, B.A.; Ajamie, R.T.; Sawada, G.A.; Gelbert, L.M.; Shannon, H.E.; Sanchez-Martinez, C.; De Dios, A. Brain Exposure of Two Selective Dual CDK4 and CDK6 Inhibitors and the Antitumor Activity of CDK4 and CDK6 Inhibition in Combination with Temozolomide in an Intracranial Glioblastoma Xenograft. Drug Metab. Dispos. 2015, 43, 1360–1371. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [Green Version]
- Hashizume, R.; Zhang, A.; Mueller, S.; Prados, M.D.; Lulla, R.R.; Goldman, S.; Saratsis, A.M.; Mazar, A.P.; Stegh, A.H.; Cheng, S.-Y.; et al. Inhibition of DNA damage repair by the CDK4/6 inhibitor palbociclib delays irradiated intracranial atypical teratoid rhabdoid tumor and glioblastoma xenograft regrowth. Neuro-Oncology 2016, 18, 1519–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, S.; Madani, D.; Joshi, S.; Chung, S.A.; Johns, T.; Day, B.; Khasraw, M.; McDonald, K.L. Combination of palbociclib and radiotherapy for glioblastoma. Cell Death Discov. 2017, 3, 17033. [Google Scholar] [CrossRef] [PubMed]
- Veldman-Jones, M.H.; Brant, R.; Rooney, C.; Geh, C.; Emery, H.; Harbron, C.G.; Wappett, M.; Sharpe, A.; Dymond, M.; Barrett, J.C.; et al. Evaluating Robustness and Sensitivity of the NanoString Technologies nCounter Platform to Enable Multiplexed Gene Expression Analysis of Clinical Samples. Cancer Res. 2015, 75, e2587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Park, K.; Lee, E.; Ahn, T.; Jung, H.H.; Lim, S.H.; Hong, M.; Do, I.-G.; Cho, E.Y.; Kim, D.-H.; et al. Gene Expression Profiling of Breast Cancer Brain Metastasis. Sci. Rep. 2016, 6, e28623. [Google Scholar] [CrossRef] [Green Version]
- Kassab, C.; Zamler, D.; Gupta, P.; Srinivasan, V.; Rao, G.; Prabhu, S.; Huse, J.; Fuller, G.; Bruner, J.; Langford, L.; et al. TMIC-60. COMPREHENSIVE SPATIAL CHARACTERIZATION OF IMMUNE CELLS IN THE CNS BRAIN TUMOR MICROENVIRONMENT. Neuro-Oncology 2019, 21, vi261. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. 2019. Available online: https://www.R-project.org/ (accessed on 11 November 2020).
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2006, 8, 118–127. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Yu, G.; He, Q.Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Variable | Number of Subjects |
|---|---|
| Age (Years, median (range)) | 64 (9–79) |
| Year specimen collected | 2003–2016 |
| Sex | |
| Male | 21 |
| Female | 20 |
| Unknown | 7 |
| Race | |
| White | 34 |
| AA | 7 |
| Unknown | 7 |
| WHO 2016 Classification | |
| Oligoastrocytoma, NOS | 3 |
| Diffuse Astrocytoma, IDH-mutant | 1 |
| Diffuse Astrocytoma, NOS | 5 |
| Anaplastic Astrocytoma, NOS | 3 |
| Anaplastic Astrocytoma, IDH-wildtype | 1 |
| Astrocytoma, fibrillary | 1 |
| Glioblastoma, IDH-wildtype | 6 |
| Glioblastoma, NOS | 21 |
| Oligodendroglioma, IDH-mutant and 1p/19q-codeleted | 5 |
| Oligodendroglioma, NOS | 2 |
| Transcriptome Profile | |
| TP1 | 30 |
| TP2 | 4 |
| TP3 | 1 |
| TP4 | 4 |
| Ambiguous | 9 |
| Preservation | |
| FFPE 1 | 41 |
| Frozen | 7 |
| IDH1 | |
| Mutated | 1 |
| WT | 7 |
| Unknown | 40 |
| 1p/19q Co-deletion | |
| Present | 3 |
| WT | 11 |
| Unknown | 34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, P.M.H.; Tran, L.K.H.; Satter, K.b.; Purohit, S.; Nechtman, J.; Hopkins, D.I.; dos Santos, B.; Bollag, R.; Kolhe, R.; Sharma, S.; et al. Retrospective Validation of a 168-Gene Expression Signature for Glioma Classification on a Single Molecule Counting Platform. Cancers 2021, 13, 439. https://doi.org/10.3390/cancers13030439
Tran PMH, Tran LKH, Satter Kb, Purohit S, Nechtman J, Hopkins DI, dos Santos B, Bollag R, Kolhe R, Sharma S, et al. Retrospective Validation of a 168-Gene Expression Signature for Glioma Classification on a Single Molecule Counting Platform. Cancers. 2021; 13(3):439. https://doi.org/10.3390/cancers13030439
Chicago/Turabian StyleTran, Paul Minh Huy, Lynn Kim Hoang Tran, Khaled bin Satter, Sharad Purohit, John Nechtman, Diane I. Hopkins, Bruno dos Santos, Roni Bollag, Ravindra Kolhe, Suash Sharma, and et al. 2021. "Retrospective Validation of a 168-Gene Expression Signature for Glioma Classification on a Single Molecule Counting Platform" Cancers 13, no. 3: 439. https://doi.org/10.3390/cancers13030439
APA StyleTran, P. M. H., Tran, L. K. H., Satter, K. b., Purohit, S., Nechtman, J., Hopkins, D. I., dos Santos, B., Bollag, R., Kolhe, R., Sharma, S., & She, J. X. (2021). Retrospective Validation of a 168-Gene Expression Signature for Glioma Classification on a Single Molecule Counting Platform. Cancers, 13(3), 439. https://doi.org/10.3390/cancers13030439