CD44 Targeted Nanomaterials for Treatment of Triple-Negative Breast Cancer

 , ,
, ,  , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
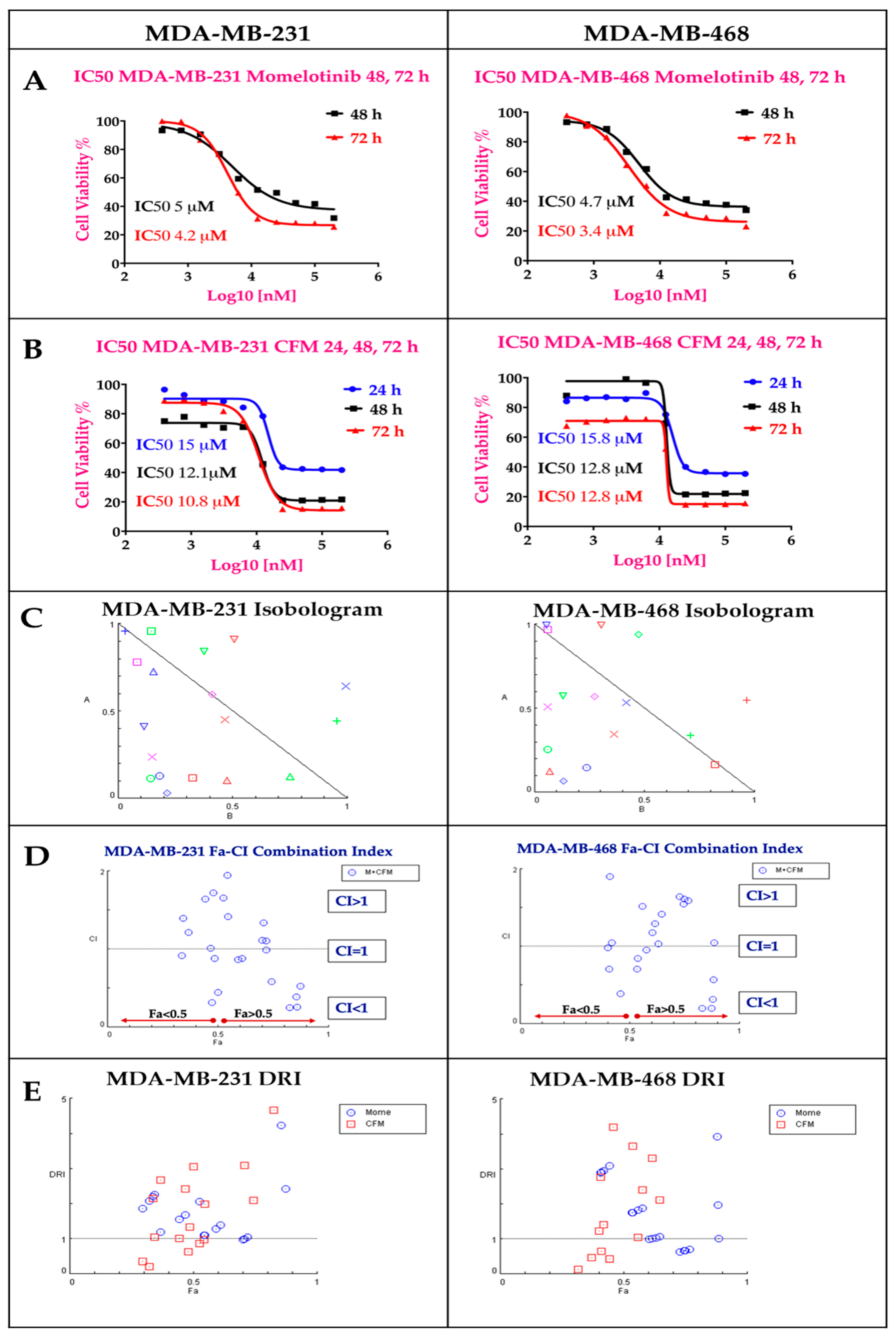
2.1. The Cytotoxicity of The Individual Drugs and Their Combinations Studies
2.2. Polymeric Nanoparticles (PNPs) Formulations
2.2.1. Synthesis of the Non-Targeted SMA-TPGS Carrier and Targeted HA-SMA-TPGS-Carrier
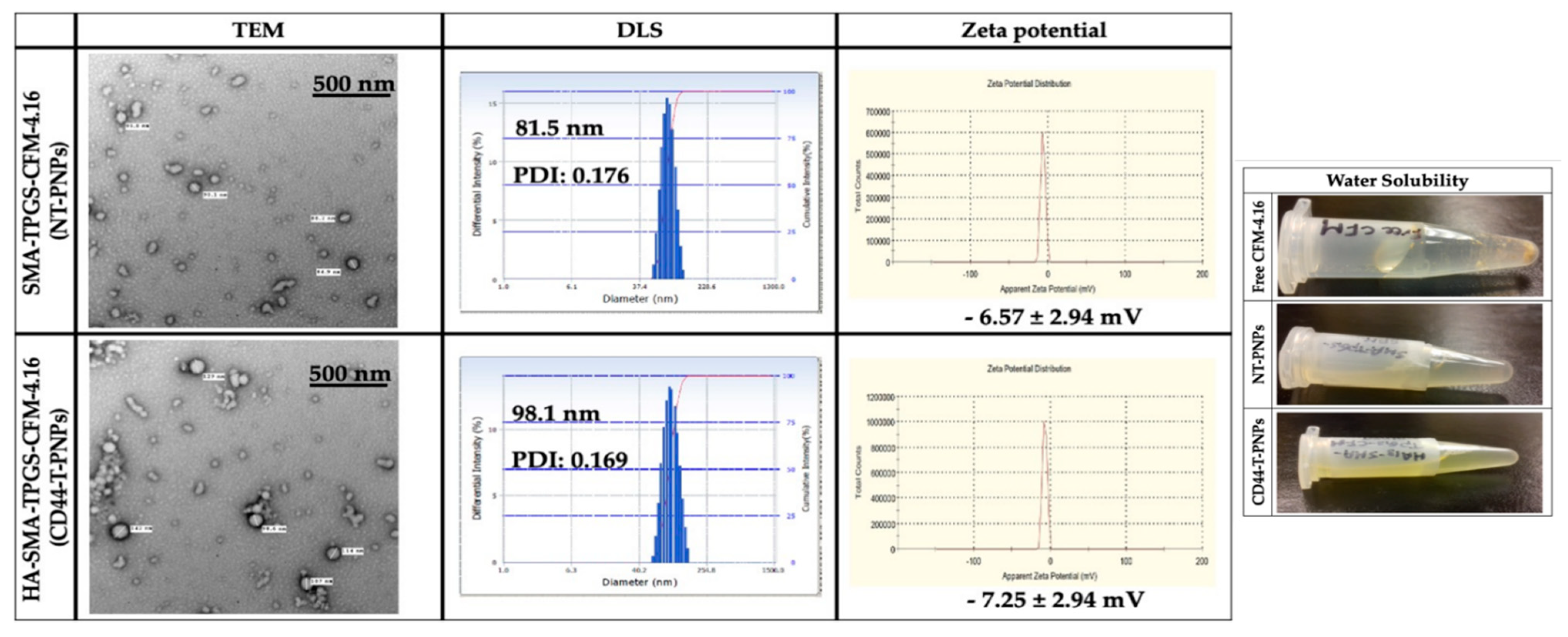
2.2.2. Preparation and Characterization of CFM-4.16 Loaded Polymeric Nanoparticles (PNPs)
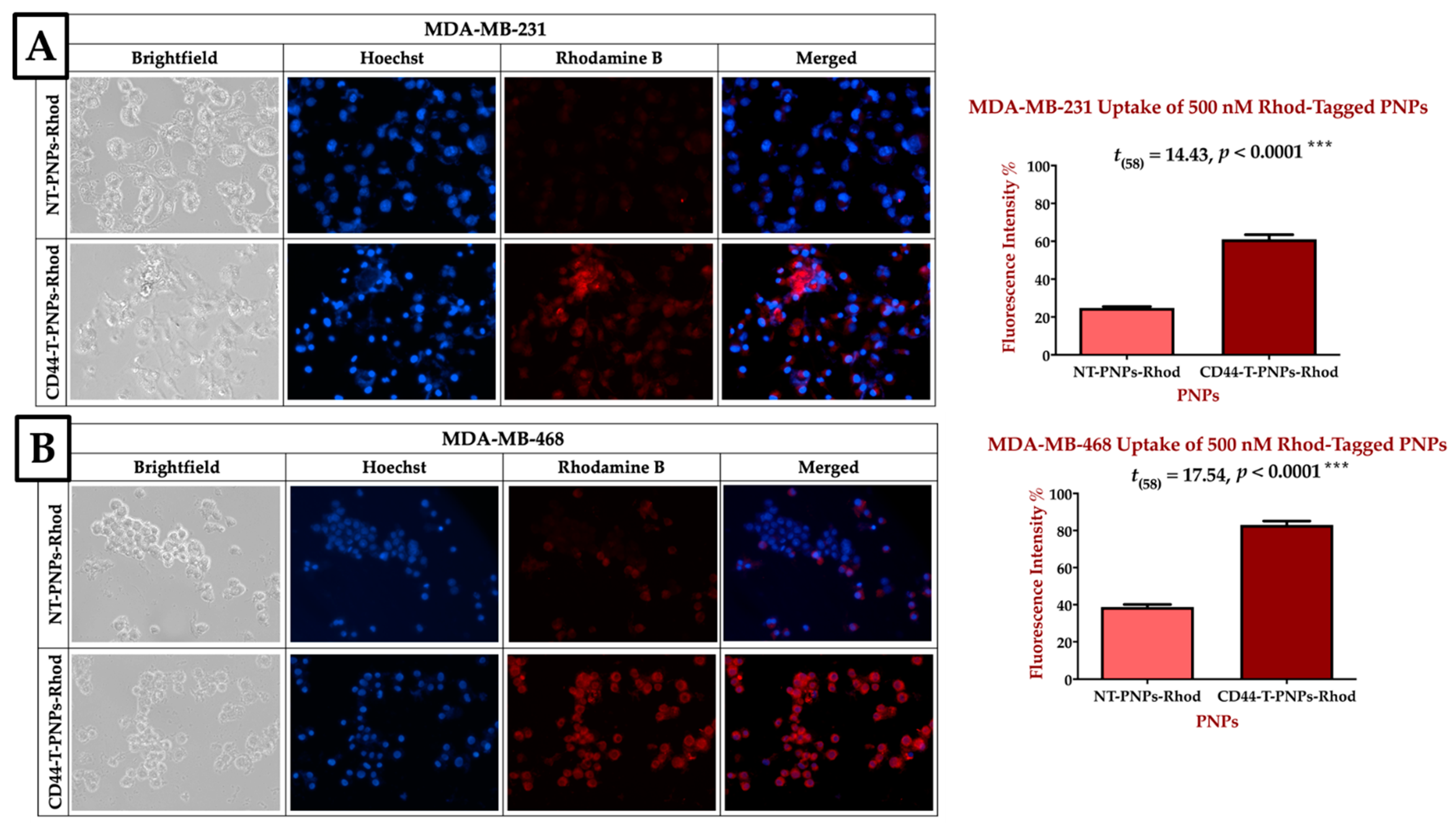
2.2.3. CD44 Targeted Polymeric NPs Mediates Cellular Uptake via CD-44 Overexpression on TNBC Cell Lines
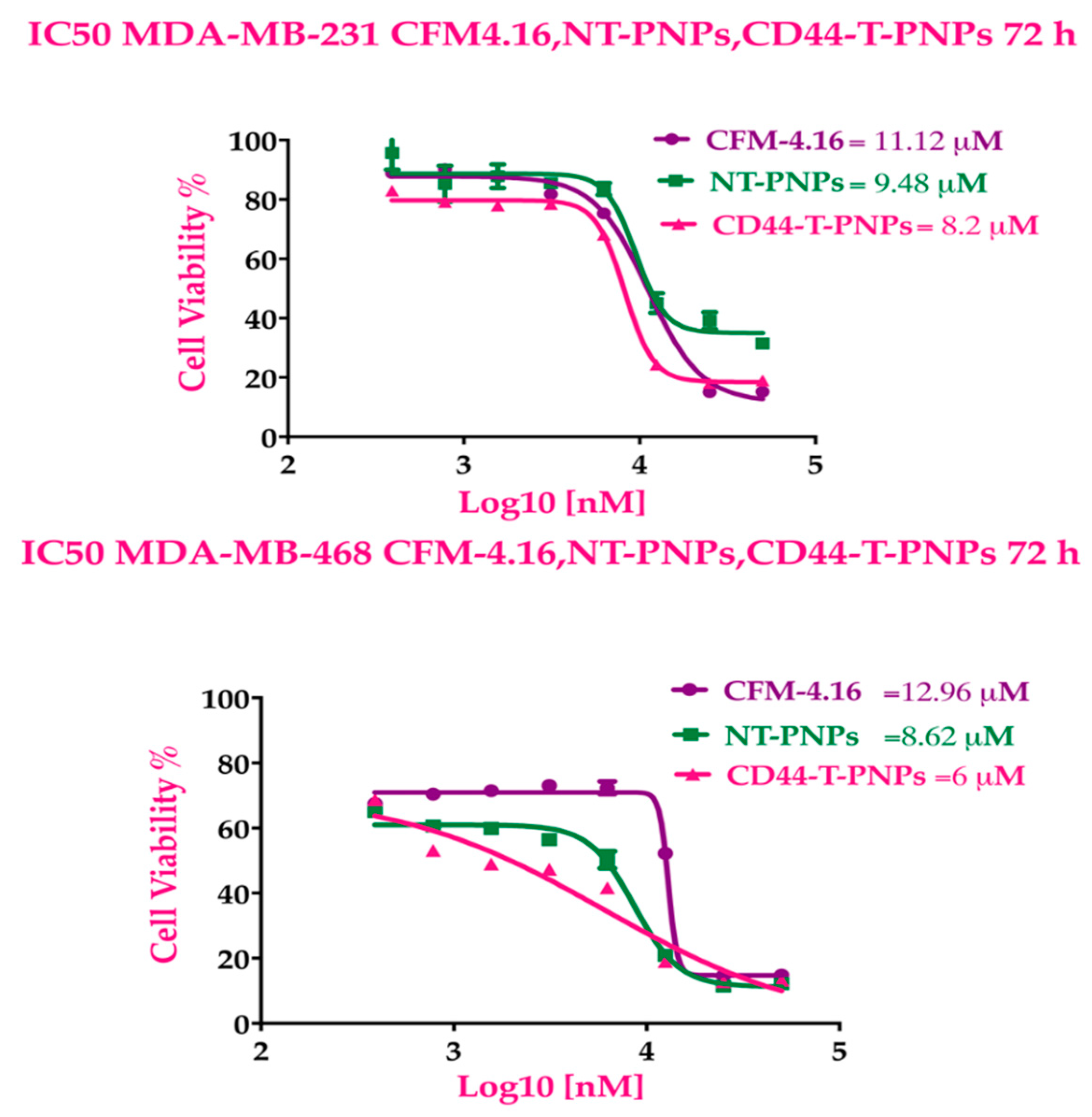
2.2.4. CD44-Targeted PNPs Increases Cytotoxicity Against TNBC Cell Lines
2.3. Momelotinib + CFM-4.16 Combination Studies and the Cause of Synergism
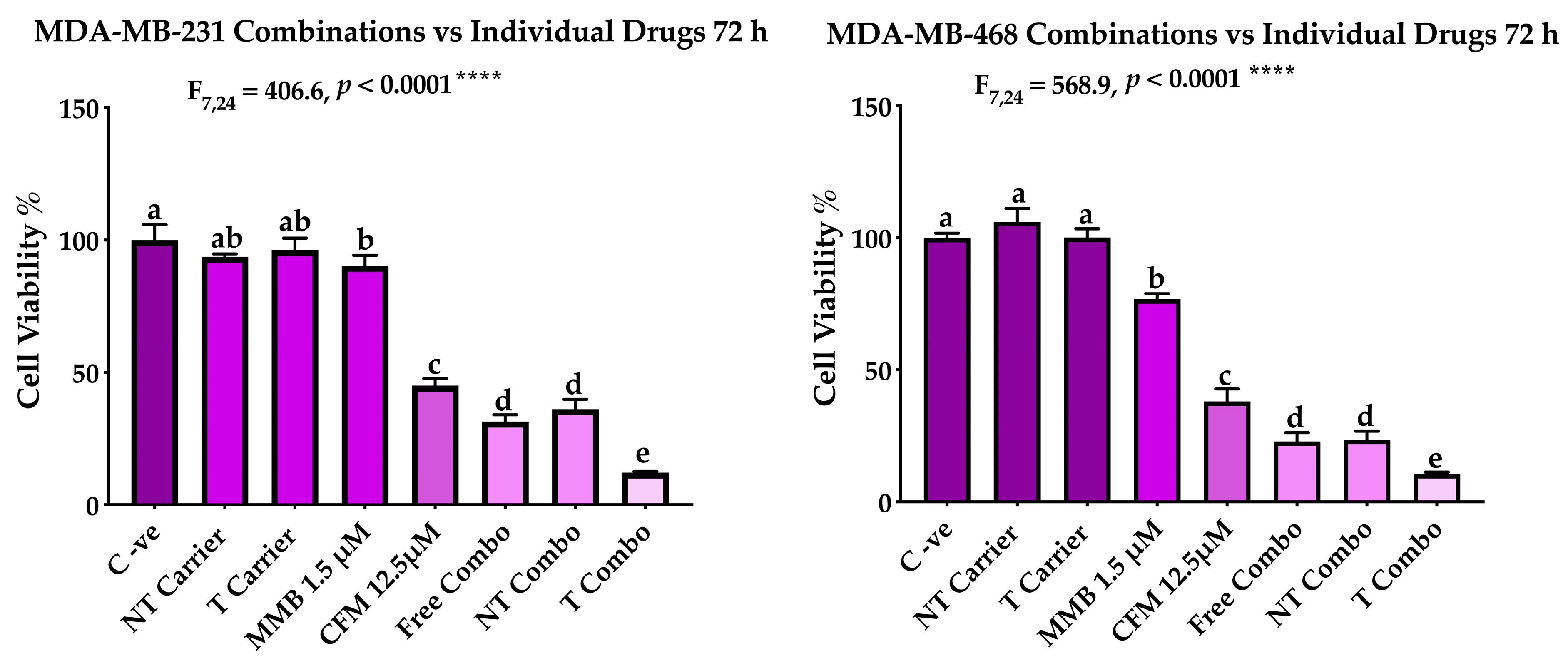
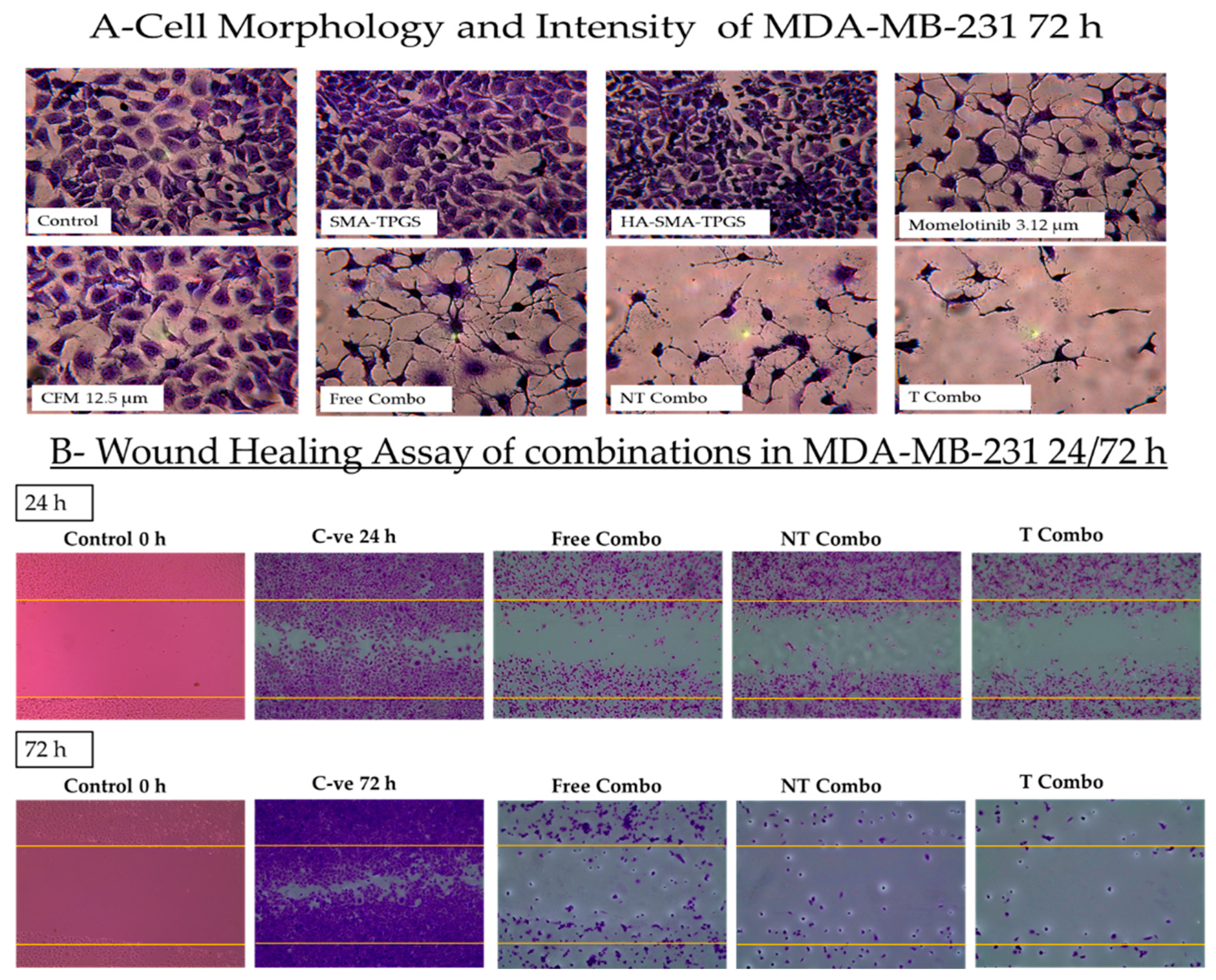
2.3.1. Targeted PNPs Combination Has Exhibited Remarkable Anticancer Activity Compared to Free Drugs Against TNBC Cell Lines
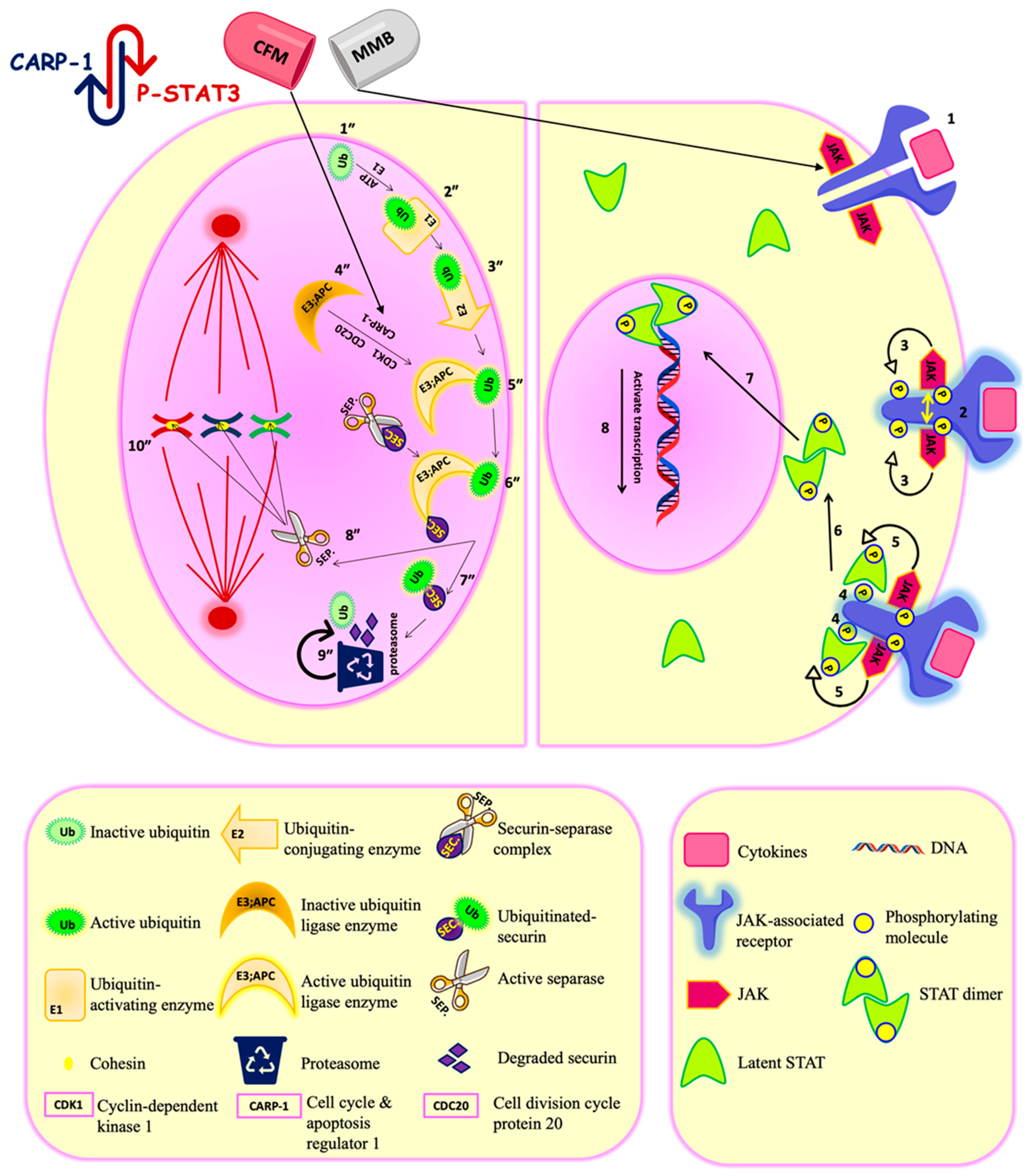
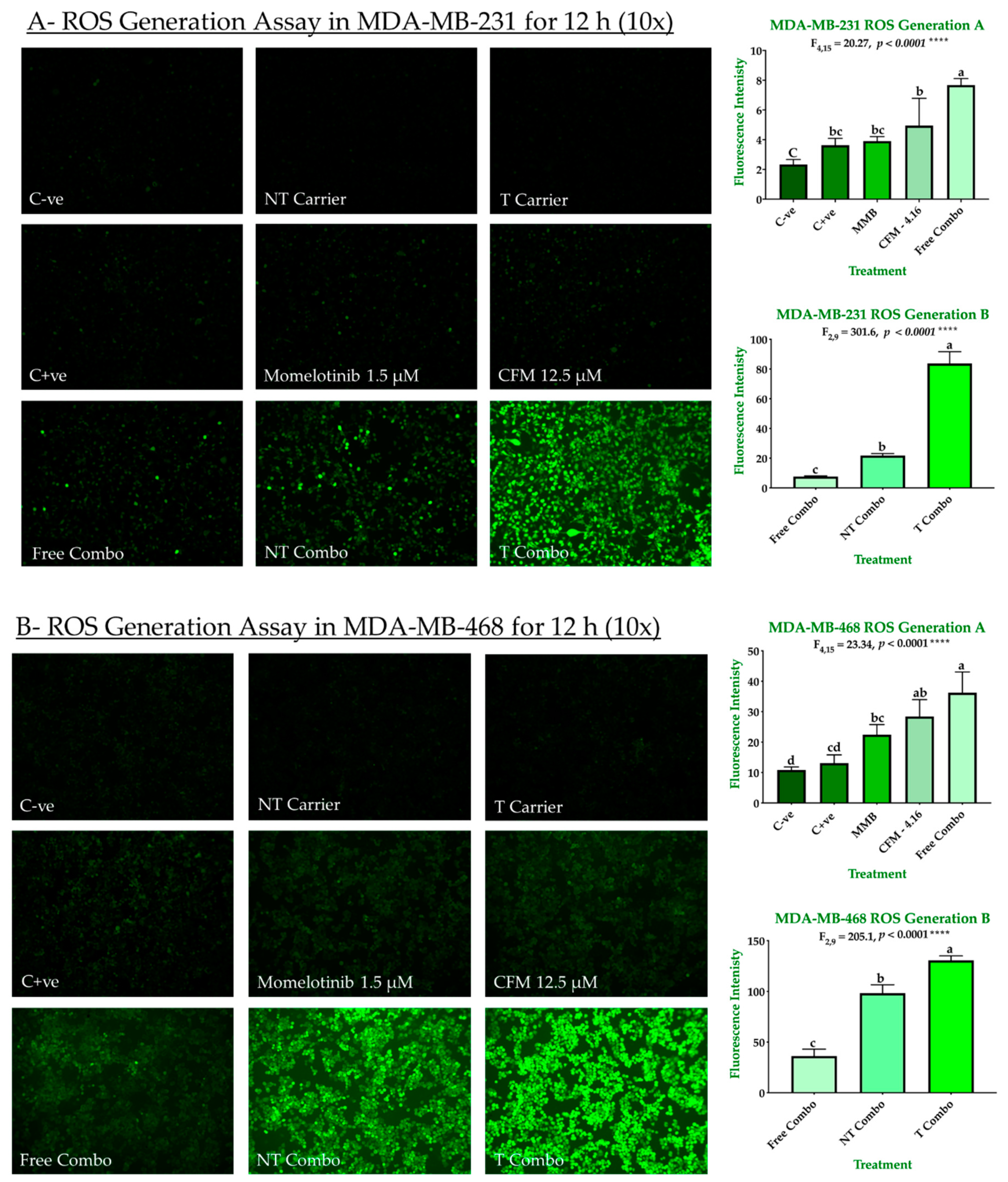
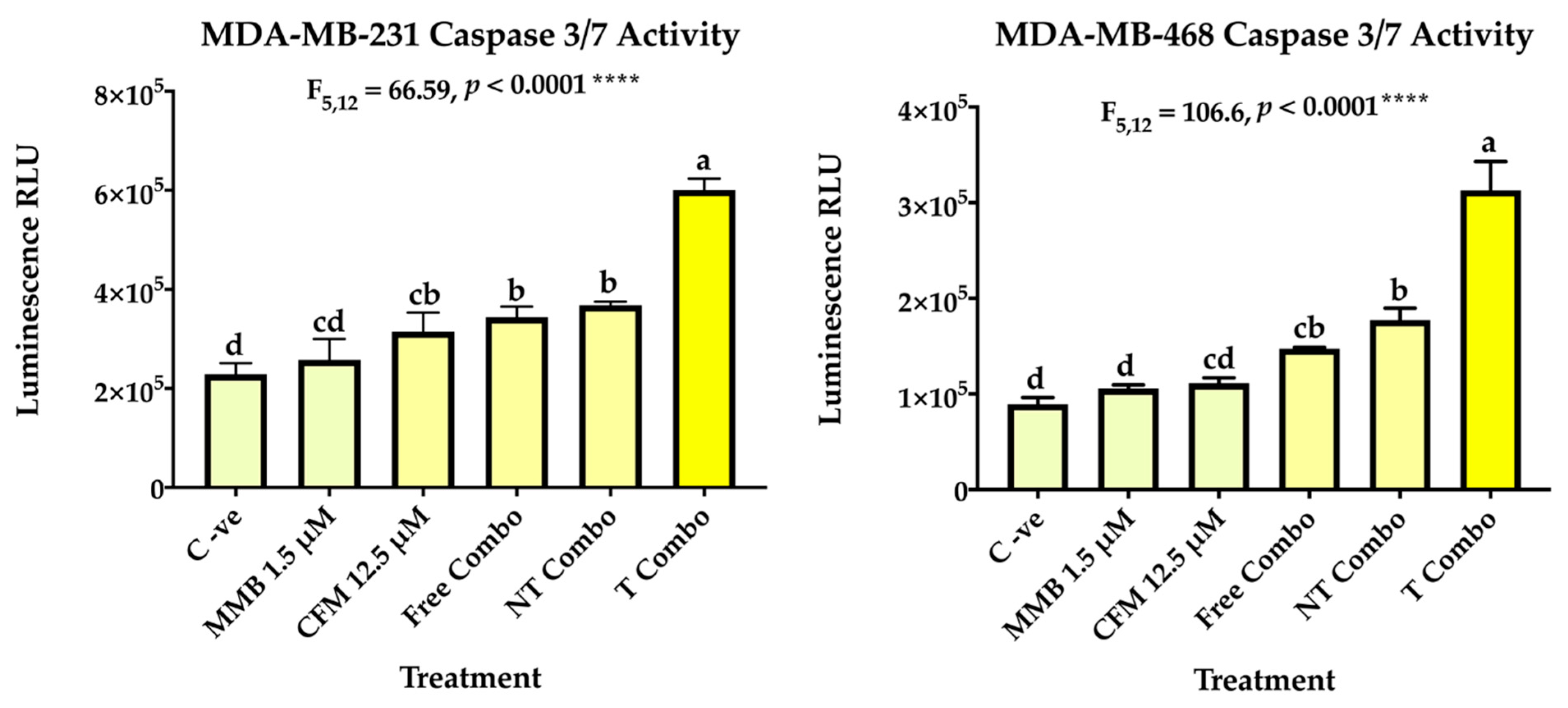
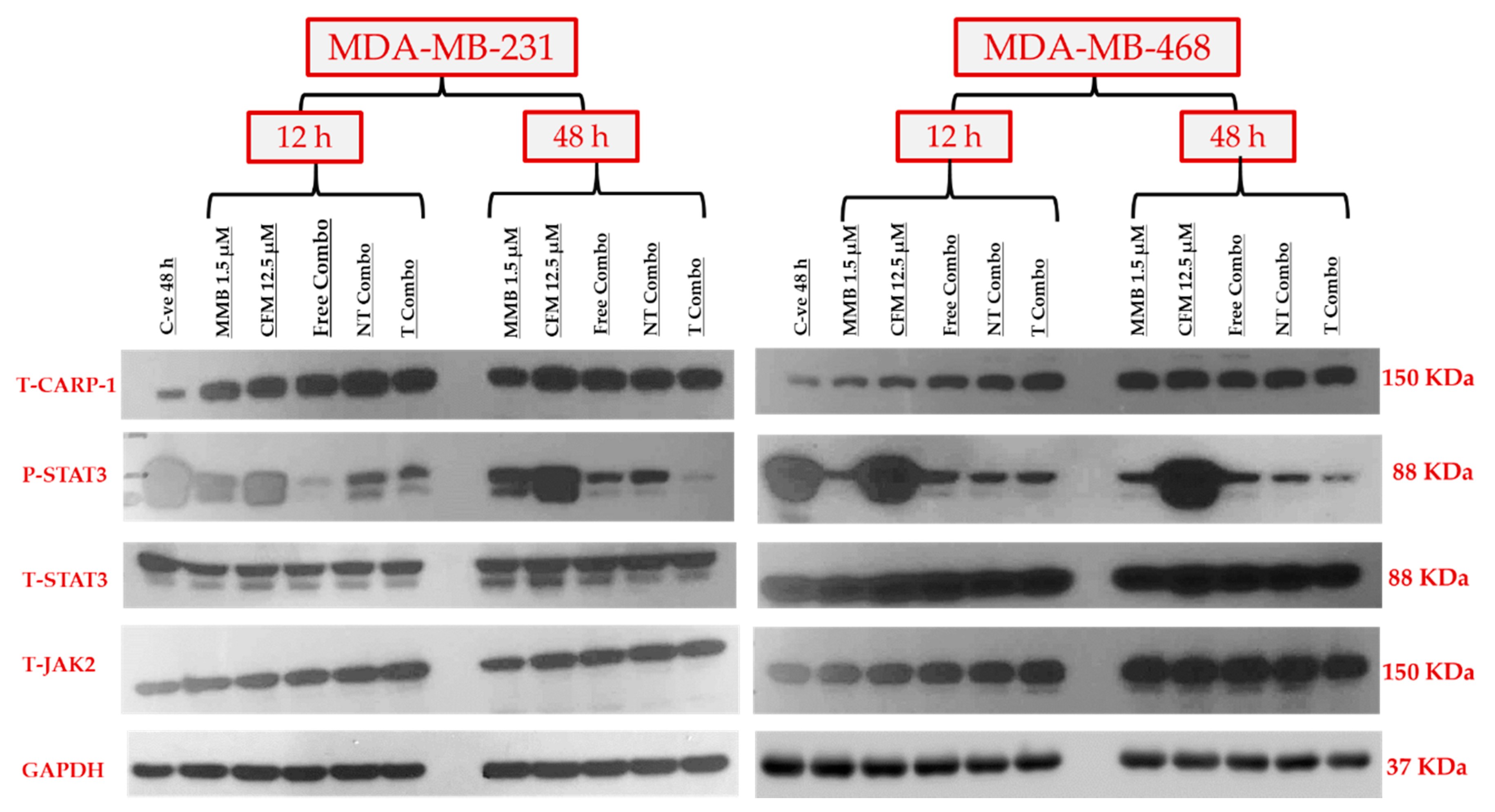
2.3.2. Combination Therapy Has Synergistic Effect Due to ROS Generation, Elevated Caspase 3/7Activity, and Downregulation of P-STAT3
2.4. Animal Studies
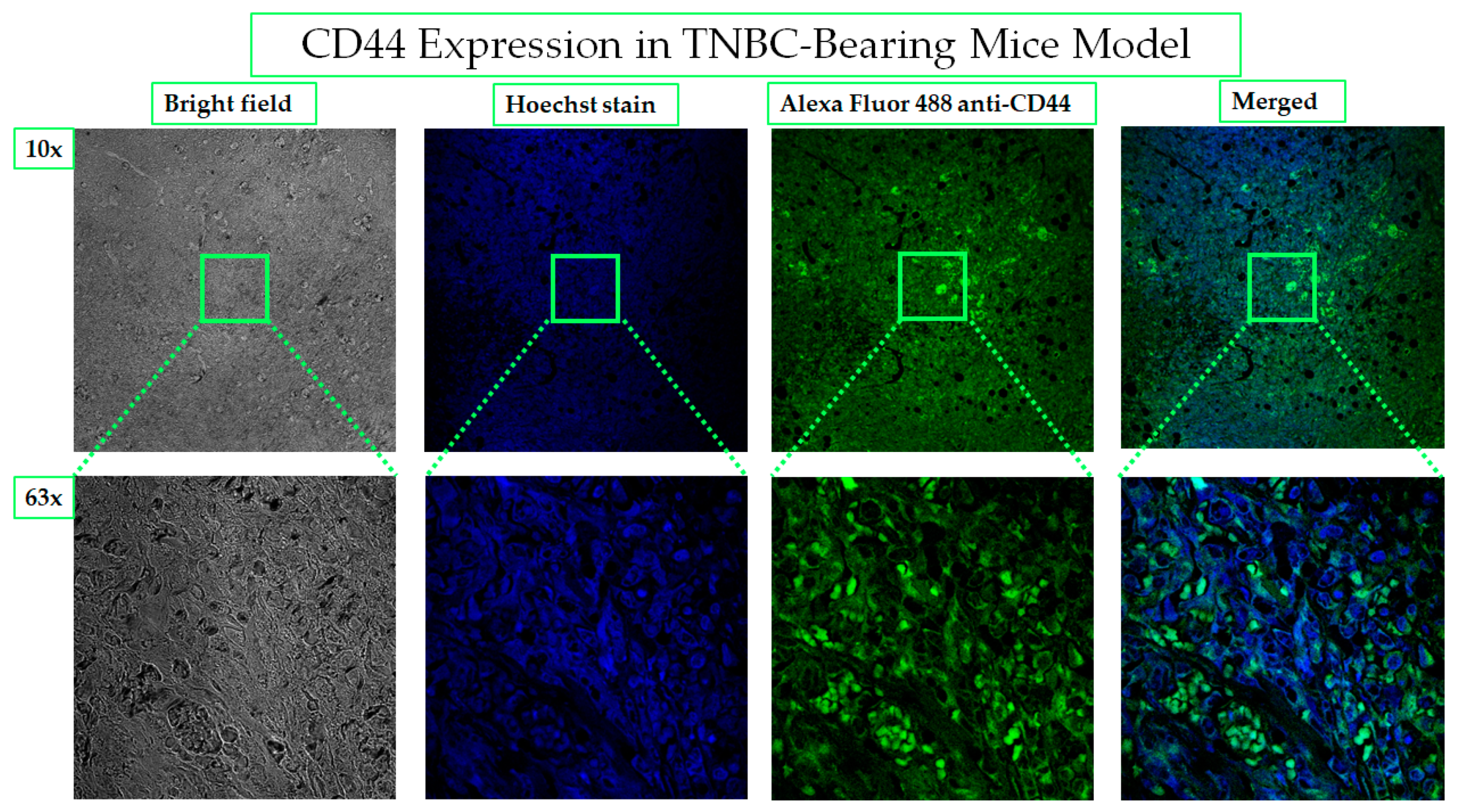
2.4.1. CD44 Receptors Are Overexpressed in Tumors of TNBC-Bearing Mice Model
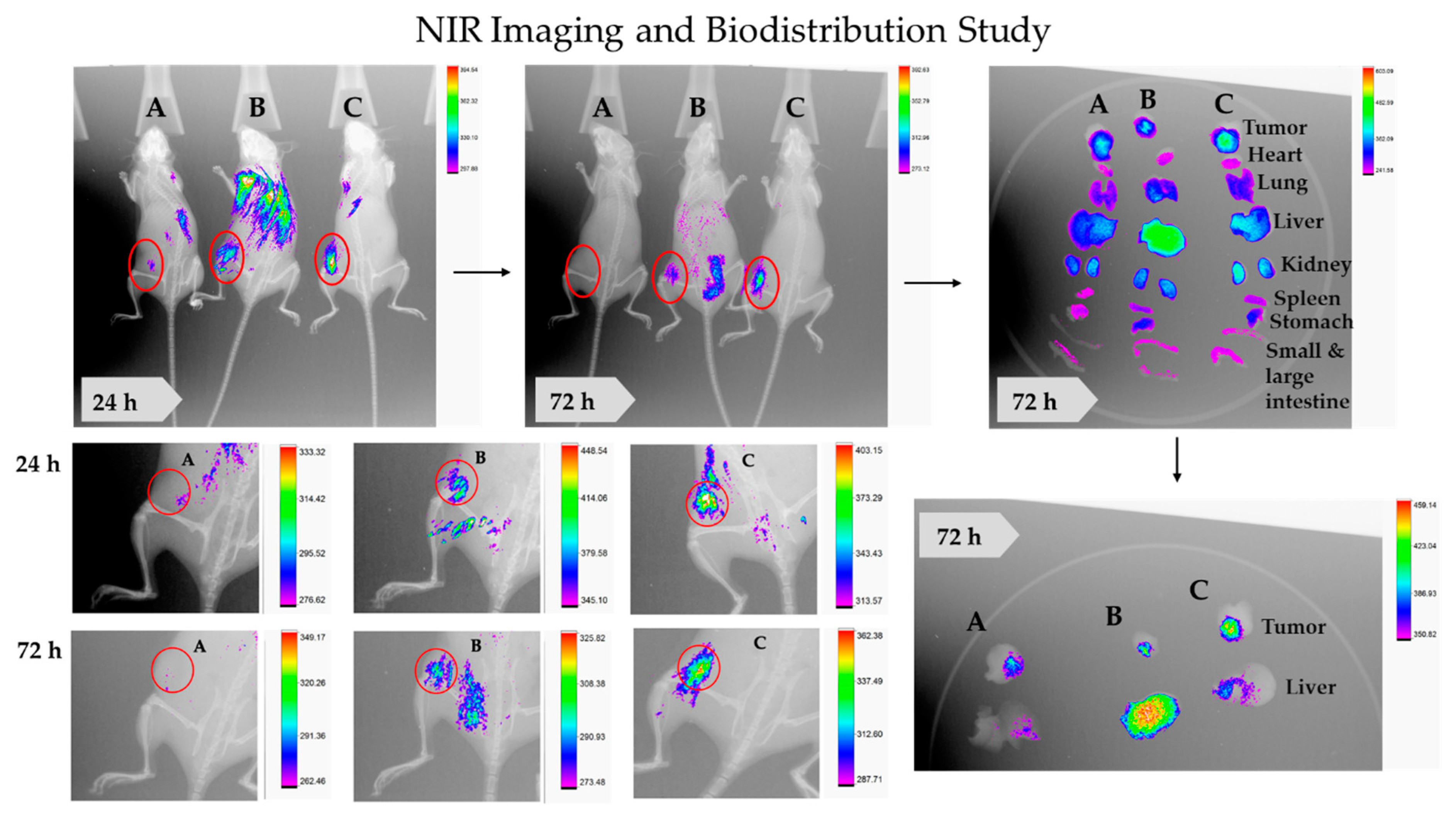
2.4.2. NIR Imaging and Biodistribution and Inducible DNA-DSBs
3. Discussion
4. Materials and Methods
4.1. Materials (Cell Lines and Chemicals)
4.2. Screening of In Vitro Cell Viability (MTT Assay) and Combination Study
4.3. Polymeric Nanoparticle Formulations
4.3.1. Synthesis of the Non-Targeted (NT) SMA-TPGS and Targeted (T) HA-SMA-TPGS-Carriers
4.3.2. Preparation and Characterization of CFM-4.16 Loaded Polymeric NPs (PNPs)
4.3.3. Loading Capacity (LC%) and Encapsulation Efficiency (EE%)
4.3.4. Cellular Uptake of and In Vitro Cytotoxicity PNPs
4.4. Combination Studies of Momelotinib (JAK/STAT inhibitor) + CFM-4.16
4.4.1. In Vitro Cytotoxicity of Combinations vs. Free Drugs
4.4.2. Morphological Alterations and Wound Healing Assay
4.4.3. Detection of ROS Generation
4.4.4. Caspase 3/7Activity Assay
4.4.5. Western Blot Analysis
4.5. Animal Studies
4.5.1. Animal Husbandry and Tumor Induction
4.5.2. CD44 Expression in TNBC Bearing Mice Model by Immunohistochemistry
4.5.3. TUNEL Assay
4.5.4. NIR Imaging and Biodistribution Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Tseng, L.M.; Hsu, N.C.; Chen, S.C.; Lu, Y.S.; Lin, C.H.; Chang, D.Y.; Li, H.; Lin, Y.C.; Chang, H.K.; Chao, T.C.; et al. Distant metastasis in triple-negative breast cancer. Neoplasma 2013, 60, 290–294. [Google Scholar] [CrossRef]
- Khosravi-Shahi, P.; Cabezón-Gutiérrez, L.; Custodio-Cabello, S. Metastatic triple negative breast cancer: Optimizing treatment options, new and emerging targeted therapies. Asia Pac. J. Clin. Oncol. 2018, 14, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Ahn, J.H.; Kim, S.B. How shall we treat early triple-negative breast cancer (TNBC): From the current standard to upcoming immuno-molecular strategies. ESMO Open 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106–116. [Google Scholar]
- Fukuoka, M.; Tsuchiya, R. Principles for Adjuvant and Neoadjuvant Chemotherapy. Gan To Kagaku Ryoho 1994, 3, 333–337. [Google Scholar]
- Miles, D.; von Minckwitz, G.; Seidman, A.D. Combination versus sequential single-agent therapy in metastatic breast cancer. Oncologist 2002, 7, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT Pathway: Impact on Human Disease and Therapeutic Intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [Green Version]
- Leonard, W.J.; O′Shea, J.J. JAKS AND STATS: Biological Implications. Annu. Rev. Immunol. 1998, 16, 293–322. [Google Scholar] [CrossRef] [Green Version]
- Miklossy, G.; Hilliard, T.S.; Turkson, J. Therapeutic modulators of STAT signalling for human diseases. Nat. Rev. Drug Discov. 2013, 12, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Gao, F.H.; Wang, J.Y.; Liu, F.; Yuan, H.H.; Zhang, W.Y.; Jiang, B. JAK2/STAT3 signaling pathway activation mediates tumor angiogenesis by upregulation of VEGF and bFGF in non-small-cell lung cancer. Lung Cancer 2011, 73, 366–374. [Google Scholar] [CrossRef]
- Chan, E.; Luwor, R.; Burns, C.; Kannourakis, G.; Findlay, J.K.; Ahmed, N. Momelotinib decreased cancer stem cell associated tumor burden and prolonged disease-free remission period in a mouse model of human ovarian cancer. Oncotarget 2018, 9, 16599–16618. [Google Scholar] [CrossRef]
- McLean, J.R.; Chaix, D.; Ohi, M.D.; Gould, K.L. State of the APC/C: Organization, function, and structure. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Sun, Y. SCF E3 Ubiquitin Ligases as Anticancer Targets. Curr. Cancer Drug Targets 2011, 11, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Muthu, M.; Somagoni, J.; Cheriyan, V.T.; Munie, S.; Levi, E.; Ashour, A.E.; Yassin, A.E.B.; Alafeefy, A.M.; Sochacki, P.; Polin, L.A.; et al. Identification and Testing of Novel CARP-1 Functional Mimetic Compounds as Inhibitors of Non-Small Cell Lung and Triple Negative Breast Cancers. J. Biomed. Nanotechnol. 2015, 11, 1608–1627. [Google Scholar] [CrossRef] [Green Version]
- Cheriyan, V.T.; Muthu, M.; Patel, K.; Sekhar, S.; Rajeswaran, W.; Larsen, S.D.; Polin, L.; Levi, E.; Singh, M.; Rishi, A.K. CARP-1 functional mimetics are novel inhibitors of drug-resistant triple negative breast cancers. Oncotarget 2016, 7, 73370–73388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabil, G.; Bhise, K.; Sau, S.; Atef, M.; El-Banna, H.A.; Iyer, A.K. Nano-engineered delivery systems for cancer imaging and therapy: Recent advances, future direction and patent evaluation. Drug Discov. Today 2019, 24, 462–491. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wu, T.; Qi, Y.; Zhang, Z. Recent advances in the application of vitamin E TPGS for drug delivery. Theranostics 2018, 8, 464–485. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, V.B.; Henry, S.M.; Hoffman, A.S.; Stayton, P.S.; Li, X.; Pun, S.H. Encapsulation and stabilization of indocyanine green within poly(styrene-alt-maleic anhydride) block-poly(styrene) micelles for near-infrared imaging. J. Biomed. Opt. 2008, 13, 014025. [Google Scholar] [CrossRef] [PubMed]
- Larson, N.; Greish, K.; Bauer, H.; Maeda, H.; Ghandehari, H. Synthesis and evaluation of poly(styrene-co-maleic acid) micellar nanocarriers for the delivery of tanespimycin. Int. J. Pharm. 2011, 420, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yan, L.; Tang, E.K.Y.; Zhang, Z.; Chen, W.; Liu, G.; Mo, J. Synthesis of TPGS/Curcumin Nanoparticles by Thin-Film Hydration and Evaluation of Their Anti-Colon Cancer Efficacy In Vitro and In Vivo. Front. Pharmacol. 2019, 10, 769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakha, E.A.; Chan, S. Metastatic Triple-negative Breast Cancer. Clin. Oncol. 2011, 23, 587–600. [Google Scholar] [CrossRef]
- Hardy, D.; Cormier, J.N.; Xing, Y.; Liu, C.C.; Xia, R.; Du, X.L. Chemotherapy-associated toxicity in a large cohort of elderly patients with non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Probin, V.; Zhou, D. Cancer therapy-induced residual bone marrow injury-Mechanisms of induction and implication for therapy. Curr. Cancer Ther. Rev. 2006, 2, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Boussios, S.; Pentheroudakis, G.; Katsanos, K.; Pavlidis, N. Systemic treatment-induced gastrointestinal toxicity: Incidence, clinical presentation and management. Ann. Gastroenterol. 2012, 25, 106–118. [Google Scholar] [PubMed]
- Rivera, E.; Gomez, H. Chemotherapy resistance in metastatic breast cancer: The evolving role of ixabepilone. Breast Cancer Res. 2010, 12, S2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coley, H.M. Mechanisms and strategies to overcome chemotherapy resistance in metastatic breast cancer. Cancer Treat. Rev. 2008, 34, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.; Sau, S.; Alzhrani, R. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Gros, L.; Ringsdorf, H.; Schupp, H. Polymeric Antitumor Agents on a Molecular and on a Cellular Level? Angew. Chemie Int. Ed. Engl. 1981, 20, 305–325. [Google Scholar] [CrossRef]
- Greish, K.; Fang, J.; Inutsuka, T.; Nagamitsu, A.; Maeda, H. Macromolecular Therapeutics: Advantages and Prospects with Special Emphasis on Solid Tumour Targeting. Clin. Pharmacokinet. 2003, 42, 1089–1105. [Google Scholar] [CrossRef]
- Current Nanotechnology Treatments—National Cancer Institute. Posted August 8, 2017. Available online: https://www.cancer.gov/nano/cancer-nanotechnology/current-treatments (accessed on 3 December 2020).
- Suzuki, F.; Munakata, T.; Maeda, H. Interferon Induction by SMANCS: A Polymer-Conjugated Derivative of Neocarzinostatin. Anticancer Res. 1988, 8, 97–103. [Google Scholar] [PubMed]
- Suzuki, F.; Pollard, R.B.; Uchimura, S.; Munakata, T.; Maeda, H. Role of Natural Killer Cells and Macrophages in the Nonspecific Resistance to Tumors in Mice Stimulated With SMANCS, a Polymer-Conjugated Derivative of Neocarzinostatin. Cancer Res. 1990, 50, 3897–3904. [Google Scholar]
- Maeda, H. SMANCS/Lipiodol. Gan To Kagaku Ryoho. 1994, 21, 907–913. [Google Scholar]
- Maeda, H.; Konno, T. Metamorphosis of Neocarzinostatin to SMANCS: Chemistry, Biology, Pharmacology, and Clinical Effect of the First Prototype Anticancer Polymer Therapeutic. In Neocarzinostatin: The Past, Present, and Future of an Anticancer Drug; Maeda, H., Edo, K., Ishida, N., Eds.; Springer: Tokyo, Japan, 1997; pp. 227–267. [Google Scholar]
- Wang, Z.; Sau, S.; Alsaab, H.O.; Iyer, A.K. CD44 directed nanomicellar payload delivery platform for selective anticancer effect and tumor specific imaging of triple negative breast cancer. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Muthu, M.S.; Avinash Kulkarni, S.; Liu, Y.; Feng, S.S. Development of docetaxel-loaded vitamin e TPGS micelles: Formulation optimization, effects on brain cancer cells and biodistribution in rats. Nanomedicine 2012, 7, 353–364. [Google Scholar] [CrossRef]
- Fan, Z.; Chen, C.; Pang, X.; Yu, Z.; Qi, Y.; Chen, X.; Liang, H.; Fang, X.; Sha, X. Adding Vitamin E-TPGS to the Formulation of Genexol-PM: Specially Mixed Micelles Improve Drug-Loading Ability and Cytotoxicity against Multidrug-Resistant Tumors Significantly. PLoS ONE 2015, 10, e0120129. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Wickens, J.M.; Alsaab, H.O.; Kesharwani, P.; Bhise, K.; Amin, M.C.I.M.; Tekade, R.K.; Gupta, U.; Iyer, A.K. Recent advances in hyaluronic acid-decorated nanocarriers for targeted cancer therapy. Drug Discov. Today 2017, 22, 665–680. [Google Scholar] [CrossRef] [Green Version]
- Necas, J.; Bartosikova, L.; Brauner, P.; Kolar, J. Hyaluronic acid (hyaluronan): A review. Vet. Med. 2008, 53, 397–411. [Google Scholar] [CrossRef] [Green Version]
- Ricardo, S.; Vieira, A.F.; Gerhard, R.; Leitão, D.; Pinto, R.; Cameselle-Teijeiro, J.F.; Milanezi, F.; Schmitt, F.; Paredes, J. Breast cancer stem cell markers CD44, CD24 and ALDH1: Expression distribution within intrinsic molecular subtype. J. Clin. Pathol. 2011, 64, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, H.; Wang, H.; Shi, X.; Fan, Y.; Ding, X.; Lin, C.; Zhan, Q.; Qian, H.; Xu, B. Enriched CD44+/CD24- population drives the aggressive phenotypes presented in triple-negative breast cancer (TNBC). Cancer Lett. 2014, 353, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Kesharwani, P.; Banerjee, S.; Padhye, S.; Sarkar, F.H.; Iyer, A.K. Hyaluronic Acid Engineered Nanomicelles Loaded with 3,4-Difluorobenzylidene Curcumin for Targeted Killing of CD44+ Stem-Like Pancreatic Cancer Cells. Biomacromolecules 2015, 16, 3042–3053. [Google Scholar] [CrossRef]
- Su, Z.; Chen, M.; Xiao, Y.; Sun, M.; Zong, L.; Asghar, S.; Dong, M.; Li, H.; Ping, Q.; Zhang, C. ROS-triggered and regenerating anticancer nanosystem: An effective strategy to subdue tumor′s multidrug resistance. J. Control. Release 2014, 196, 370–383. [Google Scholar] [CrossRef]
- Chithrani, B.D.; Chan, W.C.W. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 2007, 7, 1542–1550. [Google Scholar] [CrossRef]
- Ganesh, K.; Archana, D. Review Article on Targeted Polymeric Nanoparticles: An Overview. Am. J. Adv. Drug Deliv. 2013, 3, 196–215. [Google Scholar]
- Wang, W.; Gaus, K.; Tilley, R.D.; Gooding, J.J. The impact of nanoparticle shape on cellular internalisation and transport: What do the different analysis methods tell us? Mater. Horizons 2019, 6, 1538–1547. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, E.; Yang, J.; Cao, Z. Strategies to improve micelle stability for drug delivery. Nano Res. 2018, 11, 4985–4998. [Google Scholar] [CrossRef]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chenthamara, D.; Subramaniam, S.; Ramakrishnan, S.G.; Krishnaswamy, S.; Essa, M.M.; Lin, F.H.; Qoronfleh, M.W. Therapeutic efficacy of nanoparticles and routes of administration. Biomater. Res. 2019, 23, 1–29. [Google Scholar] [CrossRef]
- Tsirikis, P.; Xiang, S.; Selomulya, C.; Plebanski, M. Design of Nanoparticle Structures for Cancer Immunotherapy. In Nanostructures for Cancer Therapy; Ficai, A., Grumezescu, A.M., Eds.; Matthew Deans: Philadelphia, PA, USA, 2017; pp. 307–321. ISBN 9780323461504. [Google Scholar]
- Schäfer, T.; Noseck, U.; Huber, F.; Bouby, M.; Brendlé, J.; Büchner, S.; Darbha, G.K.; Goetz, R.; Flügge, J.; Hauser, W.; et al. Colloid/Nanoparticle Formation and Mobility in the Context of Deep Geological Nuclear Waste Disposal (Project KOLLORADO-2; Final Report); Institut für Nukleare Entsorgung (INE): Weisslingen, Switzerland, 2013. [Google Scholar]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Bose, T.; Latawiec, D.; Mondal, P.P.; Mandal, S. Overview of nano-drugs characteristics for clinical application: The journey from the entry to the exit point. J. Nanoparticle Res. 2014, 16, 1–25. [Google Scholar] [CrossRef]
- Nag, O.K.; Delehanty, J.B. Active cellular and subcellular targeting of nanoparticles for drug delivery. Pharmaceutics 2019, 11, 543. [Google Scholar] [CrossRef] [Green Version]
- Honary, S.; Zahir, F. Effect of Zeta Potential on the Properties of Nano-Drug Delivery Systems-A Review (Part 2). Trop. J. Pharm. Res. 2013, 12, 265–273. [Google Scholar] [CrossRef]
- McConnell, K.I.; Shamsudeen, S.; Meraz, I.M.; Mahadevan, T.S.; Ziemys, A.; Rees, P.; Summers, H.D.; Serda, R.E. Reduced Cationic Nanoparticle Cytotoxicity Based on Serum Masking of Surface Potential. J. Biomed. Nanotechnol. 2016, 12, 154–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jokerst, J.V.; Lobovkina, T.; Zare, R.N.; Gambhir, S.S. Nanoparticle PEGylation for imaging and therapy. Nanomedicine 2011, 6, 715–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Çalış, S.; Öztürk Atar, K.; Arslan, F.B.; Eroğlu, H.; Çapan, Y. Nanopharmaceuticals as Drug-Delivery Systems: For, Against, and Current Applications. In Nanocarriers for Drug Delivery; Mohapatra, S., Ranjan, S., Dasgupta, N., Kumar, R., Thomas, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 133–154. [Google Scholar]
- Dong, H.; Pang, L.; Cong, H.; Shen, Y.; Yu, B. Application and design of esterase-responsive nanoparticles for cancer therapy. Drug Deliv. 2019, 26, 416–432. [Google Scholar] [CrossRef] [Green Version]
- Pawar, A.; Prabhu, P. Nanosoldiers: A promising strategy to combat triple negative breast cancer. Biomed. Pharmacother. 2019, 110, 319–341. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, A.S.; Peres, L.L.; Faria, A.V.S.; Milani, R.; Silva, R.A.; da Costa Fernandes, C.J.; Peppelenbosch, M.P.; Ferreira-Halder, C.V.; Zambuzzi, W.F. Phosphoproteome profiling reveals critical role of JAK-STAT signaling in maintaining chemoresistance in breast cancer. Oncotarget 2017, 8, 114756–114768. [Google Scholar] [CrossRef] [Green Version]
- Marotta, L.L.C.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44 +CD24- stem cell-like breast cancer cells in human tumors. J. Clin. Invest. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Lu, L.; Dong, J.; Wang, L.; Xia, Q.; Zhang, D.; Kim, H.; Yin, T.; Fan, S.; Shen, Q. Activation of STAT3 and Bcl-2 and reduction of reactive oxygen species (ROS) promote radioresistance in breast cancer and overcome of radioresistance with niclosamide. Oncogene 2018, 37, 5292–5304. [Google Scholar] [CrossRef]
- Choi, S.M.; Kim, D.H.; Chun, K.S.; Choi, J.S. Carnosol induces apoptotic cell death through ROS-dependent inactivation of STAT3 in human melanoma G361 cells. Appl. Biol. Chem. 2019, 62, 1–11. [Google Scholar] [CrossRef]
- Cai, W.; Yang, X.; Han, S.; Guo, H.; Zheng, Z.; Wang, H.; Guan, H.; Jia, Y.; Gao, J.; Yang, T.; et al. Notch1 Pathway Protects against Burn-Induced Myocardial Injury by Repressing Reactive Oxygen Species Production through JAK2/STAT3 Signaling. Oxid. Med. Cell. Longev. 2015, 2016, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef] [PubMed]
- Brentnall, M.; Rodriguez-Menocal, L.; De Guevara, R.L.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 14, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhou, C.; Fu, Y.; Chen, T.; Liu, X.; Zhang, Z.; Gong, T. Targeted delivery of hyaluronic acid nanomicelles to hepatic stellate cells in hepatic fibrosis rats. Acta Pharm. Sin. B 2020, 10, 693–710. [Google Scholar] [CrossRef]
- Almalik, A.; Benabdelkamel, H.; Masood, A.; Alanazi, I.O.; Alradwan, I.; Majrashi, M.A.; Alfadda, A.A.; Alghamdi, W.M.; Alrabiah, H.; Tirelli, N.; et al. Hyaluronic Acid Coated Chitosan Nanoparticles Reduced the Immunogenicity of the Formed Protein Corona. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.; Kutty, R.V. Recent advances in nanotheranostics for triple negative breast cancer treatment. J. Exp. Clin. Cancer Res. 2019, 38, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheriyan, V.T.; Alsaab, H.O.; Sekhar, S.; Stieber, C.; Kesharwani, P.; Sau, S.; Muthu, M.; Polin, L.A.; Levi, E.; Iyer, A.K.; et al. A CARP-1 functional mimetic loaded vitamin E-TPGS micellar nano-formulation for inhibition of renal cell carcinoma. Oncotarget 2017, 8, 104928–104945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidieh, T.; Smith, J.R.; Ball, K.E.; An, Q. ROS as a novel indicator to predict anticancer drug efficacy. BMC Cancer 2019, 19, 1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measurements | NT-PNPs | CD44-T-PNPs |
|---|---|---|
| LC% | 25.26 ± 1.22% | 17 ± 1.45% |
| EE% | 89.45 ± 4.89% | 71 ± 6.87% |
| Yield% | 81.53 ± 2.65% | 95.89 ± 2.22% |
| Combo | Dose | CI MDA-MB-231 | CI MDA-MB-468 | |
|---|---|---|---|---|
| MMB | CFM-4.16 | |||
| MMB + CFM-4.16 | 1.5 µM | 12.5 µM | 0.58 | 0.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nabil, G.; Alzhrani, R.; Alsaab, H.O.; Atef, M.; Sau, S.; Iyer, A.K.; Banna, H.E. CD44 Targeted Nanomaterials for Treatment of Triple-Negative Breast Cancer. Cancers 2021, 13, 898. https://doi.org/10.3390/cancers13040898
Nabil G, Alzhrani R, Alsaab HO, Atef M, Sau S, Iyer AK, Banna HE. CD44 Targeted Nanomaterials for Treatment of Triple-Negative Breast Cancer. Cancers. 2021; 13(4):898. https://doi.org/10.3390/cancers13040898
Chicago/Turabian StyleNabil, Ghazal, Rami Alzhrani, Hashem O. Alsaab, Mohammed Atef, Samaresh Sau, Arun K. Iyer, and Hossny El Banna. 2021. "CD44 Targeted Nanomaterials for Treatment of Triple-Negative Breast Cancer" Cancers 13, no. 4: 898. https://doi.org/10.3390/cancers13040898
APA StyleNabil, G., Alzhrani, R., Alsaab, H. O., Atef, M., Sau, S., Iyer, A. K., & Banna, H. E. (2021). CD44 Targeted Nanomaterials for Treatment of Triple-Negative Breast Cancer. Cancers, 13(4), 898. https://doi.org/10.3390/cancers13040898