
Fatty Acid Synthase Confers Tamoxifen Resistance to ER+/HER2+ Breast Cancer

 ,
,  , ,
, ,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
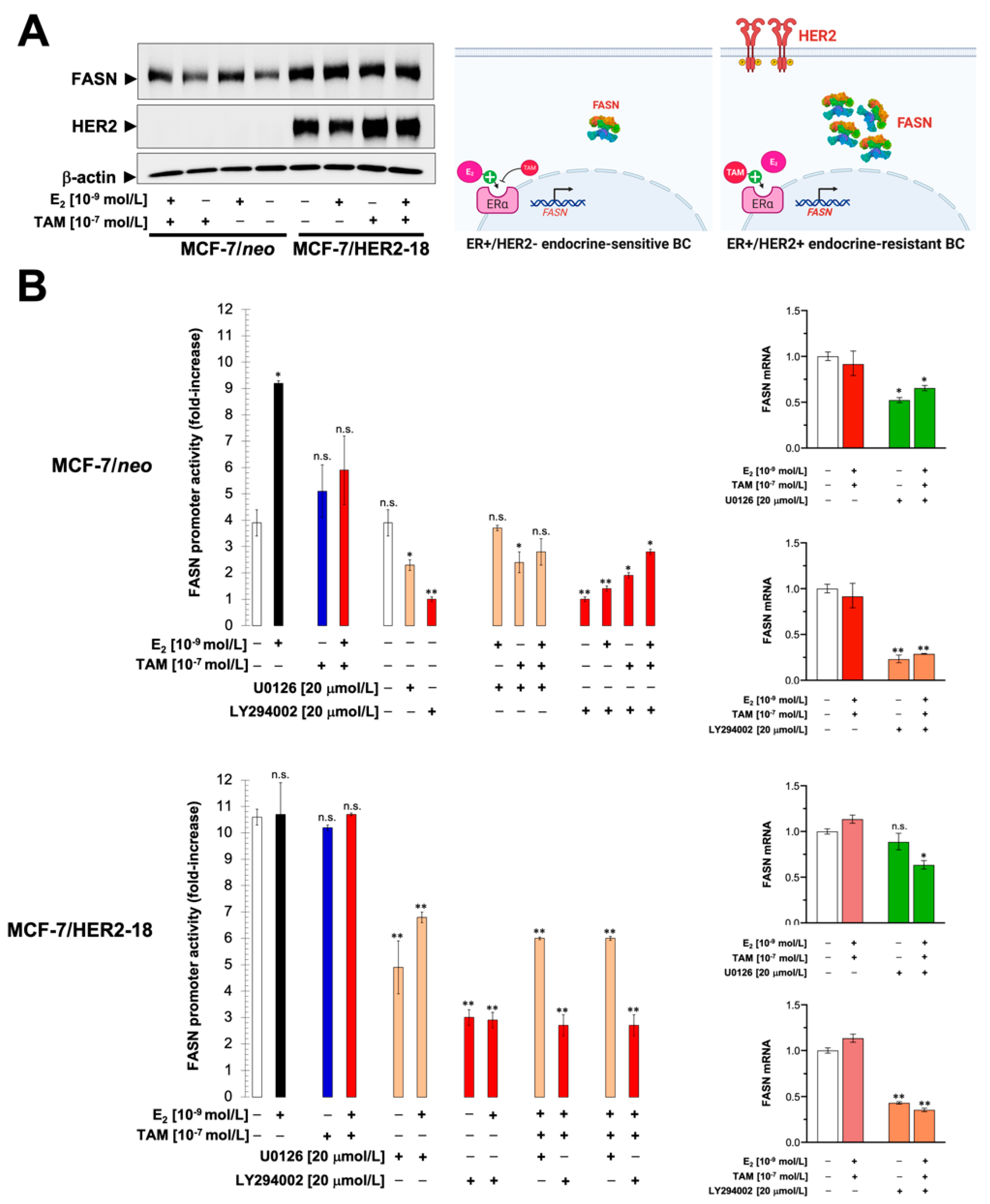
2.1. Estradiol Stimulates FASN Protein Expression via ERα in ER+ Luminal A-Like Breast Cancer Cells
2.2. Estradiol/ERα Signaling Stimulates the FASN Gene Promoter in ER+ Luminal A-Like Breast Cancer Cells
2.3. The Stimulatory Effects of Estradiol on FASN Gene Promoter Activity Involve the PI3K/AKT/SREBP Signaling Cascade
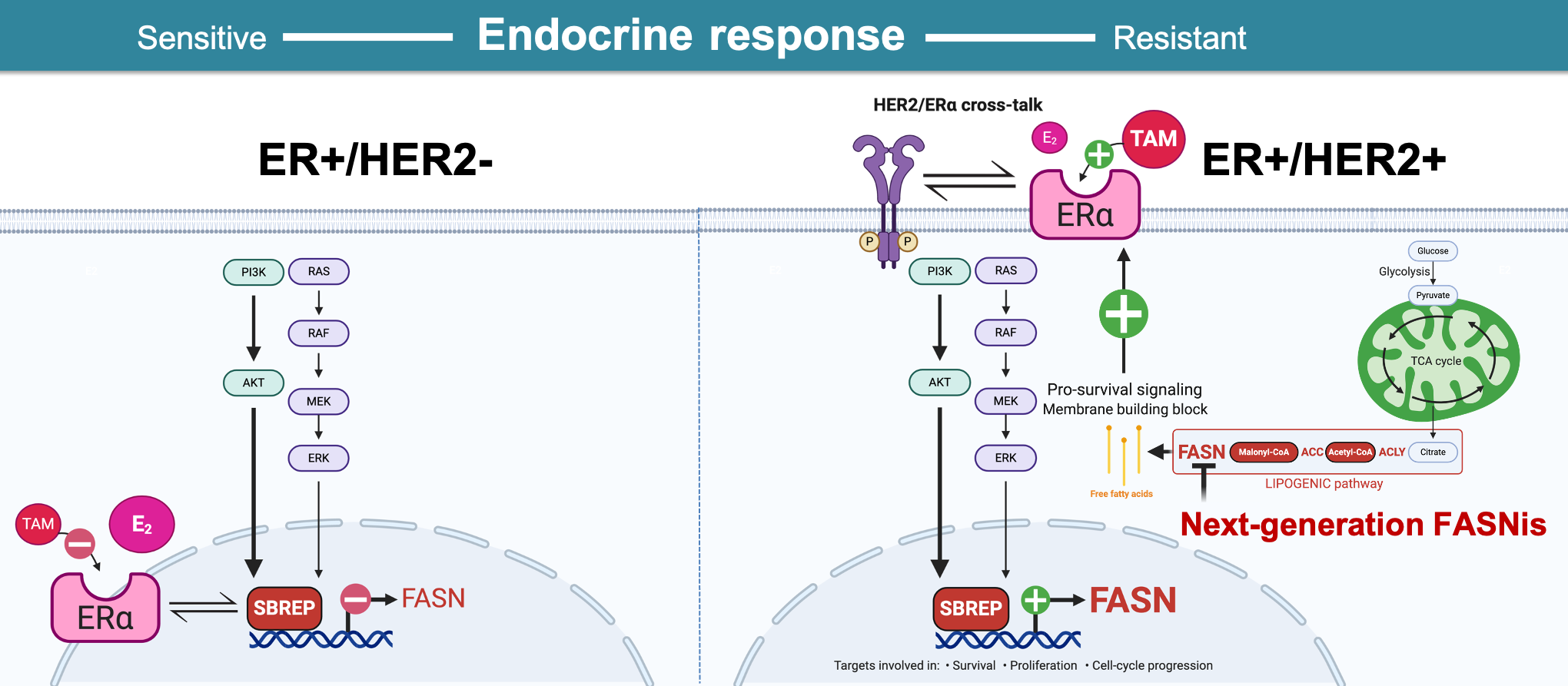
2.4. HER2 Overexpression Suppresses the Estrogen-Inducible Phenotype of FASN Expression in ER+ Breast Cancer Cells
2.5. Pharmacological Targeting of FASN Suppresses HER2-Driven Resistance to Tamoxifen In Vitro
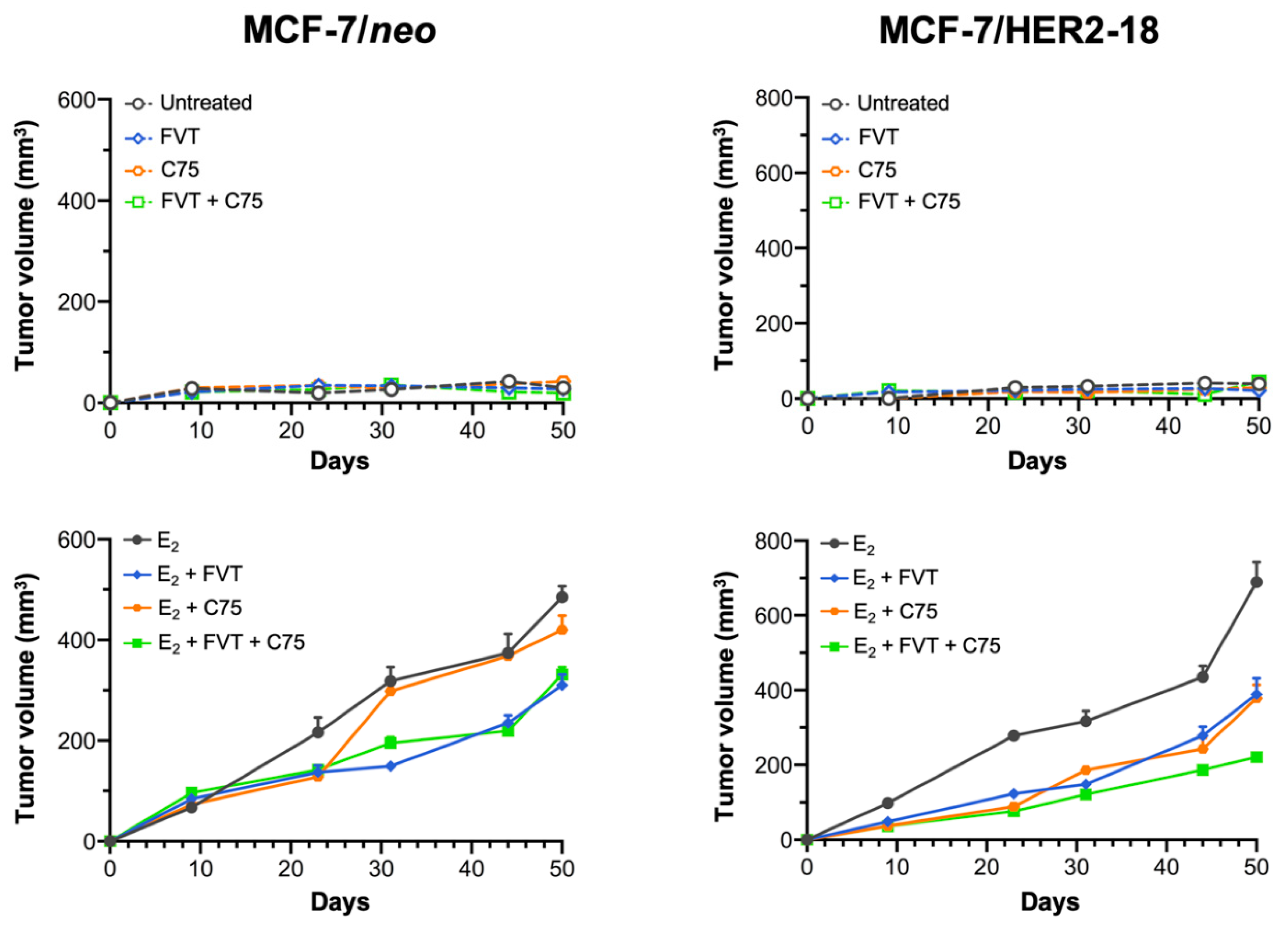
2.6. FASN Inhibition Reverses HER2-Mediated Resistance to Tamoxifen In Vivo
2.7. ER+/HER2+ Breast Carcinomas Express High Levels of FASN Protein
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Lines and Culture Conditions
4.3. In Situ Immunofluorescent Staining of FASN
4.4. FASN Promoter Activity
4.5. Immunoblotting Analyses of FASN and ERα
4.6. Soft-Agar Colony Formation Assays
4.7. Xenograft Studies
4.8. Analyses of Clinical Data
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Santen, R.J.; Fan, P.; Zhang, Z.; Bao, Y.; Song, R.X.-D.; Yue, W. Estrogen signals via an extra-nuclear pathway involving IGF-1R and EGFR in tamoxifen-sensitive and -resistant breast cancer cells. Steroids 2009, 74, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Arpino, G.; De Angelis, C.; Giuliano, M.; Giordano, A.; Falato, C.; De Laurentiis, M.; De Placido, S. Molecular Mechanism and Clinical Implications of Endocrine Therapy Resistance in Breast Cancer. Oncology 2009, 77, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Alluri, P.G.; Speers, C.; Chinnaiyan, A.M. Estrogen receptor mutations and their role in breast cancer progression. Breast Cancer Res. 2014, 16, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, W.; Chang, J.; Fu, P. Endocrine therapy resistance in breast cancer: Current status, possible mechanisms and overcoming strategies. Future Med. Chem. 2015, 7, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- Haque, M.; Desai, K.V. Pathways to Endocrine Therapy Resistance in Breast Cancer. Front. Endocrinol. 2019, 10, 573. [Google Scholar] [CrossRef] [PubMed]
- Benz, C.C.; Scott, G.K.; Sarup, J.C.; Johnson, R.M.; Tripathy, D.; Coronado, E.; Shepard, H.M.; Osborne, C.K. Estrogen-dependent, tamoxifen-resistant tumorigenic growth of MCF-7 cells transfected with HER2/neu. Breast Cancer Res. Treat. 1992, 24, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.-L.; Sheu, M.-L.; Yang, S.-C.; Lin, C.-H.; Yen, S.-H. Resistance to tamoxifen-induced apoptosis is associated with direct interaction between Her2/neu and cell membrane estrogen receptor in breast cancer. Int. J. Cancer 2002, 97, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Knowlden, J.M.; Hutcheson, I.R.; Jones, H.E.; Madden, T.; Gee, J.M.; Harper, M.E.; Barrow, D.; Wakeling, A.E.; Nicholson, R. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology 2003, 144, 1032–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Barnes, C.J.; Kumar, R. Human Epidermal Growth Factor Receptor 2 Status Modulates Subcellular Localization of and Interaction with Estrogen Receptor α in Breast Cancer Cells. Clin. Cancer Res. 2004, 10, 3621–3628. [Google Scholar] [CrossRef] [Green Version]
- Lipton, A.; Ali, S.; Leitzel, K.; Demers, L.; Harvey, H.; Chaudri-Ross, H.; Brady, C.; Wyld, P.; Carney, W. Serum HER-2/neu and Response to the Aromatase Inhibitor Letrozole Versus Tamoxifen. J. Clin. Oncol. 2003, 21, 1967–1972. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Massarweh, S.; Osborne, C.K.; Wakeling, A.E.; Ali, S.; Weiss, H.; Schiff, R. Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 2004, 96, 926–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Wang, Y.; Kane, S.E.; Chen, S. Improvement of sensitivity to tamoxifen in estrogen receptor-positive and Herceptin-resistant breast cancer cells. J. Mol. Endocrinol. 2008, 41, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, B.; Mackey, J.R.; Clemens, M.R.; Bapsy, P.P.; Vaid, A.; Wardley, A.; Tjulandin, S.; Jahn, M.; Lehle, M.; Feyereislova, A.; et al. Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2-positive, hormone receptor-positive metastatic breast cancer: Results from the randomized phase III TAnDEM study. J. Clin. Oncol. 2009, 27, 5529–5537. [Google Scholar] [CrossRef] [PubMed]
- Marcom, P.K.; Isaacs, C.; Harris, L.; Wong, Z.W.; Kommarreddy, A.; Novielli, N.; Mann, G.; Tao, Y.; Ellis, M.J. The combination of letrozole and trastuzumab as first or second-line biological therapy produces durable responses in a subset of HER2 positive and ER positive advanced breast cancers. Breast Cancer Res. Treat. 2006, 102, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.; Pippen, J.; Pivot, X.; Lichinitser, M.; Sadeghi, S.; Dieras, V.; Gomez, H.L.; Romieu, G.; Manikhas, A.; Kennedy, M.J.; et al. Lapatinib Combined With Letrozole versus Letrozole and Placebo as First-Line Therapy for Postmenopausal Hormone Receptor–Positive Metastatic Breast Cancer. J. Clin. Oncol. 2009, 27, 5538–5546. [Google Scholar] [CrossRef] [Green Version]
- Boulay, A.; Rudloff, J.; Ye, J.; Zumstein-Mecker, S.; O’Reilly, T.; Evans, D.B.; Chen, S.; Lane, H.A. Dual Inhibition of mTOR and Estrogen Receptor Signaling In vitro Induces Cell Death in Models of Breast Cancer. Clin. Cancer Res. 2005, 11, 5319–5328. [Google Scholar] [CrossRef] [Green Version]
- Ciruelos, G.E.M. Targeting the PI3K/AKT/mTOR pathway in estrogen receptor-positive breast cancer. Cancer Treat Rev. 2014, 40, 862–871. [Google Scholar] [CrossRef]
- Mills, J.N.; Rutkovsky, A.C.; Giordano, A. Mechanisms of resistance in estrogen receptor positive breast cancer: Overcoming resistance to tamoxifen/aromatase inhibitors. Curr. Opin. Pharmacol. 2018, 41, 59–65. [Google Scholar] [CrossRef]
- Araki, K.; Miyoshi, Y. Mechanism of resistance to endocrine therapy in breast cancer: The important role of PI3K/Akt/mTOR in estrogen receptor-positive, HER2-negative breast cancer. Breast Cancer 2017, 25, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Semiglazov, V.; van Dam, P.; Manikhas, A.; Bellet, M.; Mayordomo, J.; Campone, M.; Kubista, E.; Greil, R.; Bianchi, G.; et al. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plusletrozole in patients with estrogen receptor-positive breast cancer. J. Clin. Oncol. 2009, 27, 2630–2637. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Vazquez-Martin, A.; Ortega, F.J.; Fernández-Real, J.M. Fatty Acid Synthase: Association with Insulin Resistance, Type 2 Diabetes, and Cancer. Clin. Chem. 2009, 55, 425–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Corominas-Faja, B.; Vellon, L.; Cuyàs, E.; Buxó, M.; Martin-Castillo, B.; Serra, L.; García, J.; Lupu, R.; Menendez, J.A. Clinical and therapeutic relevance of the metabolic oncogene fatty acid synthase in HER2+ breast cancer. Histol. Histopathol. 2016, 32, 687–698. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin. Ther. Targets 2017, 21, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Colomer, R.; Brunet, J.; Menendez, J.A. Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab (Herceptin by transcriptionally inhibiting ’HER2 super-expression’ occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells. Int. J. Oncol. 2007, 31, 769–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavin, R.; Peluso, S.; Nguyen, P.L.; Loda, M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 2010, 6, 551–562. [Google Scholar] [CrossRef] [Green Version]
- Oliveras-Ferraros, C.; Cufí, S.; Sauri-Nadal, T.; Del Barco, S.; Martin-Castilló, B.; Vazquez-Martin, A.; Menendez, J.A. Circulating fatty acid synthase: An exploratory biomarker to predict efficacy of the dual HER1/HER2 tyrosine kinase inhibitor lapatinib. Breast Cancer Res. 2011, 13, 401. [Google Scholar] [CrossRef]
- Liu, H.; Wu, X.; Dong, Z.; Luo, Z.; Zhao, Z.; Xu, Y.; Zhang, J.-T. Fatty acid synthase causes drug resistance by inhibiting TNF-α and ceramide production. J. Lipid Res. 2013, 54, 776–785. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Qin, L.; Fako, V.; Zhang, J.-T. Molecular mechanisms of fatty acid synthase (FASN)-mediated resistance to anti-cancer treatments. Adv. Biol. Regul. 2014, 54, 214–221. [Google Scholar] [CrossRef]
- Bauerschlag, D.D.; Maass, N.; Leonhardt, P.; Verburg, F.A.; Pecks, U.; Zeppernick, F.; Morgenroth, A.; Mottaghy, F.M.; Tolba, R.; Meinhold-Heerlein, I.; et al. Fatty acid synthase overexpression: Target for therapy and reversal of chemoresistance in ovarian cancer. J. Transl. Med. 2015, 13, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Blancafort, A.; Giró-Perafita, A.; Oliveras, G.; Palomeras, S.; Turrado, C.; Campuzano, Ò.; Carrión-Salip, D.; Massaguer, A.; Brugada, R.; Palafox, M.; et al. Dual Fatty Acid Synthase and HER2 Signaling Blockade Shows Marked Antitumor Activity against Breast Cancer Models Resistant to Anti-HER2 Drugs. PLoS ONE 2015, 10, e0131241. [Google Scholar] [CrossRef] [Green Version]
- Puig, T.; Aguilar, H.; Cufí, S.; Oliveras, G.; Turrado, C.; Ortega-Gutiérrez, S.; Benhamú, B.; López-Rodríguez, M.L.; Urruticoechea, A.; Colomer, R. A novel inhibitor of fatty acid synthase shows activity against HER2+ breast cancer xenografts and is active in anti-HER2 drug-resistant cell lines. Breast Cancer Res. 2011, 13, R131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuer, T.S.; Ventura, R.; Mordec, K.; Lai, J.; Fridlib, M.; Buckley, D.; Kemble, G. FASN Inhibition and Taxane Treatment Combine to Enhance Anti-tumor Efficacy in Diverse Xenograft Tumor Models through Disruption of Tubulin Palmitoylation and Microtubule Organization and FASN Inhibition-Mediated Effects on Oncogenic Signaling and Gene Expression. EBioMedicine 2017, 16, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaevangelou, E.; Almeida, G.S.; Box, C.; DeSouza, N.M.; Chung, Y.-L. The effect of FASN inhibition on the growth and metabolism of a cisplatin-resistant ovarian carcinoma model. Int. J. Cancer 2018, 143, 992–1002. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase regulates estrogen receptor-α signaling in breast cancer cells. Oncology 2017, 6, e299. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Sikora, M.J.; Levine, K.M.; Tasdemir, N.; Riggins, R.B.; Wendell, S.G.; Van Houten, B.; Oesterreich, S. Key regulators of lipid metabolism drive endocrine resistance in invasive lobular breast cancer. Breast Cancer Res. 2018, 20, 1–15. [Google Scholar] [CrossRef]
- Gökmen-Polar, Y.; Neelamraju, Y.; Goswami, C.P.; Gu, Y.; Gu, X.; Nallamothu, G.; Vieth, E.; Janga, S.C.; Ryan, M.; Badve, S.S. Splicing factor ESRP 1 controls ER -positive breast cancer by altering metabolic pathways. EMBO Rep. 2019, 20, e46078. [Google Scholar] [CrossRef]
- Swinnen, J.V.; Ulrix, W.; Heyns, W.; Verhoeven, G. Coordinate regulation of lipogenic gene expression by androgens: Evidence for a cascade mechanism involving sterol regulatory element binding proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 12975–12980. [Google Scholar] [CrossRef] [Green Version]
- Swinnen, J.V.; Heemers, H.; Deboel, L.; Foufelle, F.; Heyns, W.; Verhoeven, G. Stimulation of tumor-associated fatty acid synthase expression by growth factor activation of the sterol regulatory element-binding protein pathway. Oncogene 2000, 19, 5173–5181. [Google Scholar] [CrossRef] [Green Version]
- Heemers, H.; Maes, B.; Foufelle, F.; Heyns, W.; Verhoeven, G.; Swinnen, J.V. Androgens stimulate lipogenic gene expression in prostate cancer cells by activation of the sterol regulatory element-binding protein cleavage activating protein/sterol regulatory element-binding protein pathway. Mol. Endocrinol. 2001, 15, 1817–1828. [Google Scholar] [CrossRef]
- Menendez, J.A.; Oza, B.P.; Atlas, E.; Verma, V.A.; Mehmi, I.; Lupu, R. Inhibition of tumor-associated fatty acid synthase activity antagonizes estradiol- and tamoxifen-induced agonist transactivation of estrogen receptor (ER) in human endometrial adenocarcinoma cells. Oncogene 2004, 23, 4945–4958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menendez, J.A.; Vellon, L.; Espinoza, I.; Lupu, R. The metastasis inducer CCN1 (CYR61) activates the fatty acid synthase (FASN)-driven lipogenic phenotype in breast cancer cells. Oncoscience 2016, 3, 242–257. [Google Scholar] [CrossRef] [Green Version]
- Menéndez, J.; Oza, B.; Colomer, R.; Lupu, R. The estrogenic activity of synthetic progestins used in oral contraceptives enhances fatty acid synthase-dependent breast cancer cell proliferation and survival. Int. J. Oncol. 2005, 26, 1507–1515. [Google Scholar] [CrossRef]
- Lupu, R.; Menendez, J.A. Targeting Fatty Acid Synthase in Breast and Endometrial Cancer: An Alternative to Selective Estrogen Receptor Modulators? Endocrinol. 2006, 147, 4056–4066. [Google Scholar] [CrossRef] [PubMed]
- Kuhajda, F.P.; Pizer, E.S.; Li, J.N.; Mani, N.S.; Frehywot, G.L.; Townsend, C.A. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc. Natl. Acad. Sci. USA 2000, 97, 3450–3454. [Google Scholar] [CrossRef] [PubMed]
- Montesdeoca, N.; López, M.; Ariza, X.; Herrero, L.; Makowski, K. Inhibitors of lipogenic enzymes as a potential therapy against cancer. FASEB J. 2020, 34, 11355–11381. [Google Scholar] [CrossRef]
- Ventura, R.; Mordec, K.; Waszczuk, J.; Wang, Z.; Lai, J.; Fridlib, M.; Buckley, D.; Kemble, G.; Heuer, T.S. Inhibition of de novo Palmitate Synthesis by Fatty Acid Synthase Induces Apoptosis in Tumor Cells by Remodeling Cell Membranes, Inhibiting Signaling Pathways, and Reprogramming Gene Expression. EBioMedicine 2015, 2, 808–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, D.; Duke, G.; Heuer, T.S.; O’Farrell, M.; Wagman, A.S.; McCulloch, W.; Kemble, G. Fatty acid synthase—Modern tumor cell biology insights into a classical oncology target. Pharmacol. Ther. 2017, 177, 23–31. [Google Scholar] [CrossRef]
- Li, J.; Lu, Y.; Akbani, R.; Ju, Z.; Roebuck, P.L.; Liu, W.; Yang, J.-Y.; Broom, B.M.; Verhaak, R.G.W.; Kane, D.W.; et al. TCPA: A resource for cancer functional proteomics data. Nat. Methods 2013, 10, 1046–1047. [Google Scholar] [CrossRef] [Green Version]
- Yiling, L.; Akbani, R.; Zhao, W.; Lu, Y.; Weinstein, J.N.; Mills, G.B.; Liang, H. Explore, Visualize, and Analyze Functional Cancer Proteomic Data Using the Cancer Proteome Atlas. Cancer Res. 2017, 77, e51–e54. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.-J.M.; Li, J.; Wang, Y.; Akbani, R.; Lu, Y.; Mills, G.B.; Liang, H. TCPA v3.0: An Integrative Platform to Explore the Pan-Cancer Analysis of Functional Proteomic Data. Mol. Cell. Proteomics 2019, 18, S15–S25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arpino, G.; Gutierrez, C.; Weiss, H.; Rimawi, M.; Massarweh, S.; Bharwani, L.; De Placido, S.; Osborne, C.K.; Schiff, R. Treatment of Human Epidermal Growth Factor Receptor 2-Overexpressing Breast Cancer Xenografts With Multiagent HER-Targeted Therapy. J. Natl. Cancer Inst. 2007, 99, 694–705. [Google Scholar] [CrossRef] [Green Version]
- Loi, S.; Haibe-Kains, B.; Desmedt, C.; Lallemand, F.; Tutt, A.M.; Gillet, C.; Ellis, P.; Harris, A.; Bergh, J.; Foekens, J.A.; et al. Definition of Clinically Distinct Molecular Subtypes in Estrogen Receptor–Positive Breast Carcinomas through Genomic Grade. J. Clin. Oncol. 2007, 25, 1239–1246. [Google Scholar] [CrossRef]
- Loi, S.; Sotiriou, C.; Haibe-Kains, B.; Lallemand, F.; Conus, N.M.; Piccart, M.J.; Speed, T.P.; McArthur, G.A. Gene expression profiling identifies activated growth factor signaling in poor prognosis (Luminal-B) estrogen receptor positive breast cancer. BMC Med Genomics 2009, 2, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.F.; Infante, J.R. Molecular Pathways: Fatty Acid Synthase. Clin. Cancer Res. 2015, 21, 5434–5438. [Google Scholar] [CrossRef] [Green Version]
- Menendez, J.A.; Mehmi, I.; Papadimitropoulou, A.; Steen, T.V.; Cuyàs, E.; Verdura, S.; Espinoza, I.; Vellon, L.; Atlas, E.; Lupu, R. Fatty Acid Synthase Is a Key Enabler for Endocrine Resistance in Heregulin-Overexpressing Luminal B-Like Breast Cancer. Int. J. Mol. Sci. 2020, 21, 7661. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menendez, J.A.; Papadimitropoulou, A.; Vander Steen, T.; Cuyàs, E.; Oza-Gajera, B.P.; Verdura, S.; Espinoza, I.; Vellon, L.; Mehmi, I.; Lupu, R. Fatty Acid Synthase Confers Tamoxifen Resistance to ER+/HER2+ Breast Cancer. Cancers 2021, 13, 1132. https://doi.org/10.3390/cancers13051132
Menendez JA, Papadimitropoulou A, Vander Steen T, Cuyàs E, Oza-Gajera BP, Verdura S, Espinoza I, Vellon L, Mehmi I, Lupu R. Fatty Acid Synthase Confers Tamoxifen Resistance to ER+/HER2+ Breast Cancer. Cancers. 2021; 13(5):1132. https://doi.org/10.3390/cancers13051132
Chicago/Turabian StyleMenendez, Javier A., Adriana Papadimitropoulou, Travis Vander Steen, Elisabet Cuyàs, Bharvi P. Oza-Gajera, Sara Verdura, Ingrid Espinoza, Luciano Vellon, Inderjit Mehmi, and Ruth Lupu. 2021. "Fatty Acid Synthase Confers Tamoxifen Resistance to ER+/HER2+ Breast Cancer" Cancers 13, no. 5: 1132. https://doi.org/10.3390/cancers13051132
APA StyleMenendez, J. A., Papadimitropoulou, A., Vander Steen, T., Cuyàs, E., Oza-Gajera, B. P., Verdura, S., Espinoza, I., Vellon, L., Mehmi, I., & Lupu, R. (2021). Fatty Acid Synthase Confers Tamoxifen Resistance to ER+/HER2+ Breast Cancer. Cancers, 13(5), 1132. https://doi.org/10.3390/cancers13051132