
T-Cell Dysfunction as a Limitation of Adoptive Immunotherapy: Current Concepts and Mitigation Strategies



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
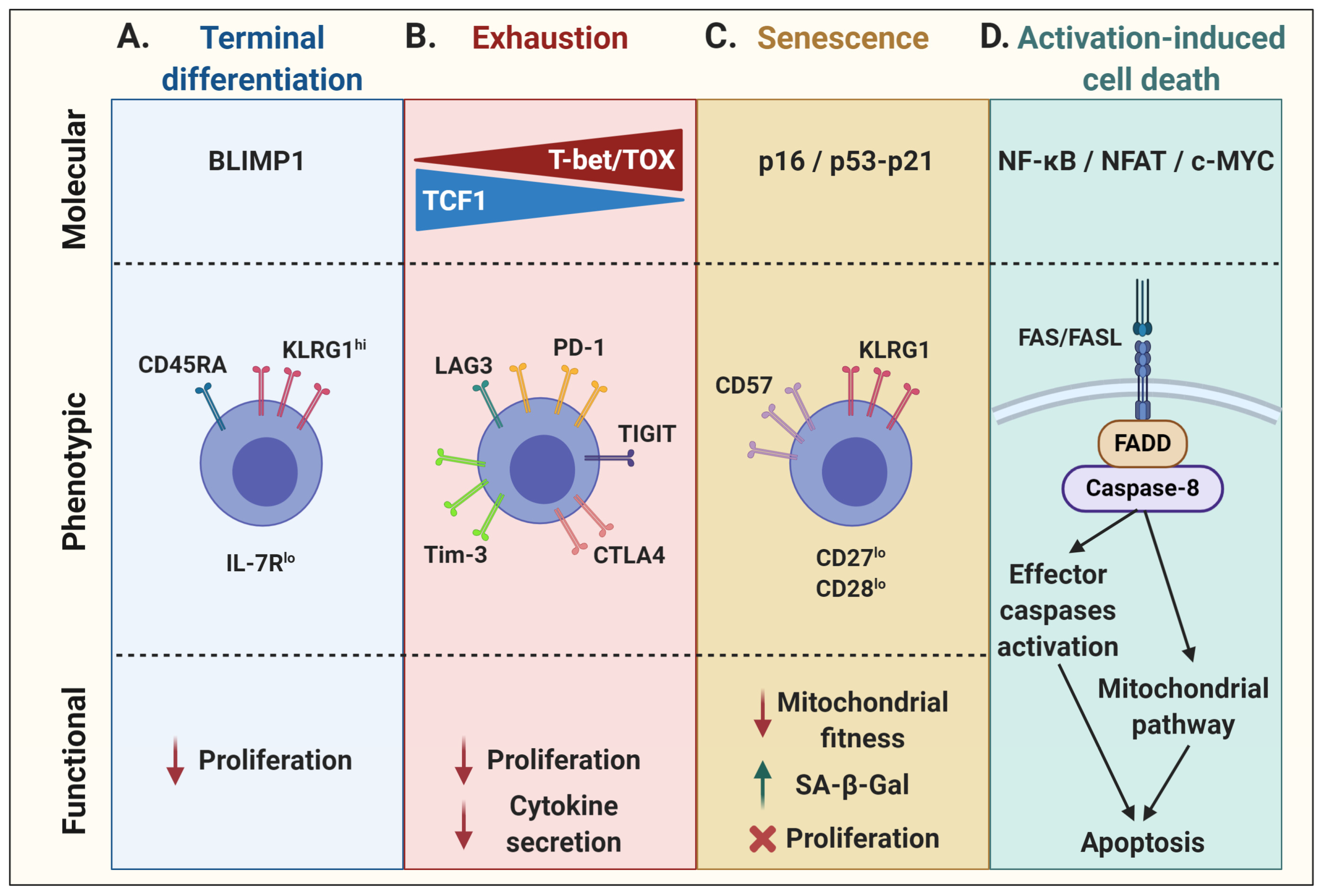
2. T-Cell Dysfunction: Multifaceted Extension of T-Cell Physiology
2.1. Terminal Effector Differentiation
2.2. Exhaustion
2.3. Senescence
2.4. Activation-Induced Cell Death
3. Biology Meets Therapy: T-Cell Dysfunction in Adoptive Cell Therapy
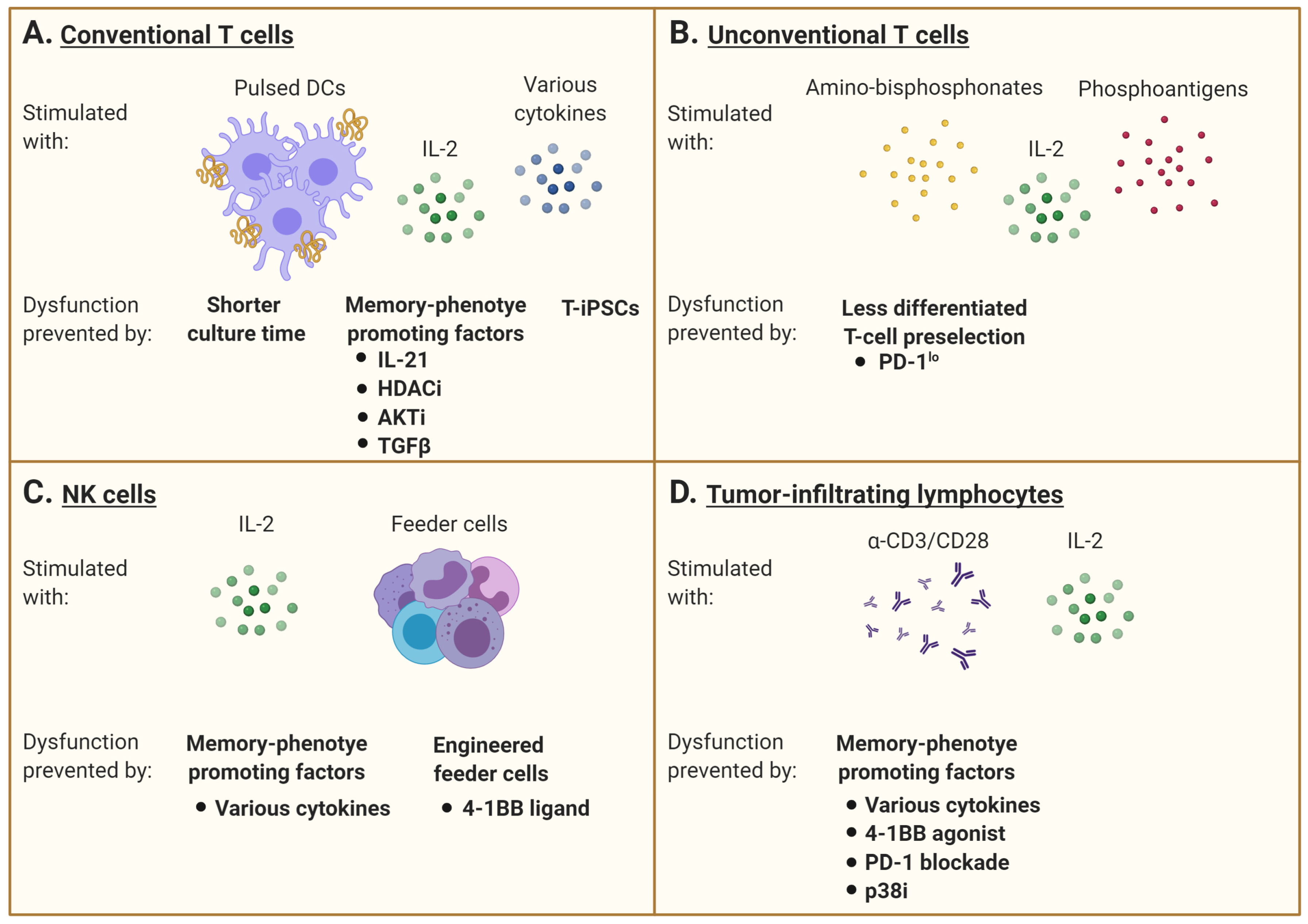
3.1. Conventional T Cells
3.2. Unconventional T Cells
3.3. NK Cells
3.4. Tumor-Infiltrating Lymphocytes
3.5. T-Cell Receptor (TCR) Transgenic Cells
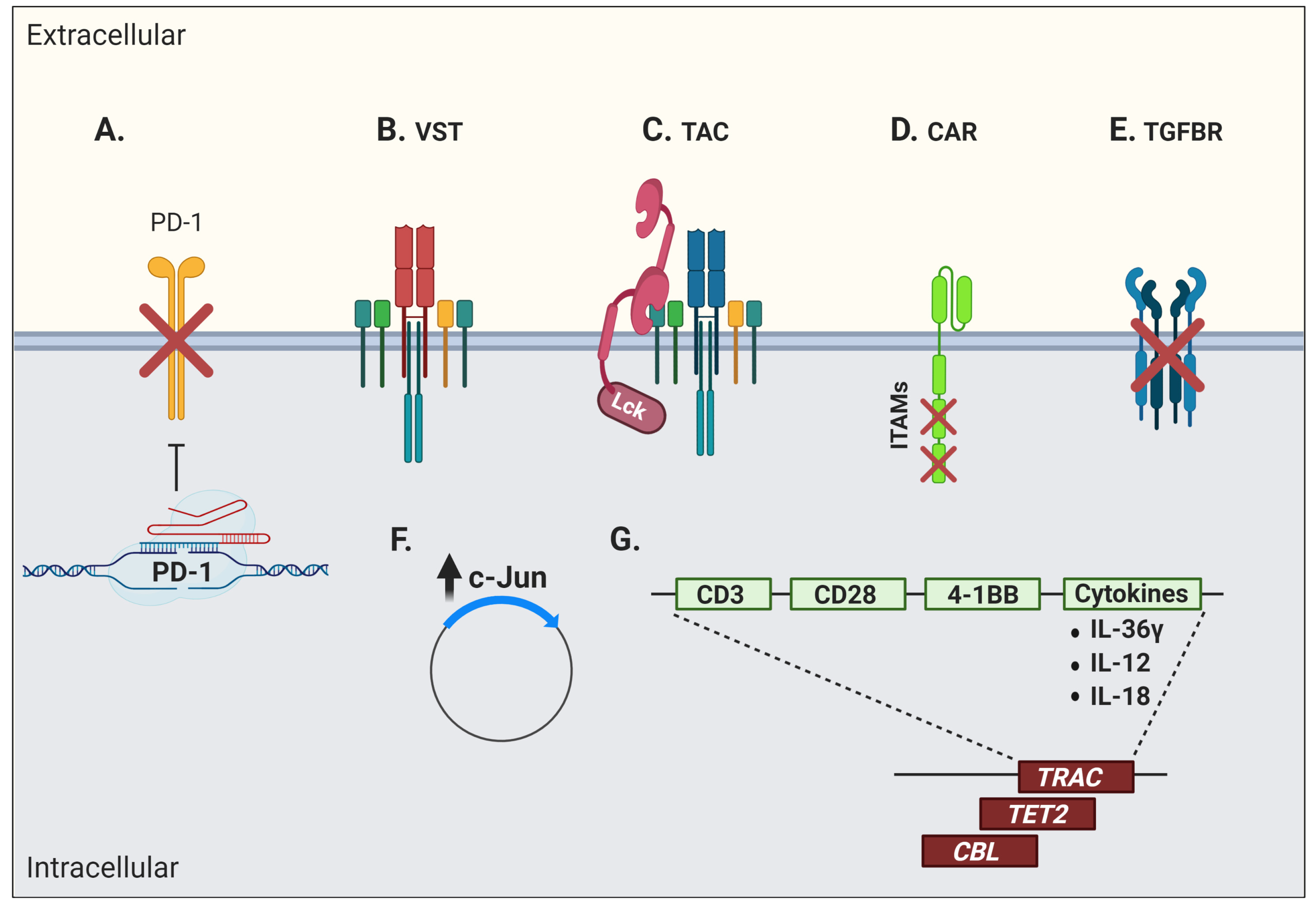
3.6. Chimeric Antigen Receptor (CAR) T Cells
3.7. Other Engineered Cell Types
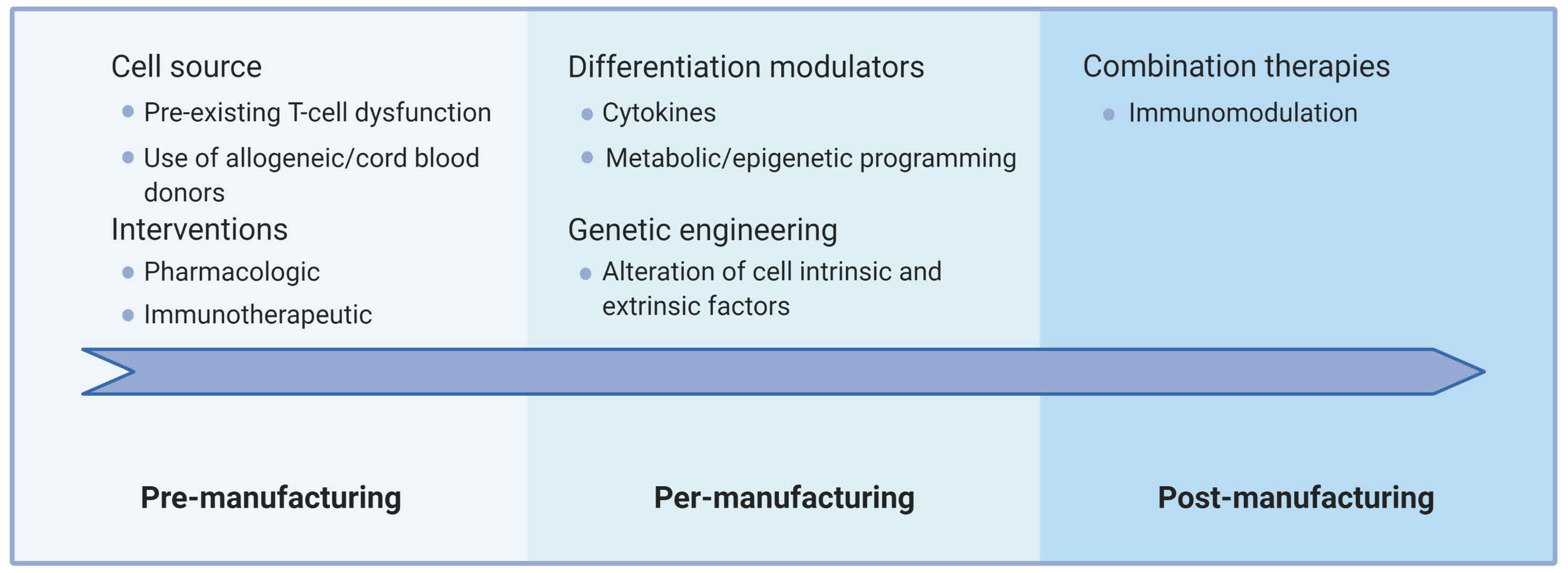
4. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Janelle, V.; Rulleau, C.; Del Testa, S.; Carli, C.; Delisle, J.-S. T-Cell Immunotherapies Targeting Histocompatibility and Tumor Antigens in Hematological Malignancies. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Janelle, V.; Carli, C.; Taillefer, J.; Orio, J.; Delisle, J.-S. Defining novel parameters for the optimal priming and expansion of minor histocompatibility antigen-specific T cells in culture. J. Transl. Med. 2015, 13, 123. [Google Scholar] [CrossRef] [Green Version]
- Gattinoni, L.; Klebanoff, C.A.; Restifo, N.P. Paths to stemness: Building the ultimate antitumour T cell. Nat. Rev. Cancer 2012, 12, 671–684. [Google Scholar] [CrossRef]
- Samelson, L.E.; Patel, M.D.; Weissman, A.M.; Harford, J.B.; Klausner, R.D. Antigen activation of murine T cells induces tyrosine phosphorylation of a polypeptide associated with the T cell antigen receptor. Cell 1986, 46, 1083–1090. [Google Scholar] [CrossRef]
- Bu, J.Y.; Shaw, A.S.; Chan, A.C. Analysis of the interaction of ZAP-70 and syk protein-tyrosine kinases with the T-cell antigen receptor by plasmon resonance. Proc. Natl. Acad. Sci. USA 1995, 92, 5106–5110. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.-R.; Byeon, Y.; Kim, D.; Park, S.-G. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp. Mol. Med. 2020, 52, 750–761. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Schietinger, A.; Greenberg, P.D. Tolerance and exhaustion: Defining mechanisms of T cell dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Hogg, G.D.; DeNardo, D.G. Rethinking immune checkpoint blockade: ‘Beyond the T cell’. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef]
- Dahlén, E.; Veitonmäki, N.; Norlén, P. Bispecific antibodies in cancer immunotherapy. Ther. Adv. Vaccines Immunother. 2018, 6, 3–17. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2015, 37, 457–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Akbar, A.N.; Henson, S.M.; Lanna, A. Senescence of T Lymphocytes: Implications for Enhancing Human Immunity. Trends Immunol. 2016, 37, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Petit, P.F.; Van den Eynde, B.J. Apoptosis of tumor-infiltrating T lymphocytes: A new immune checkpoint mechanism. Cancer Immunol. Immunother. 2019, 68, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.W.; Rutishauser, R.L.; Joshi, N.S.; Haberman, A.M.; Kaech, S.M. Differential localization of effector and memory CD8 T cell subsets in lymphoid organs during acute viral infection. J. Immunol. 2010, 185, 5315–5325. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 2012, 12, 749–761. [Google Scholar] [CrossRef]
- Henson, S.M.; Franzese, O.; Macaulay, R.; Libri, V.; Azevedo, R.I.; Kiani-Alikhan, S.; Plunkett, F.J.; Masters, J.E.; Jackson, S.; Griffiths, S.J.; et al. KLRG1 signaling induces defective Akt (ser473) phosphorylation and proliferative dysfunction of highly differentiated CD8+ T cells. Blood 2009, 113, 6619–6628. [Google Scholar] [CrossRef]
- Martin, M.D.; Badovinac, V.P. Defining Memory CD8 T Cell. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Kaech, S.M.; Tan, J.T.; Wherry, E.J.; Konieczny, B.T.; Surh, C.D.; Ahmed, R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 2003, 4, 1191–1198. [Google Scholar] [CrossRef]
- Sarkar, S.; Kalia, V.; Haining, W.N.; Konieczny, B.T.; Subramaniam, S.; Ahmed, R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J. Exp. Med. 2008, 205, 625–640. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.C.; Matthews, S.; Yap, G.S. IL-12 Signaling Drives CD8+ T Cell IFN-γ Production and Differentiation of KLRG1+ Effector Subpopulations during Toxoplasma gondii Infection. J. Immunol. 2008, 180, 5935–5945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, D.T.; Manders, P.; Wagner, S.D. Arrested Differentiation, the Self-Renewing Memory Lymphocyte, and Vaccination. Science 2001, 293, 248. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Gattinoni, L.; Restifo, N.P. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol. Rev. 2006, 211, 214–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanzavecchia, A.; Sallusto, F. Progressive differentiation and selection of the fittest in the immune response. Nat. Rev. Immunol. 2002, 2, 982–987. [Google Scholar] [CrossRef] [PubMed]
- Rutishauser, R.L.; Martins, G.A.; Kalachikov, S.; Chandele, A.; Parish, I.A.; Meffre, E.; Jacob, J.; Calame, K.; Kaech, S.M. Transcriptional Repressor Blimp-1 Promotes CD8+ T Cell Terminal Differentiation and Represses the Acquisition of Central Memory T Cell Properties. Immunity 2009, 31, 296–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukumar, M.; Kishton, R.J.; Restifo, N.P. Metabolic reprograming of anti-tumor immunity. Curr. Opin. Immunol. 2017, 46, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltra, J.C.; Manne, S.; Abdel-Hakeem, M.S.; Kurachi, M.; Giles, J.R.; Chen, Z.; Casella, V.; Ngiow, S.F.; Khan, O.; Huang, Y.J.; et al. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 2020, 52, 825–841.e828. [Google Scholar] [CrossRef]
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The Transcription Factor NFAT Promotes Exhaustion of Activated CD8+ T Cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Man, K.; Gabriel, S.S.; Liao, Y.; Gloury, R.; Preston, S.; Henstridge, D.C.; Pellegrini, M.; Zehn, D.; Berberich-Siebelt, F.; Febbraio, M.A.; et al. Transcription Factor IRF4 Promotes CD8(+) T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 2017, 47, 1129–1141.e1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennifer, P.C.; Rita, B.E. T Cell Replicative Senescence in Human Aging. Curr. Pharm. Des. 2013, 19, 1680–1698. [Google Scholar] [CrossRef]
- Akbar, A.N.; Henson, S.M. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat. Rev. Immunol. 2011, 11, 289–295. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Henson, S.M.; Akbar, A.N. KLRG1--more than a marker for T cell senescence. Age 2009, 31, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Larbi, A. Markers of T Cell Senescence in Humans. Int. J. Mol. Sci. 2017, 18, 1742. [Google Scholar] [CrossRef] [Green Version]
- Effros, R.B.; Pawelec, G. Replicative senescence of T cells: Does the Hayflick Limit lead to immune exhaustion? Immunol. Today 1997, 18, 450–454. [Google Scholar] [CrossRef]
- Effros, R.B. Replicative senescence in the immune system: Impact of the Hayflick limit on T-cell function in the elderly. Am. J. Hum. Genet. 1998, 62, 1003–1007. [Google Scholar] [CrossRef] [Green Version]
- Hodes, R.J.; Hathcock, K.S.; Weng, N.P. Telomeres in T and B cells. Nat. Rev. Immunol. 2002, 2, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sanoff, H.K.; Cho, H.; Burd, C.E.; Torrice, C.; Ibrahim, J.G.; Thomas, N.E.; Sharpless, N.E. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell 2009, 8, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Porath, I.; Weinberg, R.A. When cells get stressed: An integrative view of cellular senescence. J. Clin. Investig. 2004, 113, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Shawi, M.; Autexier, C. Telomerase, senescence and ageing. Mech. Ageing Dev. 2008, 129, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Chen, X. Tumor suppression by p53: Making cells senescent. Histol. Histopathol. 2010, 25, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [Green Version]
- Henson, S.M.; Lanna, A.; Riddell, N.E.; Franzese, O.; Macaulay, R.; Griffiths, S.J.; Puleston, D.J.; Watson, A.S.; Simon, A.K.; Tooze, S.A.; et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8⁺ T cells. J. Clin. Investig. 2014, 124, 4004–4016. [Google Scholar] [CrossRef]
- Gurusamy, D.; Henning, A.N.; Yamamoto, T.N.; Yu, Z.; Zacharakis, N.; Krishna, S.; Kishton, R.J.; Vodnala, S.K.; Eidizadeh, A.; Jia, L.; et al. Multi-phenotype CRISPR-Cas9 Screen Identifies p38 Kinase as a Target for Adoptive Immunotherapies. Cancer Cell 2020, 37, 818–833.e819. [Google Scholar] [CrossRef]
- Arakaki, R.; Yamada, A.; Kudo, Y.; Hayashi, Y.; Ishimaru, N. Mechanism of activation-induced cell death of T cells and regulation of FasL expression. Crit. Rev. Immunol. 2014, 34, 301–314. [Google Scholar] [CrossRef]
- Zheng, L.; Fisher, G.; Miller, R.E.; Peschon, J.; Lynch, D.H.; Lenardo, M.J. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature 1995, 377, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.I.; Devadas, S.; Zhang, X.; Zhang, L.; Keegan, A.; Greeneltch, K.; Solomon, J.; Wei, L.; Das, J.; Sun, E.; et al. The role of activation-induced cell death in the differentiation of T-helper-cell subsets. Immunol. Res. 2003, 28, 285–293. [Google Scholar] [CrossRef]
- Itoh, N.; Yonehara, S.; Ishii, A.; Yonehara, M.; Mizushima, S.-I.; Sameshima, M.; Hase, A.; Seto, Y.; Nagata, S. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 1991, 66, 233–243. [Google Scholar] [CrossRef]
- Suda, T.; Takahashi, T.; Golstein, P.; Nagata, S. Molecular cloning and expression of the fas ligand, a novel member of the tumor necrosis factor family. Cell 1993, 75, 1169–1178. [Google Scholar] [CrossRef]
- Karin, M.; Lin, A. NF-kappaB at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Rengarajan, J.; Mittelstadt, P.R.; Mages, H.W.; Gerth, A.J.; Kroczek, R.A.; Ashwell, J.D.; Glimcher, L.H. Sequential involvement of NFAT and Egr transcription factors in FasL regulation. Immunity 2000, 12, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Brunner, T.; Kasibhatla, S.; Pinkoski, M.J.; Frutschi, C.; Yoo, N.J.; Echeverri, F.; Mahboubi, A.; Green, D.R. Expression of Fas ligand in activated T cells is regulated by c-Myc. J. Biol. Chem. 2000, 275, 9767–9772. [Google Scholar] [CrossRef] [Green Version]
- Genestier, L.; Kasibhatla, S.; Brunner, T.; Green, D.R. Transforming growth factor beta1 inhibits Fas ligand expression and subsequent activation-induced cell death in T cells via downregulation of c-Myc. J. Exp. Med. 1999, 189, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Chinnaiyan, A.M.; O’Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of fas and initiates apoptosis. Cell 1995, 81, 505–512. [Google Scholar] [CrossRef] [Green Version]
- Thome, M.; Tschopp, J. TCR-induced NF-kappaB activation: A crucial role for Carma1, Bcl10 and MALT1. Trends Immunol. 2003, 24, 419–424. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Grinberg, M.; Schwarz, M.; Zaltsman, Y.; Eini, T.; Niv, H.; Pietrokovski, S.; Gross, A. Mitochondrial carrier homolog 2 is a target of tBID in cells signaled to die by tumor necrosis factor alpha. Mol. Cell. Biol. 2005, 25, 4579–4590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akashi, K.; Kondo, M.; von Freeden-Jeffry, U.; Murray, R.; Weissman, I.L. Bcl-2 Rescues T Lymphopoiesis in Interleukin-7 Receptor–Deficient Mice. Cell 1997, 89, 1033–1041. [Google Scholar] [CrossRef] [Green Version]
- Maraskovsky, E.; O’Reilly, L.A.; Teepe, M.; Corcoran, L.M.; Peschon, J.J.; Strasser, A. Bcl-2 Can Rescue T Lymphocyte Development in Interleukin-7 Receptor–Deficient Mice but Not in Mutant rag-1−/− Mice. Cell 1997, 89, 1011–1019. [Google Scholar] [CrossRef] [Green Version]
- Bouillet, P.; O’Reilly, L.A. CD95, BIM and T cell homeostasis. Nat. Rev. Immunol. 2009, 9, 514–519. [Google Scholar] [CrossRef]
- Hughes, P.D.; Belz, G.T.; Fortner, K.A.; Budd, R.C.; Strasser, A.; Bouillet, P. Apoptosis Regulators Fas and Bim Cooperate in Shutdown of Chronic Immune Responses and Prevention of Autoimmunity. Immunity 2008, 28, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Craddock, J.; Heslop, H.E. Adoptive cellular therapy with T cells specific for EBV-derived tumor antigens. Update Cancer Ther. 2008, 3, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Houghtelin, A.; Bollard, C.M. Virus-Specific T Cells for the Immunocompromised Patient. Front. Immunol. 2017, 8, 1272. [Google Scholar] [CrossRef] [Green Version]
- Heslop, H.E.; Slobod, K.S.; Pule, M.A.; Hale, G.A.; Rousseau, A.; Smith, C.A.; Bollard, C.M.; Liu, H.; Wu, M.F.; Rochester, R.J.; et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood 2010, 115, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Walter, E.A.; Greenberg, P.D.; Gilbert, M.J.; Finch, R.J.; Watanabe, K.S.; Thomas, E.D.; Riddell, S.R. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N. Engl. J. Med. 1995, 333, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Doubrovina, E.; Oflaz-Sozmen, B.; Prockop, S.E.; Kernan, N.A.; Abramson, S.; Teruya-Feldstein, J.; Hedvat, C.; Chou, J.F.; Heller, G.; Barker, J.N.; et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood 2012, 119, 2644–2656. [Google Scholar] [CrossRef] [PubMed]
- Gerdemann, U.; Keirnan, J.M.; Katari, U.L.; Yanagisawa, R.; Christin, A.S.; Huye, L.E.; Perna, S.K.; Ennamuri, S.; Gottschalk, S.; Brenner, M.K.; et al. Rapidly generated multivirus-specific cytotoxic T lymphocytes for the prophylaxis and treatment of viral infections. Mol. Ther. 2012, 20, 1622–1632. [Google Scholar] [CrossRef] [Green Version]
- Icheva, V.; Kayser, S.; Wolff, D.; Tuve, S.; Kyzirakos, C.; Bethge, W.; Greil, J.; Albert, M.H.; Schwinger, W.; Nathrath, M.; et al. Adoptive transfer of epstein-barr virus (EBV) nuclear antigen 1-specific t cells as treatment for EBV reactivation and lymphoproliferative disorders after allogeneic stem-cell transplantation. J. Clin. Oncol. 2013, 31, 39–48. [Google Scholar] [CrossRef]
- Hanley, P.J.; Melenhorst, J.J.; Nikiforow, S.; Scheinberg, P.; Blaney, J.W.; Demmler-Harrison, G.; Cruz, C.R.; Lam, S.; Krance, R.A.; Leung, K.S.; et al. CMV-specific T cells generated from naïve T cells recognize atypical epitopes and may be protective in vivo. Sci. Transl. Med. 2015, 7, 285ra263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudreau, G.; Carli, C.; Lamarche, C.; Rulleau, C.; Bonnaure, G.; Néron, S.; Delisle, J.-S. Leukoreduction system chambers are a reliable cellular source for the manufacturing of T-cell therapeutics. Transfusion 2019, 59, 1300–1311. [Google Scholar] [CrossRef]
- Chapuis, A.G.; Ragnarsson, G.B.; Nguyen, H.N.; Chaney, C.N.; Pufnock, J.S.; Schmitt, T.M.; Duerkopp, N.; Roberts, I.M.; Pogosov, G.L.; Ho, W.Y.; et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci. Transl. Med. 2013, 5, 174ra127. [Google Scholar] [CrossRef] [Green Version]
- Warren, E.H.; Fujii, N.; Akatsuka, Y.; Chaney, C.N.; Mito, J.K.; Loeb, K.R.; Gooley, T.A.; Brown, M.L.; Koo, K.K.W.; Rosinski, K.V.; et al. Therapy of relapsed leukemia after allogeneic hematopoietic cell transplantation with T cells specific for minor histocompatibility antigens. Blood 2010, 115, 3869–3878. [Google Scholar] [CrossRef] [Green Version]
- Meij, P.; Jedema, I.; van der Hoorn, M.A.; Bongaerts, R.; Cox, L.; Wafelman, A.R.; Marijt, E.W.A.; Willemze, R.; Falkenburg, J.H.F. Generation and administration of HA-1-specific T-cell lines for the treatment of patients with relapsed leukemia after allogeneic stem cell transplantation: A pilot study. Haematologica 2012, 97, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Granados, D.P.; Rodenbrock, A.; Laverdure, J.P.; Côté, C.; Caron-Lizotte, O.; Carli, C.; Pearson, H.; Janelle, V.; Durette, C.; Bonneil, E.; et al. Proteogenomic-based discovery of minor histocompatibility antigens with suitable features for immunotherapy of hematologic cancers. Leukemia 2016, 30, 1344. [Google Scholar] [CrossRef]
- Roopenian, D.; Choi, E.Y.; Brown, A. The immunogenomics of minor histocompatibility antigens. Immunol. Rev. 2002, 190, 86–94. [Google Scholar] [CrossRef]
- Mullally, A.; Ritz, J. Beyond HLA: The significance of genomic variation for allogeneic hematopoietic stem cell transplantation. Blood 2007, 109, 1355–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilberberg, J.; Feinman, R.; Korngold, R. Strategies for the identification of T cell-recognized tumor antigens in hematological malignancies for improved graft-versus-tumor responses after allogeneic blood and marrow transplantation. Biol. Blood Marrow Transplant. 2015, 21, 1000–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattinoni, L.; Klebanoff, C.A.; Palmer, D.C.; Wrzesinski, C.; Kerstann, K.; Yu, Z.; Finkelstein, S.E.; Theoret, M.R.; Rosenberg, S.A.; Restifo, N.P. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J. Clin. Investig. 2005, 115, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Riddell, S.R.; Schumacher, T.N. Adoptive cellular therapy: A race to the finish line. Sci. Transl. Med. 2015, 7, 280–287. [Google Scholar] [CrossRef]
- Dahmani, A.; Janelle, V.; Carli, C.; Richaud, M.; Lamarche, C.; Khalili, M.; Goupil, M.; Bezverbnaya, K.; Bramson, J.L.; Delisle, J.S. TGFβ Programs Central Memory Differentiation in Ex Vivo-Stimulated Human T Cells. Cancer Immunol Res. 2019, 7, 1426–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orio, J.; Carli, C.; Janelle, V.; Giroux, M.; Taillefer, J.; Goupil, M.; Richaud, M.; Roy, D.-C.; Delisle, J.-S. Early exposure to interleukin-21 limits rapidly generated anti–Epstein-Barr virus T-cell line differentiation. Cytotherapy 2015, 17, 496–508. [Google Scholar] [CrossRef]
- Wang, J.; Hasan, F.; Frey, A.C.; Li, H.S.; Park, J.; Pan, K.; Haymaker, C.; Bernatchez, C.; Lee, D.A.; Watowich, S.S.; et al. Histone Deacetylase Inhibitors and IL21 Cooperate to Reprogram Human Effector CD8+; T Cells to Memory T Cells. Cancer Immunol Res. 2020, 8, 794. [Google Scholar] [CrossRef] [Green Version]
- Franco, F.; Jaccard, A.; Romero, P.; Yu, Y.R.; Ho, P.C. Metabolic and epigenetic regulation of T-cell exhaustion. Nat. Metab. 2020, 2, 1001–1012. [Google Scholar] [CrossRef]
- You, L.; Han, Q.; Zhu, L.; Zhu, Y.; Bao, C.; Yang, C.; Lei, W.; Qian, W. Decitabine-Mediated Epigenetic Reprograming Enhances Anti-leukemia Efficacy of CD123-Targeted Chimeric Antigen Receptor T-Cells. Front. Immunol. 2020, 11, 1787. [Google Scholar] [CrossRef]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157.e119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Waart, A.B.; van de Weem, N.M.P.; Maas, F.; Kramer, C.S.M.; Kester, M.G.D.; Falkenburg, J.H.F.; Schaap, N.; Jansen, J.H.; van der Voort, R.; Gattinoni, L.; et al. Inhibition of Akt signaling promotes the generation of superior tumor-reactive T cells for adoptive immunotherapy. Blood 2014, 124, 3490–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilipow, K.; Scamardella, E.; Puccio, S.; Gautam, S.; De Paoli, F.; Mazza, E.M.; De Simone, G.; Polletti, S.; Buccilli, M.; Zanon, V.; et al. Antioxidant metabolism regulates CD8+ T memory stem cell formation and antitumor immunity. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapuis, A.G.; Thompson, J.A.; Margolin, K.A.; Rodmyre, R.; Lai, I.P.; Dowdy, K.; Farrar, E.A.; Bhatia, S.; Sabath, D.E.; Cao, J.; et al. Transferred melanoma-specific CD8+ T cells persist, mediate tumor regression, and acquire central memory phenotype. Proc. Natl. Acad. Sci. USA 2012, 109, 4592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, T.; Kaneko, S.; Kawana-Tachikawa, A.; Tajima, Y.; Goto, H.; Zhu, D.; Nakayama-Hosoya, K.; Iriguchi, S.; Uemura, Y.; Shimizu, T.; et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013, 12, 114–126. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, M.; Awong, G.; Sturgeon, C.M.; Ditadi, A.; LaMotte-Mohs, R.; Zúñiga-Pflücker, J.C.; Keller, G. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012, 2, 1722–1735. [Google Scholar] [CrossRef] [Green Version]
- Vizcardo, R.; Masuda, K.; Yamada, D.; Ikawa, T.; Shimizu, K.; Fujii, S.; Koseki, H.; Kawamoto, H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell 2013, 12, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Ando, M.; Nishimura, T.; Yamazaki, S.; Yamaguchi, T.; Kawana-Tachikawa, A.; Hayama, T.; Nakauchi, Y.; Ando, J.; Ota, Y.; Takahashi, S.; et al. A Safeguard System for Induced Pluripotent Stem Cell-Derived Rejuvenated T Cell Therapy. Stem Cell Rep. 2015, 5, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Timmermans, F.; Velghe, I.; Vanwalleghem, L.; De Smedt, M.; Van Coppernolle, S.; Taghon, T.; Moore, H.D.; Leclercq, G.; Langerak, A.W.; Kerre, T.; et al. Generation of T Cells from Human Embryonic Stem Cell-Derived Hematopoietic Zones. J. Immunol. 2009, 182, 6879. [Google Scholar] [CrossRef]
- Chan, J.; Kim, P.Y.; Kranz, E.; Nagaoka, Y.; Lee, Y.; Wen, J.; Elsaesser, H.J.; Qin, M.; Brooks, D.G.; Ringpis, G.E.; et al. Purging Exhausted Virus-Specific CD8 T Cell Phenotypes by Somatic Cell Reprogramming. AIDS Res. Hum. Retrovir. 2017, 33, S59–S69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Murata-Hirai, K.; Iwasaki, M.; Matsumoto, K.; Hayashi, K.; Kumagai, A.; Nada, M.H.; Wang, H.; Kobayashi, H.; Kamitakahara, H.; et al. Expansion of human γδ T cells for adoptive immunotherapy using a bisphosphonate prodrug. Cancer Sci. 2018, 109, 587–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeres, T.; Smetak, M.; Pretscher, D.; Wilhelm, M. Improving the Efficiency of Vγ9Vδ2 T-Cell Immunotherapy in Cancer. Front. Immunol. 2018, 9, 800. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Lal, G. Regulatory and effector functions of gamma–delta (γδ) T cells and their therapeutic potential in adoptive cellular therapy for cancer. Int. J. Cancer 2016, 139, 976–985. [Google Scholar] [CrossRef]
- Nada, M.H.; Wang, H.; Workalemahu, G.; Tanaka, Y.; Morita, C.T. Enhancing adoptive cancer immunotherapy with Vγ2Vδ2 T cells through pulse zoledronate stimulation. J. Immunother. Cancer 2017, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Zumwalde, N.A.; Sharma, A.; Xu, X.; Ma, S.; Schneider, C.L.; Romero-Masters, J.C.; Hudson, A.W.; Gendron-Fitzpatrick, A.; Kenney, S.C.; Gumperz, J.E. Adoptively transferred Vγ9Vδ2 T cells show potent antitumor effects in a preclinical B cell lymphomagenesis model. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Kronenberg, M. When less is more: T lymphocyte populations with restricted antigen receptor diversity. J. Immunol. 2014, 193, 975–976. [Google Scholar] [CrossRef] [Green Version]
- Van Rhijn, I.; Moody, D.B. Donor Unrestricted T Cells: A Shared Human T Cell Response. J. Immunol. 2015, 195, 1927–1932. [Google Scholar] [CrossRef] [Green Version]
- Salio, M.; Silk, J.D.; Jones, E.Y.; Cerundolo, V. Biology of CD1- and MR1-restricted T cells. Annu. Rev. Immunol. 2014, 32, 323–366. [Google Scholar] [CrossRef]
- Constantinides, M.G.; Bendelac, A. Transcriptional regulation of the NKT cell lineage. Curr. Opin. Immunol. 2013, 25, 161–167. [Google Scholar] [CrossRef]
- Brennan, P.J.; Brigl, M.; Brenner, M.B. Invariant natural killer T cells: An innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 2013, 13, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Brossay, L.; Chioda, M.; Burdin, N.; Koezuka, Y.; Casorati, G.; Dellabona, P.; Kronenberg, M. CD1d-mediated recognition of an alpha-galactosylceramide by natural killer T cells is highly conserved through mammalian evolution. J. Exp. Med. 1998, 188, 1521–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Exley, M.A.; Lynch, L.; Varghese, B.; Nowak, M.; Alatrakchi, N.; Balk, S.P. Developing understanding of the roles of CD1d-restricted T cell subsets in cancer: Reversing tumor-induced defects. Clin. Immunol. 2011, 140, 184–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motohashi, S.; Ishikawa, A.; Ishikawa, E.; Otsuji, M.; Iizasa, T.; Hanaoka, H.; Shimizu, N.; Horiguchi, S.; Okamoto, Y.; Fujii, S.; et al. A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer. Clin. Cancer. Res. 2006, 12, 6079–6086. [Google Scholar] [CrossRef] [Green Version]
- Kunii, N.; Horiguchi, S.; Motohashi, S.; Yamamoto, H.; Ueno, N.; Yamamoto, S.; Sakurai, D.; Taniguchi, M.; Nakayama, T.; Okamoto, Y. Combination therapy of in vitro-expanded natural killer T cells and alpha-galactosylceramide-pulsed antigen-presenting cells in patients with recurrent head and neck carcinoma. Cancer Sci. 2009, 100, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Horiguchi, S.; Kurosaki, M.; Kunii, N.; Nagato, K.; Hanaoka, H.; Shimizu, N.; Ueno, N.; Yamamoto, S.; Taniguchi, M.; et al. Induction of NKT cell-specific immune responses in cancer tissues after NKT cell-targeted adoptive immunotherapy. Clin. Immunol. 2011, 138, 255–265. [Google Scholar] [CrossRef]
- Exley, M.A.; Friedlander, P.; Alatrakchi, N.; Vriend, L.; Yue, S.; Sasada, T.; Zeng, W.; Mizukami, Y.; Clark, J.; Nemer, D.; et al. Adoptive Transfer of Invariant NKT Cells as Immunotherapy for Advanced Melanoma: A Phase I Clinical Trial. Clin. Cancer Res. 2017, 23, 3510–3519. [Google Scholar] [CrossRef] [Green Version]
- Exley, M.A.; Hou, R.; Shaulov, A.; Tonti, E.; Dellabona, P.; Casorati, G.; Akbari, O.; Akman, H.O.; Greenfield, E.A.; Gumperz, J.E.; et al. Selective activation, expansion, and monitoring of human iNKT cells with a monoclonal antibody specific for the TCR alpha-chain CDR3 loop. Eur. J. Immunol. 2008, 38, 1756–1766. [Google Scholar] [CrossRef]
- Exley, M.A.; Wilson, S.B.; Balk, S.P. Isolation and Functional Use of Human NKT Cells. Curr. Protoc. Immunol. 2017, 119, 14.11.11–14.11.20. [Google Scholar] [CrossRef]
- Pillai, A.B.; George, T.I.; Dutt, S.; Teo, P.; Strober, S. Host NKT Cells Can Prevent Graft-versus-Host Disease and Permit Graft Antitumor Activity after Bone Marrow Transplantation. J. Immunol. 2007, 178, 6242. [Google Scholar] [CrossRef] [Green Version]
- Wolf, B.J.; Choi, J.E.; Exley, M.A. Novel Approaches to Exploiting Invariant NKT Cells in Cancer Immunotherapy. Front. Immunol. 2018, 9, 384. [Google Scholar] [CrossRef] [PubMed]
- Chiossone, L.; Dumas, P.-Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nature 2015, 517, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Serafini, N.; Vosshenrich, C.A.; Di Santo, J.P. Transcriptional regulation of innate lymphoid cell fate. Nat. Rev. Immunol. 2015, 15, 415–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, A.; Vermi, W.; Lee, J.S.; Lonardi, S.; Gilfillan, S.; Newberry, R.D.; Cella, M.; Colonna, M. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity 2013, 38, 769–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; te Velde, A.A.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef]
- Gerbe, F.; Sidot, E.; Smyth, D.J.; Ohmoto, M.; Matsumoto, I.; Dardalhon, V.; Cesses, P.; Garnier, L.; Pouzolles, M.; Brulin, B.; et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 2016, 529, 226–230. [Google Scholar] [CrossRef] [Green Version]
- Wallrapp, A.; Riesenfeld, S.J.; Burkett, P.R.; Abdulnour, R.E.; Nyman, J.; Dionne, D.; Hofree, M.; Cuoco, M.S.; Rodman, C.; Farouq, D.; et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 2017, 549, 351–356. [Google Scholar] [CrossRef]
- Buonocore, S.; Ahern, P.P.; Uhlig, H.H.; Ivanov, I.I.; Littman, D.R.; Maloy, K.J.; Powrie, F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 2010, 464, 1371–1375. [Google Scholar] [CrossRef] [Green Version]
- Ibiza, S.; García-Cassani, B.; Ribeiro, H.; Carvalho, T.; Almeida, L.; Marques, R.; Misic, A.M.; Bartow-McKenney, C.; Larson, D.M.; Pavan, W.J.; et al. Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature 2016, 535, 440–443. [Google Scholar] [CrossRef] [Green Version]
- Withers, D.R. Lymphoid tissue inducer cells. Curr. Biol. 2011, 21, R381–R382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis E Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallmer, K.; Oxenius, A. Recognition and Regulation of T Cells by NK Cells. Front. Immunol. 2016, 7, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Bi, J.; Tian, Z. NK Cell Exhaustion. Front. Immunol. 2017, 8, 760. [Google Scholar] [CrossRef]
- Lim, O.; Lee, Y.; Chung, H.; Her, J.H.; Kang, S.M.; Jung, M.-y.; Min, B.; Shin, H.; Kim, T.M.; Heo, D.S.; et al. GMP-compliant, large-scale expanded allogeneic natural killer cells have potent cytolytic activity against cancer cells in vitro and in vivo. PLoS ONE 2013, 8, e53611. [Google Scholar] [CrossRef]
- Torelli, G.F.; Rozera, C.; Santodonato, L.; Peragine, N.; D’Agostino, G.; Montefiore, E.; Napolitano, M.R.; Monque, D.M.; Carlei, D.; Mariglia, P.; et al. A good manufacturing practice method to ex vivo expand natural killer cells for clinical use. Blood Transfus. 2015, 13, 464–471. [Google Scholar] [CrossRef]
- Alici, E.; Sutlu, T.; Björkstrand, B.; Gilljam, M.; Stellan, B.; Nahi, H.; Quezada, H.C.; Gahrton, G.; Ljunggren, H.G.; Dilber, M.S. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP-compliant components. Blood 2008, 111, 3155–3162. [Google Scholar] [CrossRef] [Green Version]
- Koehl, U.; Brehm, C.; Huenecke, S.; Zimmermann, S.Y.; Kloess, S.; Bremm, M.; Ullrich, E.; Soerensen, J.; Quaiser, A.; Erben, S.; et al. Clinical grade purification and expansion of NK cell products for an optimized manufacturing protocol. Front. Oncol. 2013, 3, 118. [Google Scholar] [CrossRef] [Green Version]
- Ortaldo, J.R.; Winkler-Pickett, R.; Wigginton, J.; Horner, M.; Bere, E.W.; Mason, A.T.; Bhat, N.; Cherry, J.; Sanford, M.; Hodge, D.L.; et al. Regulation of ITAM-positive receptors: Role of IL-12 and IL-18. Blood 2006, 107, 1468–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, S.J.; Marusina, A.I.; Pathmanathan, I.; Borrego, F.; Coligan, J.E. IL-21 down-regulates NKG2D/DAP10 expression on human NK and CD8+ T cells. J. Immunol. 2006, 176, 1490–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becknell, B.; Caligiuri, M.A. Interleukin-2, interleukin-15, and their roles in human natural killer cells. Adv. Immunol. 2005, 86, 209–239. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.P.; Trinchieri, G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002, 13, 155–168. [Google Scholar] [CrossRef]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef]
- Huenecke, S.; Zimmermann, S.Y.; Kloess, S.; Esser, R.; Brinkmann, A.; Tramsen, L.; Koenig, M.; Erben, S.; Seidl, C.; Tonn, T.; et al. IL-2-driven regulation of NK cell receptors with regard to the distribution of CD16+ and CD16- subpopulations and in vivo influence after haploidentical NK cell infusion. J. Immunother. 2010, 33, 200–210. [Google Scholar] [CrossRef]
- Wang, K.S.; Frank, D.A.; Ritz, J. Interleukin-2 enhances the response of natural killer cells to interleukin-12 through up-regulation of the interleukin-12 receptor and STAT4. Blood 2000, 95, 3183–3190. [Google Scholar] [CrossRef]
- Mao, Y.; van Hoef, V.; Zhang, X.; Wennerberg, E.; Lorent, J.; Witt, K.; Masvidal, L.; Liang, S.; Murray, S.; Larsson, O.; et al. IL-15 activates mTOR and primes stress-activated gene expression leading to prolonged antitumor capacity of NK cells. Blood 2016, 128, 1475–1489. [Google Scholar] [CrossRef] [Green Version]
- Fehniger, T.A.; Cooper, M.A.; Caligiuri, M.A. Interleukin-2 and interleukin-15: Immunotherapy for cancer. Cytokine Growth Factor Rev. 2002, 13, 169–183. [Google Scholar] [CrossRef]
- Waldmann, T.A. The biology of interleukin-2 and interleukin-15: Implications for cancer therapy and vaccine design. Nat. Rev. Immunol. 2006, 6, 595–601. [Google Scholar] [CrossRef]
- Felices, M.; Lenvik, A.; Chu, S.; McElmurry, R.; Cooley, S.; Tolar, J.; Blazar, B.R.; Miller, J.S. Continuous IL-15 Signaling Leads to Functional Exhaustion of Human Natural Killer Cells through Metabolic Changes That Alters Their In Vivo Anti-Tumor Activity. Blood 2016, 128, 551. [Google Scholar] [CrossRef]
- French, A.R.; Holroyd, E.B.; Yang, L.; Kim, S.; Yokoyama, W.M. IL-18 acts synergistically with IL-15 in stimulating natural killer cell proliferation. Cytokine 2006, 35, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Chaix, J.; Tessmer, M.S.; Hoebe, K.; Fuséri, N.; Ryffel, B.; Dalod, M.; Alexopoulou, L.; Beutler, B.; Brossay, L.; Vivier, E.; et al. Cutting edge: Priming of NK cells by IL-18. J. Immunol. 2008, 181, 1627–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMichael, E.L.; Jaime-Ramirez, A.C.; Guenterberg, K.D.; Luedke, E.; Atwal, L.S.; Campbell, A.R.; Hu, Z.; Tatum, A.S.; Kondadasula, S.V.; Mo, X.; et al. IL-21 Enhances Natural Killer Cell Response to Cetuximab-Coated Pancreatic Tumor Cells. Clin. Cancer. Res. 2017, 23, 489–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannello, A.; Boulassel, M.-R.; Samarani, S.; Tremblay, C.; Toma, E.; Routy, J.-P.; Ahmad, A. IL-21 enhances NK cell functions and survival in healthy and HIV-infected patients with minimal stimulation of viral replication. J. Leukoc. Biol. 2010, 87, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Kasaian, M.T.; Whitters, M.J.; Carter, L.L.; Lowe, L.D.; Jussif, J.M.; Deng, B.; Johnson, K.A.; Witek, J.S.; Senices, M.; Konz, R.F.; et al. IL-21 limits NK cell responses and promotes antigen-specific T cell activation: A mediator of the transition from innate to adaptive immunity. Immunity 2002, 16, 559–569. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Ye, L.J.; Ren, H.L.; Huyan, T.; Li, J.; Shi, J.L.; Huang, Q.S. Multiple effects of IL-21 on human NK cells in ex vivo expansion. Immunobiology 2015, 220, 876–888. [Google Scholar] [CrossRef]
- Granzin, M.; Stojanovic, A.; Miller, M.; Childs, R.; Huppert, V.; Cerwenka, A. Highly efficient IL-21 and feeder cell-driven ex vivo expansion of human NK cells with therapeutic activity in a xenograft mouse model of melanoma. Oncoimmunology 2016, 5, e1219007. [Google Scholar] [CrossRef] [Green Version]
- Lim, D.P.; Jang, Y.Y.; Kim, S.; Koh, S.S.; Lee, J.J.; Kim, J.S.; Thi Phan, M.T.; Shin, D.J.; Shin, M.G.; Lee, S.H.; et al. Effect of exposure to interleukin-21 at various time points on human natural killer cell culture. Cytotherapy 2014, 16, 1419–1430. [Google Scholar] [CrossRef]
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.-C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 2018, 128, 4654–4668. [Google Scholar] [CrossRef]
- Geller, M.A.; Cooley, S.; Judson, P.L.; Ghebre, R.; Carson, L.F.; Argenta, P.A.; Jonson, A.L.; Panoskaltsis-Mortari, A.; Curtsinger, J.; McKenna, D.; et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011, 13, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leivas, A.; Perez-Martinez, A.; Blanchard, M.J.; Martín-Clavero, E.; Fernández, L.; Lahuerta, J.J.; Martinez-Lopez, J. Novel treatment strategy with autologous activated and expanded natural killer cells plus anti-myeloma drugs for multiple myeloma. Oncoimmunology 2016, 5, e1250051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inozume, T.; Hanada, K.; Wang, Q.J.; Ahmadzadeh, M.; Wunderlich, J.R.; Rosenberg, S.A.; Yang, J.C. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J. Immunother. 2010, 33, 956–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Ji, Y.; Gattinoni, L.; Zhang, L.; Yu, Z.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Modulating the differentiation status of ex vivo-cultured anti-tumor T cells using cytokine cocktails. Cancer Immunol. Immunother. 2013, 62, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, S.; Hernandez, J.; Vence, L.; Hwu, P.; Radvanyi, L. MART-1-specific melanoma tumor-infiltrating lymphocytes maintaining CD28 expression have improved survival and expansion capability following antigenic restimulation in vitro. J. Immunol. 2010, 184, 452–465. [Google Scholar] [CrossRef] [Green Version]
- van den Berg, J.H.; Heemskerk, B.; van Rooij, N.; Gomez-Eerland, R.; Michels, S.; van Zon, M.; de Boer, R.; Bakker, N.A.M.; Jorritsma-Smit, A.; van Buuren, M.M.; et al. Tumor infiltrating lymphocytes (TIL) therapy in metastatic melanoma: Boosting of neoantigen-specific T cell reactivity and long-term follow-up. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Jansen, C.S.; Prokhnevska, N.; Master, V.A.; Sanda, M.G.; Carlisle, J.W.; Bilen, M.A.; Cardenas, M.; Wilkinson, S.; Lake, R.; Sowalsky, A.G.; et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 2019, 576, 465–470. [Google Scholar] [CrossRef]
- Brien, S.M.; Klampatsa, A.; Thompson, J.C.; Martinez, M.C.; Hwang, W.-T.; Rao, A.S.; Standalick, J.E.; Kim, S.; Cantu, E.; Litzky, L.A.; et al. Function of Human Tumor-Infiltrating Lymphocytes in Early-Stage Non–Small Cell Lung Cancer. Cancer Immunol. Res. 2019, 7, 896. [Google Scholar] [CrossRef]
- Kumar, B.V.; Ma, W.; Miron, M.; Granot, T.; Guyer, R.S.; Carpenter, D.J.; Senda, T.; Sun, X.; Ho, S.-H.; Lerner, H.; et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep. 2017, 20, 2921–2934. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhu, B.; Son, Y.M.; Wang, Z.; Jiang, L.; Xiang, M.; Ye, Z.; Beckermann, K.E.; Wu, Y.; Jenkins, J.W.; et al. The Transcription Factor Bhlhe40 Programs Mitochondrial Regulation of Resident CD8(+) T Cell Fitness and Functionality. Immunity 2019, 51, 491–507.e497. [Google Scholar] [CrossRef]
- Woroniecka, K.I.; Rhodin, K.E.; Dechant, C.; Cui, X.; Chongsathidkiet, P.; Wilkinson, D.; Waibl-Polania, J.; Sanchez-Perez, L.; Fecci, P.E. 4-1BB Agonism Averts TIL Exhaustion and Licenses PD-1 Blockade in Glioblastoma and Other Intracranial Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 1349–1358. [Google Scholar] [CrossRef]
- Hall, M.; Liu, H.; Malafa, M.; Centeno, B.; Hodul, P.J.; Pimiento, J.; Pilon-Thomas, S.; Sarnaik, A.A. Expansion of tumor-infiltrating lymphocytes (TIL) from human pancreatic tumors. J. Immunother. Cancer 2016, 4, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Stachura, P.; Xu, H.C.; Bhatia, S.; Borkhardt, A.; Lang, P.A.; Pandyra, A.A. Senescent Tumor CD8(+) T Cells: Mechanisms of Induction and Challenges to Immunotherapy. Cancers 2020, 12, 2828. [Google Scholar] [CrossRef]
- Roninson, I.B.; Broude, E.V.; Chang, B.D. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resistance Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2001, 4, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; Andersen, J.; Kapahi, P.; Melov, S. Cellular senescence: A link between cancer and age-related degenerative disease? Semin. Cancer Biol. 2011, 21, 354–359. [Google Scholar] [CrossRef] [Green Version]
- Montes, C.L.; Chapoval, A.I.; Nelson, J.; Orhue, V.; Zhang, X.; Schulze, D.H.; Strome, S.E.; Gastman, B.R. Tumor-Induced Senescent T Cells with Suppressor Function: A Potential Form of Tumor Immune Evasion. Cancer Res. 2008, 68, 870. [Google Scholar] [CrossRef] [Green Version]
- Dossett, M.L.; Teague, R.M.; Schmitt, T.M.; Tan, X.; Cooper, L.J.; Pinzon, C.; Greenberg, P.D. Adoptive immunotherapy of disseminated leukemia with TCR-transduced, CD8+ T cells expressing a known endogenous TCR. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 742–749. [Google Scholar] [CrossRef]
- Zhang, L.; Morgan, R.A. Genetic engineering with T cell receptors. Adv. Drug Deliv. Rev. 2012, 64, 756–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadi, M.; King, J.W.; Xue, S.A.; Voisine, C.; Holler, A.; Wright, G.P.; Waxman, J.; Morris, E.; Stauss, H.J. CD3 limits the efficacy of TCR gene therapy in vivo. Blood 2011, 118, 3528–3537. [Google Scholar] [CrossRef] [PubMed]
- Legut, M.; Dolton, G.; Mian, A.A.; Ottmann, O.G.; Sewell, A.K. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood 2018, 131, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, L.T.; Reijmers, R.M.; Wouters, A.K.; Kweekel, C.; Remst, D.F.G.; Pothast, C.R.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Simultaneous Deletion of Endogenous TCRαβ for TCR Gene Therapy Creates an Improved and Safe Cellular Therapeutic. Mol. Ther. 2020, 28, 64–74. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Heemskerk, M.H.M.; Hoogeboom, M.; Hagedoorn, R.; Kester, M.G.D.; Willemze, R.; Falkenburg, J.H.F. Reprogramming of Virus-specific T Cells into Leukemia-reactive T Cells Using T Cell Receptor Gene Transfer. J. Exp. Med. 2004, 199, 885–894. [Google Scholar] [CrossRef] [Green Version]
- Marieke, G.; van Egmond, H.M.E.; Helen, B.-P.; van der Hoorn, M.A.W.G.; Renate, S.H.; Michel, G.D.K.; Nikolai, S.; Roel, W.; Falkenburg, J.H.F.; Mirjam, H.M.H. Genetic engineering of virus-specific T cells with T-cell receptors recognizing minor histocompatibility antigens for clinical application. Haematologica 2008, 93, 1535–1543. [Google Scholar] [CrossRef]
- Marleen, M.V.L.; de Renate, B.; van Ellis, L.; Pauline, M.; Inge, J.; Falkenburg, J.H.F.; Mirjam, H.M.H. A Good Manufacturing Practice procedure to engineer donor virus-specific T cells into potent anti-leukemic effector cells. Haematologica 2014, 99, 759–768. [Google Scholar] [CrossRef]
- Chapuis, A.G.; Egan, D.N.; Bar, M.; Schmitt, T.M.; McAfee, M.S.; Paulson, K.G.; Voillet, V.; Gottardo, R.; Ragnarsson, G.B.; Bleakley, M.; et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat. Med. 2019, 25, 1064–1072. [Google Scholar] [CrossRef]
- van Loenen, M.M.; Hagedoorn, R.S.; Kester, M.G.; Hoogeboom, M.; Willemze, R.; Falkenburg, J.H.; Heemskerk, M.H. Kinetic preservation of dual specificity of coprogrammed minor histocompatibility antigen-reactive virus-specific T cells. Cancer Res. 2009, 69, 2034–2041. [Google Scholar] [CrossRef] [Green Version]
- Hart, D.P.; Xue, S.A.; Thomas, S.; Cesco-Gaspere, M.; Tranter, A.; Willcox, B.; Lee, S.P.; Steven, N.; Morris, E.C.; Stauss, H.J. Retroviral transfer of a dominant TCR prevents surface expression of a large proportion of the endogenous TCR repertoire in human T cells. Gene Ther. 2008, 15, 625–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, J.A.; Bajwa, G.; Carreres, B.; Hoyer, E.; Gruber, I.; Martínez-Paniagua, M.A.; Yu, Y.-R.; Nouraee, N.; Sadeghi, F.; Wu, M.; et al. Single-cell transcriptomics identifies multiple pathways underlying antitumor function of TCR- and CD8αβ-engineered human CD4+ T cells. Sci. Adv. 2020, 6, eaaz7809. [Google Scholar] [CrossRef] [PubMed]
- Arber, C.; Feng, X.; Abhyankar, H.; Romero, E.; Wu, M.F.; Heslop, H.E.; Barth, P.; Dotti, G.; Savoldo, B. Survivin-specific T cell receptor targets tumor but not T cells. J. Clin. Investig. 2015, 125, 157–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajwa, G.; Lanz, I.; Cardenas, M.; Brenner, M.K.; Arber, C. Transgenic CD8αβ co-receptor rescues endogenous TCR function in TCR-transgenic virus-specific T cells. J. Immunother. Cancer 2020, 8, e001487. [Google Scholar] [CrossRef]
- Laugel, B.; van den Berg, H.A.; Gostick, E.; Cole, D.K.; Wooldridge, L.; Boulter, J.; Milicic, A.; Price, D.A.; Sewell, A.K. Different T Cell Receptor Affinity Thresholds and CD8 Coreceptor Dependence Govern Cytotoxic T Lymphocyte Activation and Tetramer Binding Properties. J. Biol. Chem. 2007, 282, 23799–23810. [Google Scholar] [CrossRef] [Green Version]
- Laugel, B.; Cole, D.K.; Clement, M.; Wooldridge, L.; Price, D.A.; Sewell, A.K. The multiple roles of the CD8 coreceptor in T cell biology: Opportunities for the selective modulation of self-reactive cytotoxic T cells. J. Leukoc. Biol. 2011, 90, 1089–1099. [Google Scholar] [CrossRef]
- Helsen, C.W.; Hammill, J.A.; Lau, V.W.C.; Mwawasi, K.A.; Afsahi, A.; Bezverbnaya, K.; Newhook, L.; Hayes, D.L.; Aarts, C.; Bojovic, B.; et al. The chimeric TAC receptor co-opts the T cell receptor yielding robust anti-tumor activity without toxicity. Nat. Commun. 2018, 9, 3049. [Google Scholar] [CrossRef] [Green Version]
- Baeuerle, P.A.; Ding, J.; Patel, E.; Thorausch, N.; Horton, H.; Gierut, J.; Scarfo, I.; Choudhary, R.; Kiner, O.; Krishnamurthy, J.; et al. Synthetic TRuC receptors engaging the complete T cell receptor for potent anti-tumor response. Nat. Commun. 2019, 10, 2087. [Google Scholar] [CrossRef] [Green Version]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.R.; Flaswinkel, H.; Reth, M.; Scott, D.W. Immunoreceptor tyrosine-based activation motif is required to signal pathways of receptor-mediated growth arrest and apoptosis in murine B lymphoma cells. J. Immunol. 1995, 155, 652–661. [Google Scholar]
- Brocker, T.; Karjalainen, K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J. Exp. Med. 1995, 181, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ /CD28 receptor. Nat. Biotechnol. 2002, 20, 70. [Google Scholar] [CrossRef] [PubMed]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.-J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef]
- Riches, J.C.; Gribben, J.G. Understanding the immunodeficiency in chronic lymphocytic leukemia: Potential clinical implications. Hematol. Oncol. Clin. N. Am. 2013, 27, 207–235. [Google Scholar] [CrossRef]
- te Raa, G.D.; Tonino, S.H.; Remmerswaal, E.B.; van Houte, A.J.; Koene, H.R.; van Oers, M.H.; Kater, A.P. Chronic lymphocytic leukemia specific T-cell subset alterations are clone-size dependent and not present in monoclonal B lymphocytosis. Leuk. Lymphoma 2012, 53, 2321–2325. [Google Scholar] [CrossRef]
- Tonino, S.H.; van de Berg, P.J.; Yong, S.L.; ten Berge, I.J.; Kersten, M.J.; van Lier, R.A.; van Oers, M.H.; Kater, A.P. Expansion of effector T cells associated with decreased PD-1 expression in patients with indolent B cell lymphomas and chronic lymphocytic leukemia. Leuk. Lymphoma 2012, 53, 1785–1794. [Google Scholar] [CrossRef] [PubMed]
- Forconi, F.; Moss, P. Perturbation of the normal immune system in patients with CLL. Blood 2015, 126, 573–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsay, A.G.; Johnson, A.J.; Lee, A.M.; Gorgün, G.; Le Dieu, R.; Blum, W.; Byrd, J.C.; Gribben, J.G. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J. Clin. Invest. 2008, 118, 2427–2437. [Google Scholar] [CrossRef] [PubMed]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.G.; Gribben, J.G. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood 2013, 121, 1612–1621. [Google Scholar] [CrossRef]
- Görgün, G.; Holderried, T.A.; Zahrieh, D.; Neuberg, D.; Gribben, J.G. Chronic lymphocytic leukemia cells induce changes in gene expression of CD4 and CD8 T cells. J. Clin. Investig. 2005, 115, 1797–1805. [Google Scholar] [CrossRef]
- Gorgun, G.; Ramsay, A.G.; Holderried, T.A.; Zahrieh, D.; Le Dieu, R.; Liu, F.; Quackenbush, J.; Croce, C.M.; Gribben, J.G. E(mu)-TCL1 mice represent a model for immunotherapeutic reversal of chronic lymphocytic leukemia-induced T-cell dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 6250–6255. [Google Scholar] [CrossRef] [Green Version]
- van Bruggen, J.A.C.; Martens, A.W.J.; Fraietta, J.A.; Hofland, T.; Tonino, S.H.; Eldering, E.; Levin, M.D.; Siska, P.J.; Endstra, S.; Rathmell, J.C.; et al. Chronic lymphocytic leukemia cells impair mitochondrial fitness in CD8(+) T cells and impede CAR T-cell efficacy. Blood 2019, 134, 44–58. [Google Scholar] [CrossRef]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Investig. 2019, 129, 2210–2221. [Google Scholar] [CrossRef] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Garfall, A.L.; Dancy, E.K.; Cohen, A.D.; Hwang, W.T.; Fraietta, J.A.; Davis, M.M.; Levine, B.L.; Siegel, D.L.; Stadtmauer, E.A.; Vogl, D.T.; et al. T-cell phenotypes associated with effective CAR T-cell therapy in postinduction vs relapsed multiple myeloma. Blood Adv. 2019, 3, 2812–2815. [Google Scholar] [CrossRef] [Green Version]
- Ghassemi, S.; Nunez-Cruz, S.; O’Connor, R.S.; Fraietta, J.A.; Patel, P.R.; Scholler, J.; Barrett, D.M.; Lundh, S.M.; Davis, M.M.; Bedoya, F.; et al. Reducing Ex Vivo Culture Improves the Antileukemic Activity of Chimeric Antigen Receptor (CAR) T Cells. Cancer Immunol Res. 2018, 6, 1100–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stüber, T.; Monjezi, R.; Wallstabe, L.; Kühnemundt, J.; Nietzer, S.L.; Dandekar, G.; Wöckel, A.; Einsele, H.; Wischhusen, J.; Hudecek, M. Inhibition of TGF-β-receptor signaling augments the antitumor function of ROR1-specific CAR T-cells against triple-negative breast cancer. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Dahmani, A.; Delisle, J.-S. TGF-β in T Cell Biology: Implications for Cancer Immunotherapy. Cancers 2018, 10, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [Green Version]
- Ankri, C.; Shamalov, K.; Horovitz-Fried, M.; Mauer, S.; Cohen, C.J. Human T Cells Engineered To Express a Programmed Death 1/28 Costimulatory Retargeting Molecule Display Enhanced Antitumor Activity. J. Immunol. 2013, 1203085. [Google Scholar] [CrossRef] [Green Version]
- Angel, P.; Karin, M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1991, 1072, 129–157. [Google Scholar] [CrossRef]
- Samelson, L.E. Signal transduction mediated by the T cell antigen receptor: The role of adapter proteins. Annu. Rev. Immunol. 2002, 20, 371–394. [Google Scholar] [CrossRef] [Green Version]
- Murphy, T.L.; Tussiwand, R.; Murphy, K.M. Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat. Rev. Immunol. 2013, 13, 499–509. [Google Scholar] [CrossRef]
- Quigley, M.; Pereyra, F.; Nilsson, B.; Porichis, F.; Fonseca, C.; Eichbaum, Q.; Julg, B.; Jesneck, J.L.; Brosnahan, K.; Imam, S.; et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat. Med. 2010, 16, 1147–1151. [Google Scholar] [CrossRef]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Daniyan, A.F.; Lopez, A.V.; Purdon, T.J.; Brentjens, R.J. Cytokine IL-36γ improves CAR T-cell functionality and induces endogenous antitumor response. Leukemia 2020. [Google Scholar] [CrossRef] [PubMed]
- Avanzi, M.P.; Yeku, O.; Li, X.; Wijewarnasuriya, D.P.; van Leeuwen, D.G.; Cheung, K.; Park, H.; Purdon, T.J.; Daniyan, A.F.; Spitzer, M.H.; et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep. 2018, 23, 2130–2141. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [Green Version]
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017, 7, 10541. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Morgan, R.A.; Beane, J.D.; Zheng, Z.; Dudley, M.E.; Kassim, S.H.; Nahvi, A.V.; Ngo, L.T.; Sherry, R.M.; Phan, G.Q.; et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin. Cancer. Res. 2015, 21, 2278–2288. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Davies, J.S.; Serna, C.; Yu, Z.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A.; Hinrichs, C.S. Enhanced efficacy and limited systemic cytokine exposure with membrane-anchored interleukin-12 T-cell therapy in murine tumor models. J. Immunother. Cancer 2020, 8, e000210. [Google Scholar] [CrossRef] [Green Version]
- Lapteva, N.; Gilbert, M.; Diaconu, I.; Rollins, L.A.; Al-Sabbagh, M.; Naik, S.; Krance, R.A.; Tripic, T.; Hiregange, M.; Raghavan, D.; et al. T-Cell Receptor Stimulation Enhances the Expansion and Function of CD19 Chimeric Antigen Receptor–Expressing T Cells. Clin. Cancer. Res. 2019, 25, 7340. [Google Scholar] [CrossRef] [Green Version]
- von Kalle, C.; Deichmann, A.; Schmidt, M. Vector Integration and Tumorigenesis. Hum. Gene Ther. 2014, 25, 475–481. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.L. Activation of T cells: Releasing the brakes by proteolytic elimination of Cbl-b. Sci. Signal. 2009, 2, pe38. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Qin, H.; Yates, B.; Su, L.; Shalabi, H.; Raffeld, M.; Ahlman, M.A.; Stetler-Stevenson, M.; Yuan, C.; Guo, S.; et al. Clonal expansion of CAR T cells harboring lentivector integration in the CBL gene following anti-CD22 CAR T-cell therapy. Blood Adv. 2019, 3, 2317–2322. [Google Scholar] [CrossRef] [PubMed]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nianias, A.; Themeli, M. Induced Pluripotent Stem Cell (iPSC)-Derived Lymphocytes for Adoptive Cell Immunotherapy: Recent Advances and Challenges. Curr. Hematol. Malig. Rep. 2019, 14, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59. [Google Scholar] [CrossRef]
- Zhang, Y.; Wallace, D.L.; de Lara, C.M.; Ghattas, H.; Asquith, B.; Worth, A.; Griffin, G.E.; Taylor, G.P.; Tough, D.F.; Beverley, P.C.; et al. In vivo kinetics of human natural killer cells: The effects of ageing and acute and chronic viral infection. Immunology 2007, 121, 258–265. [Google Scholar] [CrossRef]
- Klingemann, H. Are natural killer cells superior CAR drivers? Oncoimmunology 2014, 3, e28147. [Google Scholar] [CrossRef] [Green Version]
- Oei, V.Y.S.; Siernicka, M.; Graczyk-Jarzynka, A.; Hoel, H.J.; Yang, W.; Palacios, D.; Almåsbak, H.; Bajor, M.; Clement, D.; Brandt, L.; et al. Intrinsic Functional Potential of NK-Cell Subsets Constrains Retargeting Driven by Chimeric Antigen Receptors. Cancer Immunol. Res. 2018, 6, 467–480. [Google Scholar] [CrossRef] [Green Version]
- Ueda, T.; Kumagai, A.; Iriguchi, S.; Yasui, Y.; Miyasaka, T.; Nakagoshi, K.; Nakane, K.; Saito, K.; Takahashi, M.; Sasaki, A.; et al. Non–clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell-derived anti–glypican-3 chimeric antigen receptor-expressing natural killer/innate lymphoid cells. Cancer Sci. 2020, 111, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Badeti, S.; Tseng, H.-c.; Ma, M.T.; Liu, T.; Jiang, J.-G.; Liu, C.; Liu, D. Superior Expansion and Cytotoxicity of Human Primary NK and CAR-NK Cells from Various Sources via Enriched Metabolic Pathways. Mol. Ther. Methods Clin. Dev. 2020, 18, 428–445. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Beckwith, K.A.; Patel, P.R.; Ruella, M.; Zheng, Z.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; McGettigan, S.E.; Cook, D.R.; et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016, 127, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Qasim, W. Allogeneic CAR T cell therapies for leukemia. Am. J. Hematol. 2019, 94, S50–S54. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, R.J.; Prockop, S.; Hasan, A.N.; Koehne, G.; Doubrovina, E. Virus-specific T-cell banks for ‘off the shelf’ adoptive therapy of refractory infections. Bone Marrow Transplant. 2016, 51, 1163–1172. [Google Scholar] [CrossRef] [Green Version]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.C.; Kapoor, V.; Predina, J.; Powell, D.J., Jr.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer. Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.-Z.; Zhu, Y.-Y.; Ruan, M.; Chen, L.; Zhang, Q.-Y. Local Irradiation Sensitized Tumors to Adoptive T Cell Therapy via Enhancing the Cross-Priming, Homing, and Cytotoxicity of Antigen-Specific CD8 T Cells. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Bezu, L.; Gomes-da-Silva, L.C.; Dewitte, H.; Breckpot, K.; Fucikova, J.; Spisek, R.; Galluzzi, L.; Kepp, O.; Kroemer, G. Combinatorial Strategies for the Induction of Immunogenic Cell Death. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef]
- Guedan, S.; Alemany, R. CAR-T Cells and Oncolytic Viruses: Joining Forces to Overcome the Solid Tumor Challenge. Front. Immunol. 2018, 9, 2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Lu, W.; Sun, R.; Jin, X.; Cheng, L.; He, X.; Wang, L.; Yuan, T.; Lyu, C.; Zhao, M. Anti-CD19 Chimeric Antigen Receptor T Cells in Combination with Nivolumab Are Safe and Effective Against Relapsed/Refractory B-Cell Non-hodgkin Lymphoma. Front. Oncol. 2019, 9, 767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odorizzi, P.M.; Pauken, K.E.; Paley, M.A.; Sharpe, A.; Wherry, E.J. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med. 2015, 212, 1125–1137. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janelle, V.; Delisle, J.-S. T-Cell Dysfunction as a Limitation of Adoptive Immunotherapy: Current Concepts and Mitigation Strategies. Cancers 2021, 13, 598. https://doi.org/10.3390/cancers13040598
Janelle V, Delisle J-S. T-Cell Dysfunction as a Limitation of Adoptive Immunotherapy: Current Concepts and Mitigation Strategies. Cancers. 2021; 13(4):598. https://doi.org/10.3390/cancers13040598
Chicago/Turabian StyleJanelle, Valérie, and Jean-Sébastien Delisle. 2021. "T-Cell Dysfunction as a Limitation of Adoptive Immunotherapy: Current Concepts and Mitigation Strategies" Cancers 13, no. 4: 598. https://doi.org/10.3390/cancers13040598
APA StyleJanelle, V., & Delisle, J. -S. (2021). T-Cell Dysfunction as a Limitation of Adoptive Immunotherapy: Current Concepts and Mitigation Strategies. Cancers, 13(4), 598. https://doi.org/10.3390/cancers13040598