
Neuroblastoma Invasion Strategies Are Regulated by the Extracellular Matrix

 ,
,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Cell Invasion Strategies of NB Organoids in ECM-Mimicking Matrices
2.2. Invasion of NB Organoids Depends on the PDX of Origin and ECM Composition
2.3. Invasion, Growth and Viability of NB Organoids Depend on Soluble Factors
2.4. Neuroblastoma Cell Lines Display ECM-Independent Invasion Strategies
2.5. Expression Levels of MYCN Are Associated with Aggressive NB Behaviour
2.6. The In Vivo Tumour Microenvironment Fosters a Distinct Invasion Strategy in NB Cells
3. Discussion
3.1. NB Organoids Isolated from PDXs Are Phenotypically Heterogenous during Local ECM Invasion
3.2. The Phenotypic Heterogeneity Seen in PDX Organoids Was Not Recapitulated in These Cell Lines
3.3. Matrigel Is the Preferred Substratum for NB Invasion
3.4. Local Invasion of NB Cells Is Dependent on the Sample of Origin
3.5. Repression of MYCN Transcription Promotes Less Aggressive Cell Behaviour
3.6. Benefits of 3D NB Cell Assays over Current Models of Local Invasion
4. Materials and Methods
4.1. NB Cell Lines and Culture Conditions
4.2. Patient-Derived Xenograft Generation and Harvesting
4.3. Isolation of Neuroblastoma PDX Organoids
4.4. Neuroblastoma Organoid Culture
4.5. Time-Lapse Differential Interference (DIC) Contrast Microscopy
4.6. Quantification of Organoid Growth and Invasion
4.7. Fluorescence Staining of 3D Hydrogels
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Steliarova-Foucher, E.; Colombet, M.; Ries, L.A.G.; Moreno, F.; Dolya, A.; Bray, F.; Hesseling, P.; Shin, H.Y.; Stiller, C.A.; IICC-3 Contributors. International incidence of childhood cancer, 2001–2010: A population-based registry study. Lancet Oncol. 2017, 18, 719–731. [Google Scholar] [CrossRef]
- Force, L.M.; Abdollahpour, I.; Advani, S.M.; Agius, D.; Ahmadian, E.; Alahdab, F.; Alam, T.; Alebel, A.; Alipour, V.; Allen, C.A.; et al. The global burden of childhood and adolescent cancer in 2017: An analysis of the Global Burden of Disease Study 2017. Lancet Oncol. 2019, 20, 1211–1225. [Google Scholar] [CrossRef] [Green Version]
- Smith, V.; Foster, J. High-Risk Neuroblastoma Treatment Review. Children 2018, 5, 114. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, A.E.; Danner-Koptik, K.; Golden, S.; Schneiderman, J.; Kletzel, M.; Reichek, J.; Gosiengfiao, Y. Late Effects in Pediatric High-risk Neuroblastoma Survivors After Intensive Induction Chemotherapy Followed by Myeloablative Consolidation Chemotherapy and Triple Autologous Stem Cell Transplants. J. Pediatr. Hematol. 2018, 40, 31–35. [Google Scholar] [CrossRef]
- Nolan, J.; Frawley, T.; Tighe, J.; Soh, H.; Curtin, C.; Piskareva, O. Preclinical models for neuroblastoma: Advances and challenges. Cancer Lett. 2020, 474, 53–62. [Google Scholar] [CrossRef]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta BBA Bioenerg. 2010, 1805, 105–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gengenbacher, N.; Singhal, M.; Augustin, H.G. Preclinical mouse solid tumour models: Status quo, challenges and perspectives. Nat. Rev. Cancer 2017, 17, 751–765. [Google Scholar] [CrossRef]
- Kamili, A.; Atkinson, C.; Trahair, T.N.; Fletcher, J.I. Mouse models of high-risk neuroblastoma. Cancer Metastasis Rev. 2020, 39, 261–274. [Google Scholar] [CrossRef]
- Braekeveldt, N.; Bexell, D. Patient-derived xenografts as preclinical neuroblastoma models. Cell Tissue Res. 2018, 372, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.M.; Lin, T.; Zolkind, P.; Barnell, E.K.; Skidmore, Z.L.; Winkler, A.E.; Law, J.H.; Mardis, E.R.; Wartman, L.D.; Adkins, D.R.; et al. Oral Cavity Squamous Cell Carcinoma Xenografts Retain Complex Genotypes and Intertumor Molecular Heterogeneity. Cell Rep. 2018, 24, 2167–2178. [Google Scholar] [CrossRef] [Green Version]
- Braekeveldt, N.; Wigerup, C.; Gisselsson, D.; Mohlin, S.; Merselius, M.; Beckman, S.; Jonson, T.; Börjesson, A.; Backman, T.; Tadeo, I.; et al. Neuroblastoma patient-derived orthotopic xenografts retain metastatic patterns and geno- and phenotypes of patient tumours. Int. J. Cancer 2014, 136, E252–E261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braekeveldt, N.; Wigerup, C.; Tadeo, I.; Beckman, S.; Sandén, C.; Jönsson, J.; Erjefält, J.S.; Berbegall, A.P.; Börjesson, A.; Backman, T.; et al. Neuroblastoma patient-derived orthotopic xenografts reflect the microenvironmental hallmarks of aggressive patient tumours. Cancer Lett. 2016, 375, 384–389. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Liu, Z.; Yu, L.; Zhang, Y.; Baxter, P.; Voicu, H.; Gurusiddappa, S.; Luan, J.; Su, J.M.; Leung, H.-C.E.; et al. Global gene expression profiling confirms the molecular fidelity of primary tumor-based orthotopic xenograft mouse models of medulloblastoma. Neuro-Oncology 2012, 14, 574–583. [Google Scholar] [CrossRef] [Green Version]
- Rokita, J.L.; Rathi, K.S.; Cardenas, M.F.; Upton, K.A.; Jayaseelan, J.; Cross, K.L.; Pfeil, J.; Egolf, L.E.; Way, G.P.; Farrel, A.; et al. Genomic Profiling of Childhood Tumor Patient-Derived Xenograft Models to Enable Rational Clinical Trial Design. Cell Rep. 2019, 29, 1675–1689.e9. [Google Scholar] [CrossRef] [PubMed]
- Tomás-Bort, E.; Kieler, M.; Sharma, S.; Candido, J.B.; Loessner, D. 3D approaches to model the tumor microenvironment of pancreatic cancer. Theranostics 2020, 10, 5074–5089. [Google Scholar] [CrossRef] [PubMed]
- Ilina, O.; Campanello, L.; Gritsenko, P.G.; Vullings, M.; Wang, C.; Bult, P.; Losert, W.; Friedl, P. Intravital microscopy of collective invasion plasticity in breast cancer. Dis. Model. Mech. 2018, 11, dmm034330. [Google Scholar] [CrossRef] [Green Version]
- Wolf, K.; Wu, Y.I.; Liu, Y.; Geiger, J.; Tam, E.; Overall, C.M.; Stack, M.S.; Friedl, P. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat. Cell Biol. 2007, 9, 893–904. [Google Scholar] [CrossRef]
- Khalil, A.A.; Ilina, O.; Vasaturo, A.; Venhuizen, J.-H.; Vullings, M.; Venhuizen, V.; Bilos, A.; Figdor, C.G.; Span, P.N.; Friedl, P. Collective invasion induced by an autocrine purinergic loop through connexin-43 hemichannels. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef]
- Alexander, S.; Weigelin, B.; Winkler, F.; Friedl, P. Preclinical intravital microscopy of the tumour-stroma interface: Invasion, metastasis, and therapy response. Curr. Opin. Cell Biol. 2013, 25, 659–671. [Google Scholar] [CrossRef]
- Dwane, S.; Durack, E.; Kiely, P.A. Optimising parameters for the differentiation of SH-SY5Y cells to study cell adhesion and cell migration. BMC Res. Notes 2013, 6, 366. [Google Scholar] [CrossRef] [Green Version]
- Gritsenko, P.G.; Atlasy, N.; Dieteren, C.E.J.; Navis, A.C.; Venhuizen, J.-H.; Veelken, C.; Schubert, D.; Acker-Palmer, A.; Westerman, B.A.; Wurdinger, T.; et al. p120-catenin-dependent collective brain infiltration by glioma cell networks. Nat. Cell Biol. 2020, 22, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Serres, E.; Debarbieux, F.; Stanchi, F.; Maggiorella, L.; Grall, D.; Turchi, L.; Burelvandenbos, F.; Figarellabranger, D.; Virolle, T.; Rougon, G.; et al. Fibronectin expression in glioblastomas promotes cell cohesion, collective invasion of basement membrane in vitro and orthotopic tumor growth in mice. Oncogene 2014, 33, 3451–3462. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Gilmour, D. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2009, 10, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Ngoc, K.-V.; Shamir, E.R.; Huebner, R.J.; Beck, J.N.; Cheung, K.J.; Ewald, A. 3D Culture Assays of Murine Mammary Branching Morphogenesis and Epithelial Invasion. In Tissue Morphogenesis: Methods and Protocols; Humana Press: New York, NY, USA, 2014; ISBN 9781493911646. [Google Scholar]
- Zheng, Y.; Xue, X.; Resto-Irizarry, A.M.; Li, Z.; Shao, Y.; Zhao, G.; Fu, J. Dorsal-ventral patterned neural cyst from human pluripotent stem cells in a neurogenic niche. Sci. Adv. 2019, 5, eaax5933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbot, N.; Caperna, T.J. Proteome array identification of bioactive soluble proteins/peptides in Matrigel: Relevance to stem cell responses. Cytotechnology 2014, 67, 873–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciences Center Texas Tech University Health Childhood Cancer Repository. Available online: http://www.cccells.org/ (accessed on 5 January 2021).
- Nguyen, T.H.; Koneru, B.; Wei, S.-J.; Chen, W.H.; Makena, M.R.; Urias, E.; Kang, M.H.; Reynolds, C.P. Fenretinide via NOXA Induction, Enhanced Activity of the BCL-2 Inhibitor Venetoclax in High BCL-2–Expressing Neuroblastoma Preclinical Models. Mol. Cancer Ther. 2019, 18, 2270–2282. [Google Scholar] [CrossRef] [Green Version]
- Krytska, K.; Ryles, H.T.; Sano, R.; Raman, P.; Infarinato, N.R.; Hansel, T.D.; Makena, M.R.; Song, M.M.; Reynolds, C.P.; Mossé, Y.P. Crizotinib Synergizes with Chemotherapy in Preclinical Models of Neuroblastoma. Clin. Cancer Res. 2016, 22, 948–960. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef]
- Lutz, W.; Stöhr, M.; Schürmann, J.; Wenzel, A.; Löhr, A.; Schwab, M. Conditional expression of N-myc in human neuroblastoma cells increases expression of alpha-prothymosin and ornithine decarboxylase and accelerates progression into S-phase early after mitogenic stimulation of quiescent cells. Oncogene 1996, 13, 803–812. [Google Scholar]
- Khoja, L.; Shenjere, P.; Hodgson, C.; Hodgetts, J.; Clack, G.; Hughes, A.; Lorigan, P.; Dive, C. Prevalence and heterogeneity of circulating tumour cells in metastatic cutaneous melanoma. Melanoma Res. 2014, 24, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Zhang, B.; Zheng, Y.; Zheng, C.; Liu, B.; Zheng, S.; Dong, K.; Dong, R. Circulating tumor cells detection in neuroblastoma patients by EpCAM-independent enrichment and immunostaining-fluorescence in situ hybridization. EBioMedicine 2018, 35, 244–250. [Google Scholar] [CrossRef] [Green Version]
- Marler, K.J.M.; Kozma, R.; Ahmed, S.; Dong, J.-M.; Hall, C.; Lim, L. Outgrowth of Neurites from NIE-115 Neuroblastoma Cells Is Prevented on Repulsive Substrates through the Action of PAK. Mol. Cell. Biol. 2005, 25, 5226–5241. [Google Scholar] [CrossRef] [Green Version]
- Fayzullin, A.; Tuvnes, F.A.; Skjellegrind, H.K.; Behnan, J.; Mughal, A.A.; Langmoen, I.A.; Vik-Mo, E.O. Time-lapse phenotyping of invasive glioma cells ex vivo reveals subtype-specific movement patterns guided by tumor core signaling. Exp. Cell Res. 2016, 349, 199–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elisha, Y.; Kalchenko, V.; Kuznetsov, Y.; Geiger, B. Dual role of E-cadherin in the regulation of invasive collective migration of mammary carcinoma cells. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brabletz, T. To differentiate or not—Routes towards metastasis. Nat. Rev. Cancer 2012, 12, 425–436. [Google Scholar] [CrossRef]
- Kim, H.; Lin, Q.; Glazer, P.M.; Yun, Z. The hypoxic tumor microenvironment in vivo selects the cancer stem cell fate of breast cancer cells. Breast Cancer Res. 2018, 20, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.M.; Shaw, P.A.; Gedye, C.; Bernardini, M.Q.; Neel, B.G.; Ailles, L.E. Phenotypic heterogeneity and instability of human ovarian tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2011, 108, 6468–6473. [Google Scholar] [CrossRef] [Green Version]
- Padmanaban, V.; Grasset, E.M.; Neumann, N.M.; Fraser, A.K.; Henriet, E.; Matsui, W.; Tran, P.T.; Cheung, K.J.; Georgess, D.; Ewald, A. Organotypic culture assays for murine and human primary and metastatic-site tumors. Nat. Protoc. 2020, 15, 2413–2442. [Google Scholar] [CrossRef]
- Nguyen-Ngoc, K.-V.; Cheung, K.J.; Brenot, A.; Shamir, E.R.; Gray, R.S.; Hines, W.C.; Yaswen, P.; Werb, Z.; Ewald, A. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc. Natl. Acad. Sci. USA 2012, 109, E2595–E2604. [Google Scholar] [CrossRef] [Green Version]
- Puls, T.J.; Tan, X.; Whittington, C.F.; Voytik-Harbin, S.L. 3D collagen fibrillar microstructure guides pancreatic cancer cell phenotype and serves as a critical design parameter for phenotypic models of EMT. PLoS ONE 2017, 12, e0188870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vellinga, T.T.; den Uil, S.; Rinkes, I.H.; Marvin, D.; Ponsioen, B.; Alvarez-Varela, A.; Fatrai, S.; Scheele, C.; Zwijnenburg, D.A.; Snippert, H.; et al. Collagen-rich stroma in aggressive colon tumors induces mesenchymal gene expression and tumor cell invasion. Oncogene 2016, 35, 5263–5271. [Google Scholar] [CrossRef] [PubMed]
- Perris, R.; Perissinotto, D. Role of the extracellular matrix during neural crest cell migration. Mech. Dev. 2000, 95, 3–21. [Google Scholar] [CrossRef]
- Bilozur, M.E.; Hay, E.D. Neural crest migration in 3D extracellular matrix utilizes laminin, fibronectin, or collagen. Dev. Biol. 1988, 125, 19–33. [Google Scholar] [CrossRef]
- Tosney, K.W.; Dehnbostel, D.B.; Erickson, C.A. Neural Crest Cells Prefer the Myotome’s Basal Lamina over the Sclerotome as a Substratum. Dev. Biol. 1994, 163, 389–406. [Google Scholar] [CrossRef]
- Flanagan, L.A.; Rebaza, L.M.; Derzic, S.; Schwartz, P.H.; Monuki, E.S. Regulation of human neural precursor cells by laminin and integrins. J. Neurosci. Res. 2006, 83, 845–856. [Google Scholar] [CrossRef] [Green Version]
- Campbell, K.; Gastier-Foster, J.M.; Mann, M.; Naranjo, A.H.; Ms, C.V.R.; Bagatell, R.; Matthay, K.K.; London, W.B.; Irwin, M.S.; Shimada, H.; et al. Association ofMYCNcopy number with clinical features, tumor biology, and outcomes in neuroblastoma: A report from the Children’s Oncology Group. Cancer 2017, 123, 4224–4235. [Google Scholar] [CrossRef] [Green Version]
- Goodman, L.A.; Liu, B.C.S.; Thiele, C.J.; Schmidt, M.L.; Cohn, S.L.; Yamashiro, J.M.; Pai, D.S.M.; Ikegaki, N.; Wada, R.K. Modulation of N-myc expression alters the invasiveness of neuroblastoma. Clin. Exp. Metastasis 1997, 15, 130–139. [Google Scholar] [CrossRef]
- Selmi, A.; De Saint-Jean, M.; Jallas, A.-C.; Garin, E.; Hogarty, M.D.; Bénard, J.; Puisieux, A.; Marabelle, A.; Valsesia-Wittmann, S. TWIST1 is a direct transcriptional target of MYCN and MYC in neuroblastoma. Cancer Lett. 2015, 357, 412–418. [Google Scholar] [CrossRef]
- Planellsferrer, L.; Urresti, J.; Soriano, A.; Reix, S.; Murphy, D.M.; Ferreres, J.C.; Borras, F.E.; Gallego, S.M.; Stallings, R.L.; Moubarak, R.S.; et al. MYCN repression of Lifeguard/FAIM2 enhances neuroblastoma aggressiveness. Cell Death Dis. 2014, 5, e1401. [Google Scholar] [CrossRef] [Green Version]
- Stewart, E.; Shelat, A.; Bradley, C.; Chen, X.; Federico, S.; Thiagarajan, S.; Shirinifard, A.; Bahrami, A.; Pappo, A.; Qu, C.; et al. Development and characterization of a human orthotopic neuroblastoma xenograft. Dev. Biol. 2015, 407, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Shankar, V.; Hori, H.; Kihira, K.; Lei, Q.; Toyoda, H.; Iwamoto, S.; Komada, Y. Mesenchymal Stromal Cell Secretome Up-Regulates 47 kDa CXCR4 Expression, and Induce Invasiveness in Neuroblastoma Cell Lines. PLoS ONE 2015, 10, e0120069. [Google Scholar] [CrossRef]
- Piskareva, O.; Harvey, H.; Nolan, J.P.; Conlon, R.; Alcock, L.; Buckley, P.; Dowling, P.; Henry, M.D.; O’Sullivan, F.; Bray, I.; et al. The development of cisplatin resistance in neuroblastoma is accompanied by epithelial to mesenchymal transition in vitro. Cancer Lett. 2015, 364, 142–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraley, S.I.; Feng, Y.; Krishnamurthy, R.; Kim, D.-H.; Celedon, A.; Longmore, G.D.; Wirtz, D. A distinctive role for focal adhesion proteins in three-dimensional cell motility. Nat. Cell Biol. 2010, 12, 598–604. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.S.; Hughes-Alford, S.K.; Kay, J.E.; Castillo, A.; Wells, A.; Gertler, F.B.; Lauffenburger, D.A. 2D protrusion but not motility predicts growth factor–induced cancer cell migration in 3D collagen. J. Cell Biol. 2012, 197, 721–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtin, C.; Nolan, J.; Conlon, R.; Deneweth, L.; Gallagher, C.; Tan, Y.; Cavanagh, B.; Asraf, A.; Harvey, H.; Miller-Delaney, S.; et al. A physiologically relevant 3D collagen-based scaffold–neuroblastoma cell system exhibits chemosensitivity similar to orthotopic xenograft models. Acta Biomater. 2018, 70, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Fayzullin, A.; Sandberg, C.J.; Spreadbury, M.; Saberniak, B.M.; Grieg, Z.; Skaga, E.; Langmoen, I.A.; Vik-Mo, E.O. Phenotypic and Expressional Heterogeneity in the Invasive Glioma Cells. Transl. Oncol. 2019, 12, 122–133. [Google Scholar] [CrossRef]
- Szabó, A.; Melchionda, M.; Nastasi, G.; Woods, M.L.; Campo, S.; Perris, R.; Mayor, R. In vivo confinement promotes collective migration of neural crest cells. J. Cell Biol. 2016, 213, 543–555. [Google Scholar] [CrossRef]
- McLennan, R.; Schumacher, L.J.; Morrison, J.A.; Teddy, J.M.; Ridenour, D.A.; Box, A.C.; Semerad, C.L.; Li, H.; McDowell, W.; Kay, D.; et al. Neural crest migration is driven by a few trailblazer cells with a unique molecular signature narrowly confined to the invasive front. Development 2015, 142, 2014–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, J.; Gauert, A.; Montecinos, L.B.; Fanlo, L.; Alhashem, Z.M.; Assar, R.; Martí, E.; Kabla, A.; Härtel, S.; Linker, C. Leader Cells Define Directionality of Trunk, but Not Cranial, Neural Crest Cell Migration. Cell Rep. 2016, 15, 2076–2088. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.A.; Yamada, K.M. Contact stimulation of cell migration. J. Cell Sci. 1992, 103, 1211–1214. [Google Scholar]
- Mitchell, C.B.; O’Neill, G.M. Rac GTPase regulation of 3D invasion in neuroblastomas lacking MYCN amplification. Cell Adhes. Migr. 2016, 11, 68–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, C.S.; Postovit, L.M.; Lajoie, G.A. Matrigel: A complex protein mixture required for optimal growth of cell culture. Proteomics 2010, 10, 1886–1890. [Google Scholar] [CrossRef]
- Attieh, Y.; Vignjevic, D.M. The hallmarks of CAFs in cancer invasion. Eur. J. Cell Biol. 2016, 95, 493–502. [Google Scholar] [CrossRef]
- Hanley, C.J.; Henriet, E.; Sirka, O.K.; Thomas, G.; Ewald, A. Tumor-Resident Stromal Cells Promote Breast Cancer Invasion through Regulation of the Basal Phenotype. Mol. Cancer Res. 2020, 18, 1615–1622. [Google Scholar] [CrossRef]
- Neri, S.; Ishii, G.; Hashimoto, H.; Kuwata, T.; Nagai, K.; Date, H.; Ochiai, A. Podoplanin-expressing cancer-associated fibroblasts lead and enhance the local invasion of cancer cells in lung adenocarcinoma. Int. J. Cancer 2015, 137, 784–796. [Google Scholar] [CrossRef]
- Izar, B.; Joyce, C.E.; Goff, S.; Cho, N.L.; Shah, P.M.; Sharma, G.; Li, J.; Ibrahim, N.; Gold, J.; Hodi, F.S.; et al. Bidirectional cross talk between patient-derived melanoma and cancer-associated fibroblasts promotes invasion and proliferation. Pigment. Cell Melanoma Res. 2016, 29, 656–668. [Google Scholar] [CrossRef]
- Puls, T.J.; Tan, X.; Husain, M.; Whittington, C.F.; Fishel, M.L.; Voytik-Harbin, S.L. Development of a Novel 3D Tumor-tissue Invasion Model for High-throughput, High-content Phenotypic Drug Screening. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Smith, H.A.; Kang, Y. The metastasis-promoting roles of tumor-associated immune cells. J. Mol. Med. 2013, 91, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Liu, Q.; Dou, R.; Xiong, B. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer 2019, 18, 64. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, A.R.; Ellies, L.G.; Holme, A.L.; Pixley, F.J. A three-dimensional co-culture system to investigate macrophage-dependent tumor cell invasion. J. Biol. Methods 2016, 3, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
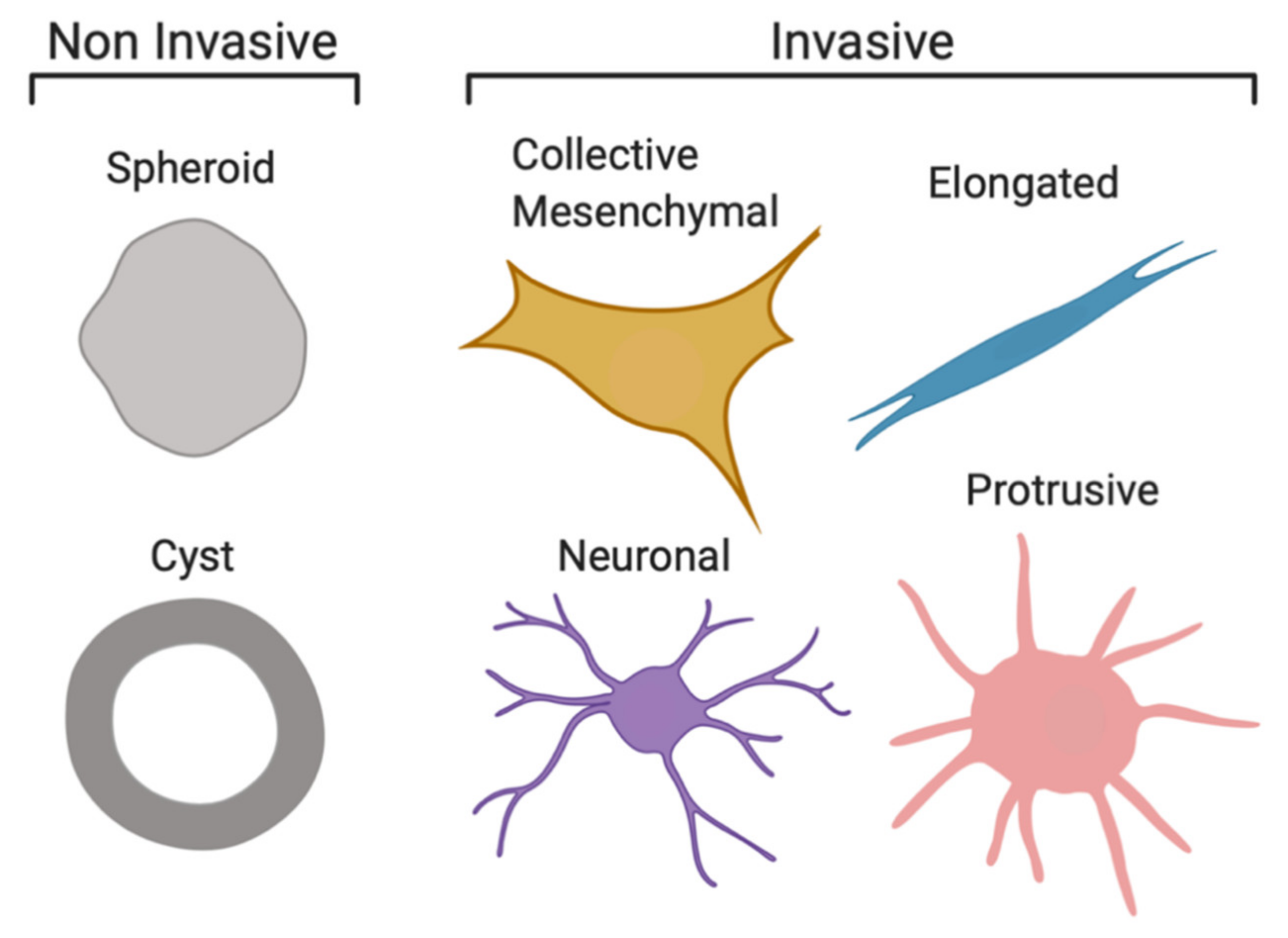{kind=link}
| PDX | Risk Classification | Phase of Therapy | Sample Type | Stage | MYCN Status | ALK Status | TERT mRNA | Ref. |
|---|---|---|---|---|---|---|---|---|
| COG-N-424x | High | DX | Tumour | 4 | Amp | WT | ++ | [15,28] |
| COG-N-573x | High | DX | BM | 4 | Amp | WT | ++ | [28] |
| COG-N-603x | High | DX | Tumour | 4 | Amp | WT | +++ | [15,28,29] |
| Felix-PDX | High | PD-PM | Blood | 4 | Non-Amp | ALK F1245C | ++ | [15,28,30] |
| Cell Line/Organoid | Time | (Hours) |
|---|---|---|
| Matrigel | Collagen | |
| SHEP-21N(MYCN-On/Off) | <24 | 24–48 |
| LAN-1 | <24 | 48–72 |
| SH-SY5Y | 24–48 | 48–72 |
| SH-SY5Y Organoids | <24 | 48–72 |
| COG-N-573x | <48 | <12 * |
| COG-N-603x | <48 | <12 * |
| COG-N-Felix | <48 | >120 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gavin, C.; Geerts, N.; Cavanagh, B.; Haynes, M.; Reynolds, C.P.; Loessner, D.; Ewald, A.J.; Piskareva, O. Neuroblastoma Invasion Strategies Are Regulated by the Extracellular Matrix. Cancers 2021, 13, 736. https://doi.org/10.3390/cancers13040736
Gavin C, Geerts N, Cavanagh B, Haynes M, Reynolds CP, Loessner D, Ewald AJ, Piskareva O. Neuroblastoma Invasion Strategies Are Regulated by the Extracellular Matrix. Cancers. 2021; 13(4):736. https://doi.org/10.3390/cancers13040736
Chicago/Turabian StyleGavin, Cian, Nele Geerts, Brenton Cavanagh, Meagan Haynes, C. Patrick Reynolds, Daniela Loessner, Andrew J. Ewald, and Olga Piskareva. 2021. "Neuroblastoma Invasion Strategies Are Regulated by the Extracellular Matrix" Cancers 13, no. 4: 736. https://doi.org/10.3390/cancers13040736
APA StyleGavin, C., Geerts, N., Cavanagh, B., Haynes, M., Reynolds, C. P., Loessner, D., Ewald, A. J., & Piskareva, O. (2021). Neuroblastoma Invasion Strategies Are Regulated by the Extracellular Matrix. Cancers, 13(4), 736. https://doi.org/10.3390/cancers13040736