
Annexin A1 Is Required for Efficient Tumor Initiation and Cancer Stem Cell Maintenance in a Model of Human Breast Cancer

 ,
,  , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results and Discussion
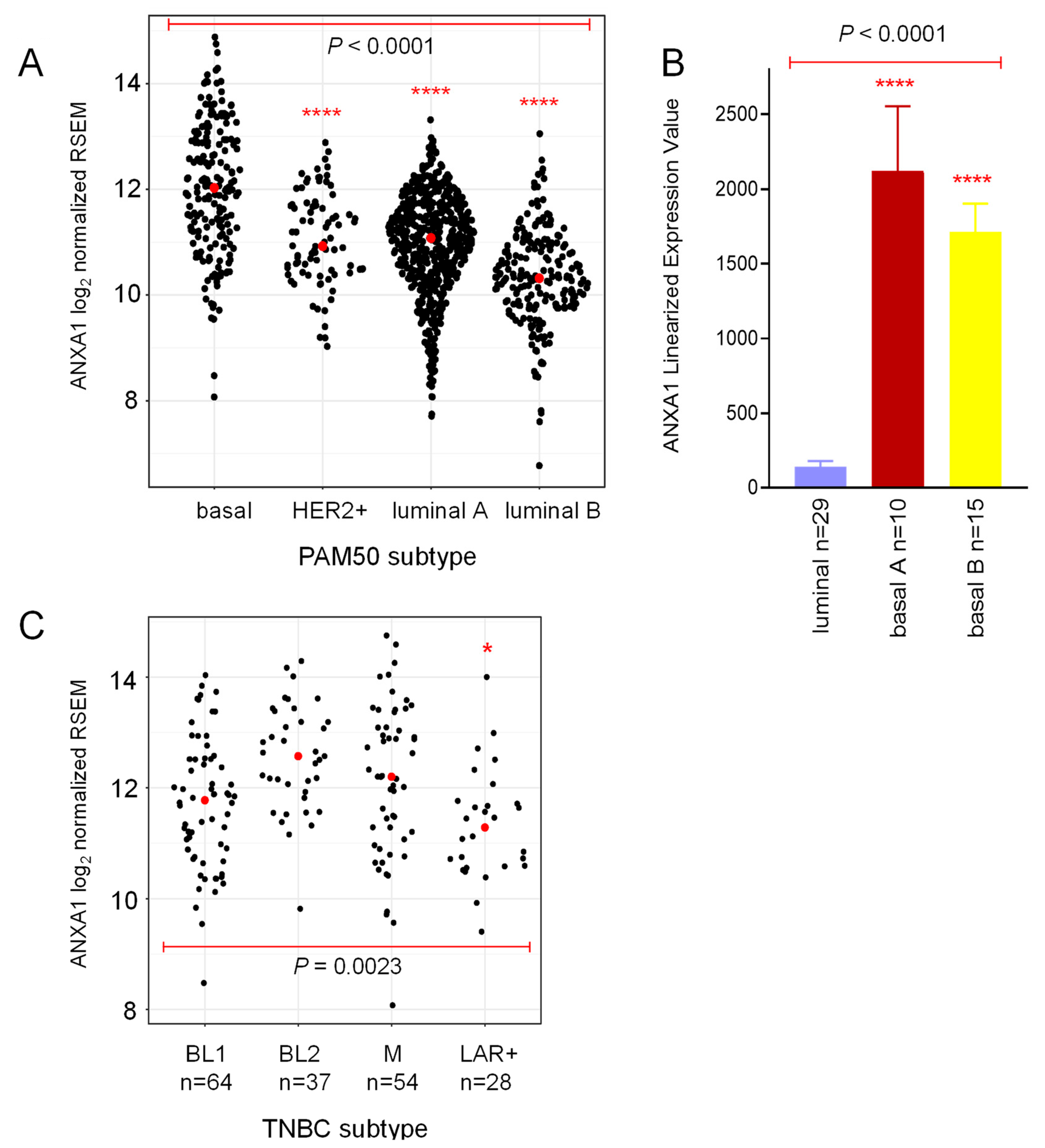
2.1. Annexin 1 Expression Is Associated with Worse Prognosis in Basal-Like Breast Cancer
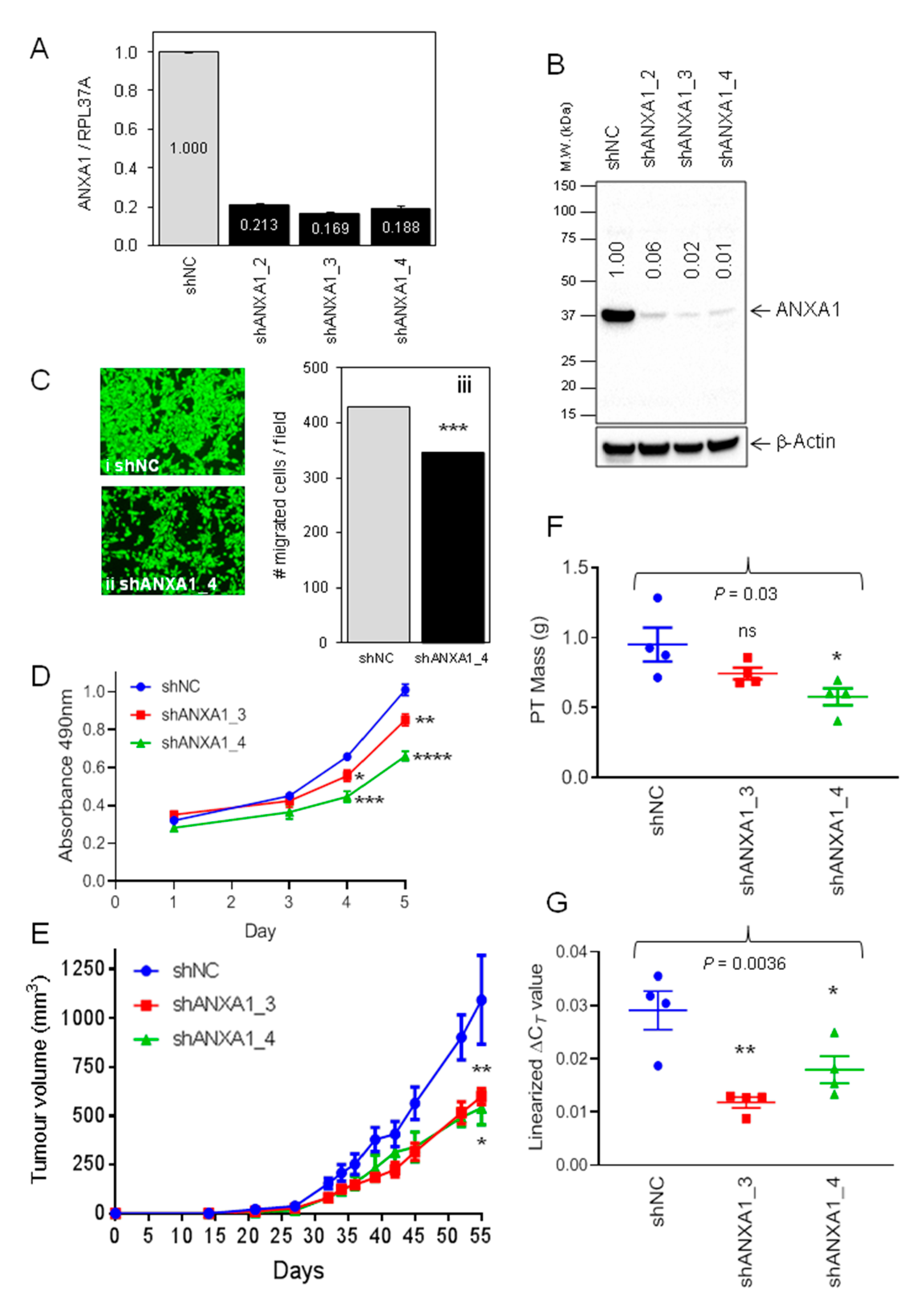
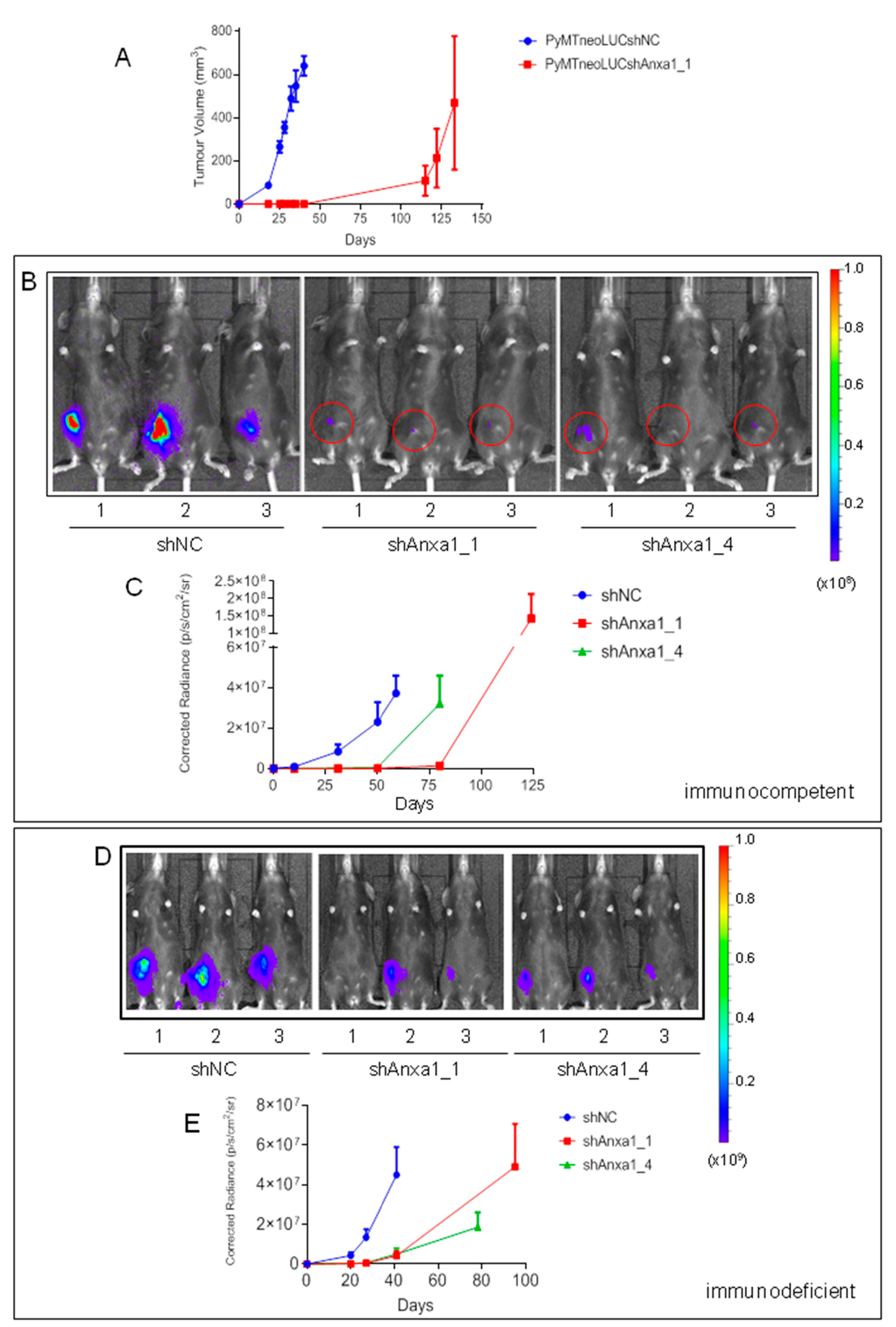
2.2. Evaluation of Annexin A1 Function in Xenograft Mouse Models of Breast Cancer
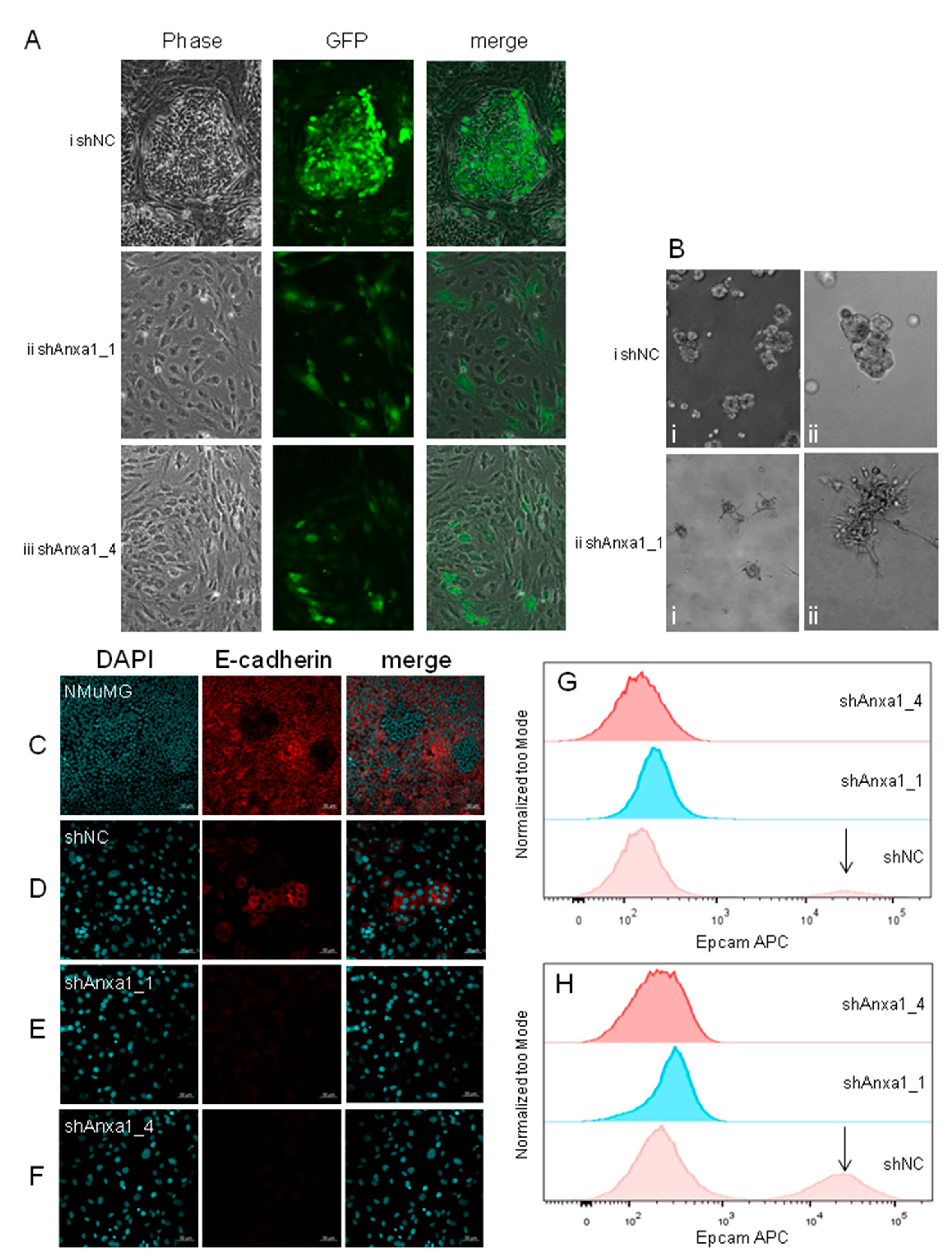
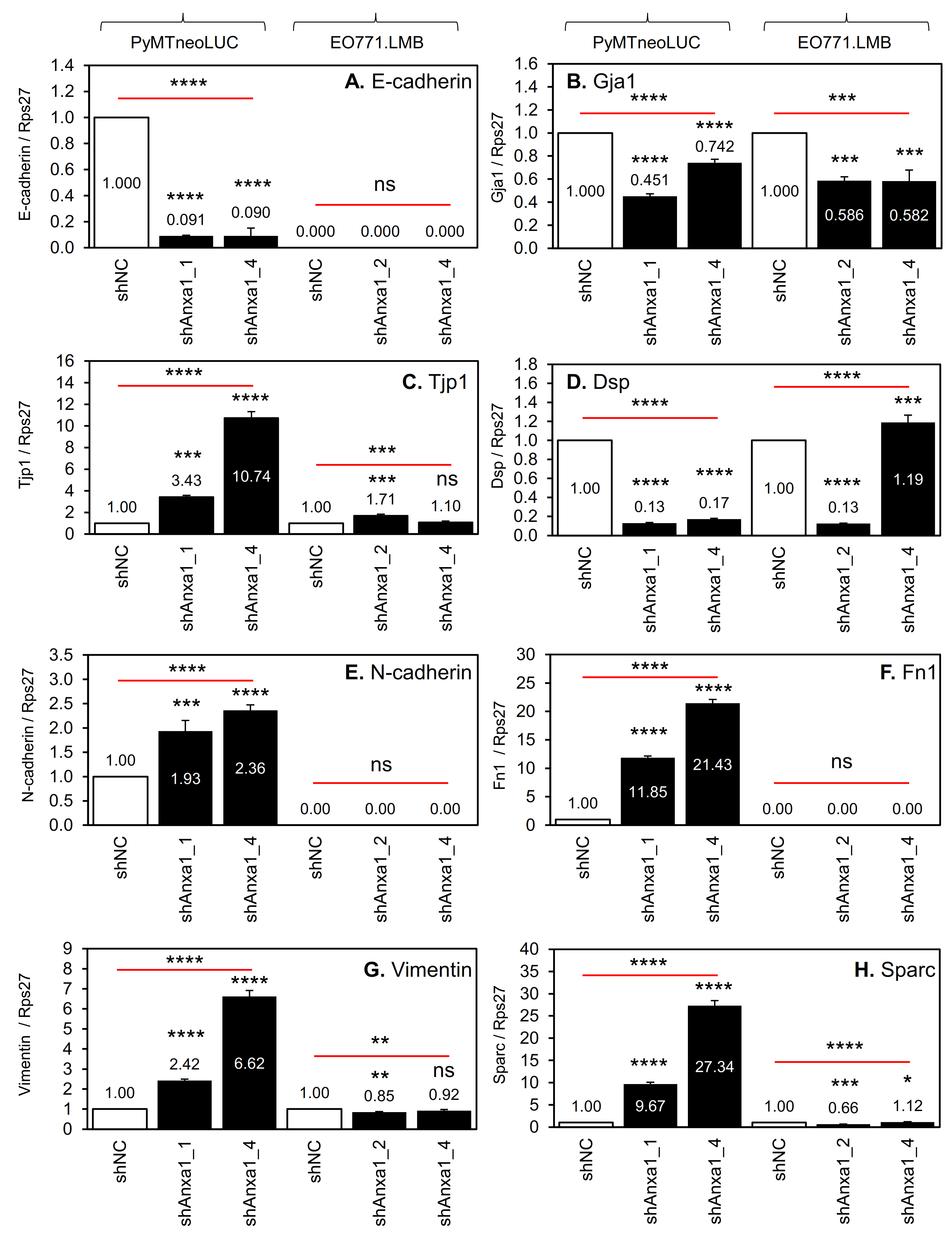
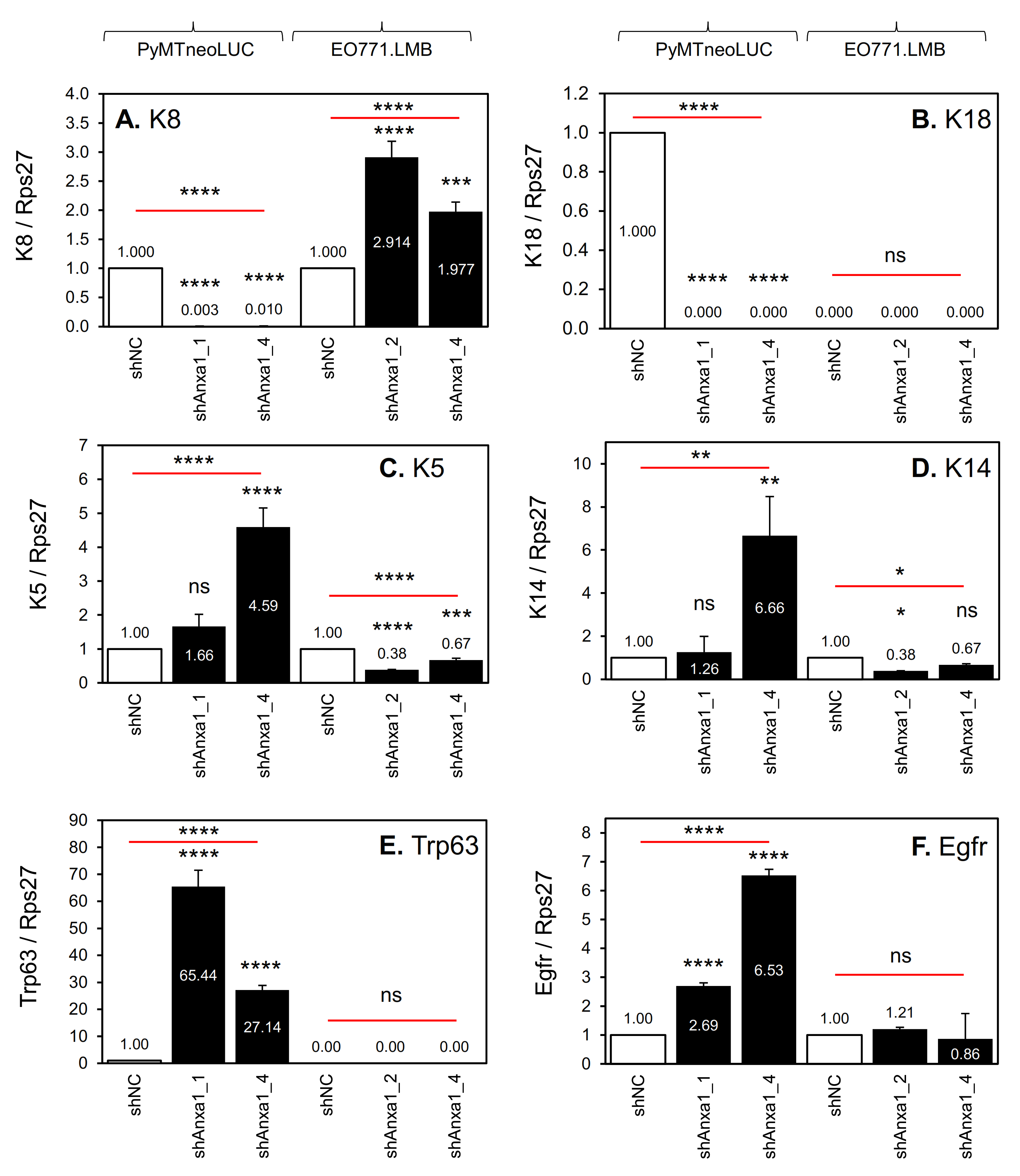
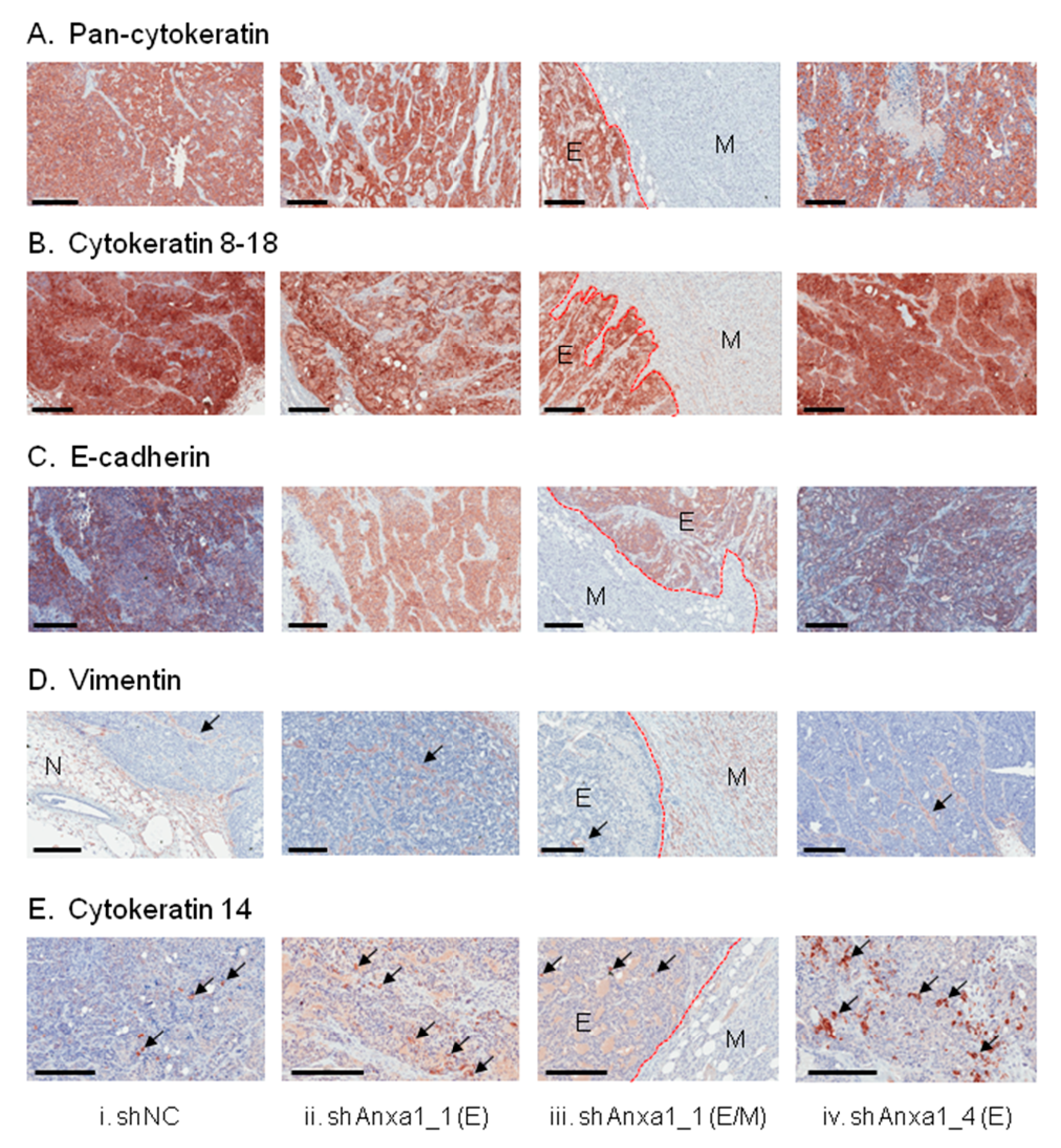
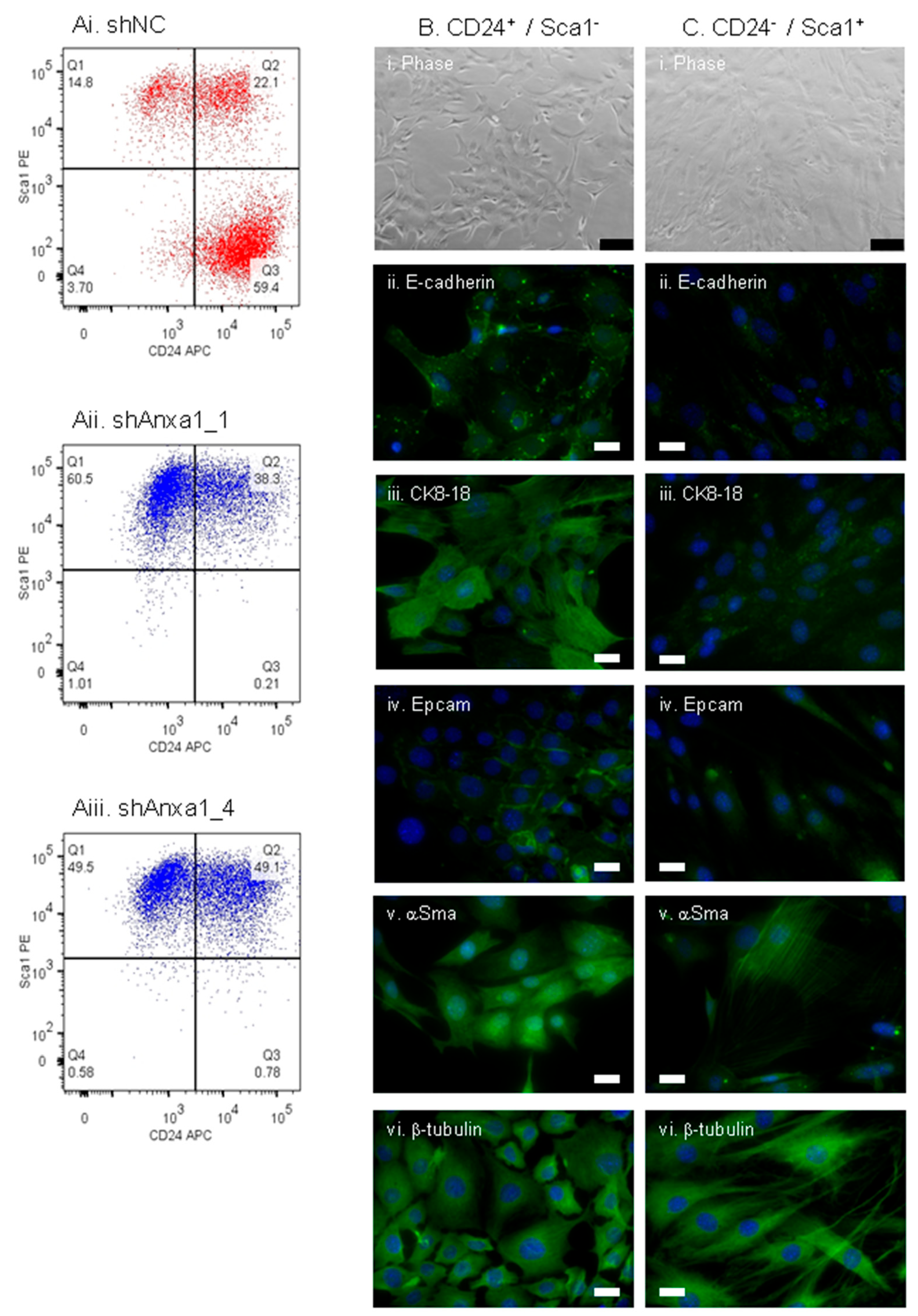
2.3. Suppression of Annexin A1 in PyMTneoLUC Cells Results in Epithelial-to-Mesenchymal Transition and Loss of a Putative Tumor-Initiating Cell Population
3. Materials and Methods
3.1. Cell Culture
3.2. Generation of Stable Annexin A1 Knockdown Cell Lines
3.3. Western Blotting
3.4. Antibodies
3.5. 2D In Vitro Proliferation Assays
3.6. In Vitro Analysis of Cell Motility
3.7. Quantitative Real-Time RT-PCR (qRT-PCR)
3.8. Flow Cytometry
3.9. Immunofluorescence and Phase Contrast Microscopy
3.10. Confocal Microscopy
3.11. Monitoring of Tumor Growth In Vivo
3.12. Histology and Immunohistochemistry
3.13. Bioinformatics
3.14. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, L.; Winer, E.; Viale, G.; Cameron, D.; Gianni, L. Triple-negative breast cancer: Disease entity or title of convenience? Nat. Rev. Clin. Oncol. 2010, 7, 683–692. [Google Scholar] [CrossRef]
- Denkert, C.; Liedtke, C.; Tutt, A.; von Minckwitz, G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet 2017, 389, 2430–2442. [Google Scholar] [CrossRef] [Green Version]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Pietenpol, J.A. Clinical implications of molecular heterogeneity in triple negative breast cancer. Breast 2015, 24, S36–S40. [Google Scholar] [CrossRef] [PubMed]
- Ring, B.Z.; Hout, D.R.; Morris, S.W.; Lawrence, K.; Schweitzer, B.L.; Bailey, D.B.; Lehmann, B.D.; Pietenpol, J.A.; Seitz, R.S. Generation of an algorithm based on minimal gene sets to clinically subtype triple negative breast cancer patients. BMC Cancer 2016, 16, 143. [Google Scholar] [CrossRef] [Green Version]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Prat, A.; Adamo, B.; Cheang, M.C.; Anders, C.K.; Carey, L.A.; Perou, C.M. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist 2013, 18, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [Green Version]
- Prat, A.; Parker, J.S.; Fan, C.; Perou, C.M. PAM50 assay and the three-gene model for identifying the major and clinically relevant molecular subtypes of breast cancer. Breast Cancer Res. Treat. 2012, 135, 301–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 2005, 6, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Perretti, M.; D′Acquisto, F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 2009, 9, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Cirino, G.; Flower, R.J.; Browning, J.L.; Sinclair, L.K.; Pepinsky, R.B. Recombinant human lipocortin 1 inhibits thromboxane release from guinea-pig isolated perfused lung. Nature 1987, 328, 270–272. [Google Scholar] [CrossRef] [PubMed]
- Foo, S.L.; Yap, G.; Cui, J.; Lim, L.H.K. Annexin-A1—A Blessing or a Curse in Cancer? Trends Mol. Med. 2019, 25, 315–327. [Google Scholar] [CrossRef]
- Boudhraa, Z.; Bouchon, B.; Viallard, C.; D′Incan, M.; Degoul, F. Annexin A1 localization and its relevance to cancer. Clin. Sci. 2016, 130, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445e.18. [Google Scholar] [CrossRef] [Green Version]
- Khau, T.; Langenbach, S.Y.; Schuliga, M.; Harris, T.; Johnstone, C.N.; Anderson, R.L.; Stewart, A.G. Annexin-1 signals mitogen-stimulated breast tumor cell proliferation by activation of the formyl peptide receptors (FPRs) 1 and 2. FASEB J. 2011, 25, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Y.; Yao, X.; Ping, Y.; Jiang, T.; Liu, Q.; Xu, S.; Huang, J.; Mou, H.; Gong, W.; et al. Annexin 1 released by necrotic human glioblastoma cells stimulates tumor cell growth through the formyl peptide receptor 1. Am. J. Pathol. 2011, 179, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Ma, Y.; Baracco, E.E.; Sistigu, A.; Enot, D.P.; Pietrocola, F.; Yang, H.; Adjemian, S.; Chaba, K.; Semeraro, M.; et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 2015, 350, 972–978. [Google Scholar] [CrossRef]
- Moraes, L.A.; Kar, S.; Foo, S.L.; Gu, T.; Toh, Y.Q.; Ampomah, P.B.; Sachaphibulkij, K.; Yap, G.; Zharkova, O.; Lukman, H.M.; et al. Annexin-A1 enhances breast cancer growth and migration by promoting alternative macrophage polarization in the tumour microenvironment. Sci. Rep. 2017, 7, 17925. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.D.; Boulay, F.; Wang, J.M.; Dahlgren, C.; Gerard, C.; Parmentier, M.; Serhan, C.N.; Murphy, P.M. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol. Rev. 2009, 61, 119–161. [Google Scholar] [CrossRef]
- Spurr, L.; Nadkarni, S.; Pederzoli-Ribeil, M.; Goulding, N.J.; Perretti, M.; D′Acquisto, F. Comparative analysis of Annexin A1-formyl peptide receptor 2/ALX expression in human leukocyte subsets. Int. Immunopharmacol. 2011, 11, 55–66. [Google Scholar] [CrossRef]
- Bai, F.; Zhang, P.; Fu, Y.; Chen, H.; Zhang, M.; Huang, Q.; Li, D.; Li, B.; Wu, K. Targeting ANXA1 abrogates Treg-mediated immune suppression in triple-negative breast cancer. J. Immunother. Cancer 2020, 8, e000169. [Google Scholar] [CrossRef]
- de Graauw, M.; van Miltenburg, M.H.; Schmidt, M.K.; Pont, C.; Lalai, R.; Kartopawiro, J.; Pardali, E.; Le Devedec, S.E.; Smit, V.T.; van der Wal, A.; et al. Annexin A1 regulates TGF-beta signaling and promotes metastasis formation of basal-like breast cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 6340–6345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yom, C.K.; Han, W.; Kim, S.W.; Kim, H.S.; Shin, H.C.; Chang, J.N.; Koo, M.; Noh, D.Y.; Moon, B.I. Clinical significance of annexin A1 expression in breast cancer. J. Breast Cancer 2011, 14, 262–268. [Google Scholar] [CrossRef]
- Wang, L.P.; Bi, J.; Yao, C.; Xu, X.D.; Li, X.X.; Wang, S.M.; Li, Z.L.; Zhang, D.Y.; Wang, M.; Chang, G.Q. Annexin A1 expression and its prognostic significance in human breast cancer. Neoplasma 2010, 57, 253–259. [Google Scholar] [CrossRef]
- Maschler, S.; Gebeshuber, C.A.; Wiedemann, E.M.; Alacakaptan, M.; Schreiber, M.; Custic, I.; Beug, H. Annexin A1 attenuates EMT and metastatic potential in breast cancer. EMBO Mol. Med. 2010, 2, 401–414. [Google Scholar] [CrossRef]
- Sobral-Leite, M.; Wesseling, J.; Smit, V.T.; Nevanlinna, H.; van Miltenburg, M.H.; Sanders, J.; Hofland, I.; Blows, F.M.; Coulson, P.; Patrycja, G.; et al. Annexin A1 expression in a pooled breast cancer series: Association with tumor subtypes and prognosis. BMC Med. 2015, 13, 156. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Li, Y.; Edelweiss, M.; Arun, B.; Rosen, D.; Resetkova, E.; Wu, Y.; Liu, J.; Sahin, A.; Albarracin, C.T. Loss of annexin A1 expression in breast cancer progression. Appl. Immunohistochem. Mol. Morphol. 2008, 16, 530–534. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Ganesan, N.; Tachibana, K.; Rajapakshe, K.; Albarracin, C.T.; Gunaratne, P.H.; Coarfa, C.; Bedrosian, I. Annexin A1 Preferentially Predicts Poor Prognosis of Basal-Like Breast Cancer Patients by Activating mTOR-S6 Signaling. PLoS ONE 2015, 10, e0127678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okano, M.; Kumamoto, K.; Saito, M.; Onozawa, H.; Saito, K.; Abe, N.; Ohtake, T.; Takenoshita, S. Upregulated Annexin A1 promotes cellular invasion in triple-negative breast cancer. Oncol. Rep. 2015, 33, 1064–1070. [Google Scholar] [CrossRef] [Green Version]
- Okano, M.; Oshi, M.; Butash, A.L.; Katsuta, E.; Tachibana, K.; Saito, K.; Okayama, H.; Peng, X.; Yan, L.; Kono, K.; et al. Triple-Negative Breast Cancer with High Levels of Annexin A1 Expression Is Associated with Mast Cell Infiltration, Inflammation, and Angiogenesis. Int. J. Mol. Sci. 2019, 20, 4197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, L.D.; Vishwanatha, J.K. Prognostic impact of AnxA1 and AnxA2 gene expression in triple-negative breast cancer. Oncotarget 2018, 9, 2697–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swa, H.L.; Shaik, A.A.; Lim, L.H.; Gunaratne, J. Mass spectrometry based quantitative proteomics and integrative network analysis accentuates modulating roles of annexin-1 in mammary tumorigenesis. Proteomics 2015, 15, 408–418. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Prat, A.; Karginova, O.; Parker, J.S.; Fan, C.; He, X.; Bixby, L.; Harrell, J.C.; Roman, E.; Adamo, B.; Troester, M.; et al. Characterization of cell lines derived from breast cancers and normal mammary tissues for the study of the intrinsic molecular subtypes. Breast Cancer Res. Treat. 2013, 142, 237–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunus, J.M.; Smart, C.E.; Kutasovic, J.R.; Johnston, R.L.; Kalita-de Croft, P.; Miranda, M.; Rozali, E.N.; Vargas, A.C.; Reid, L.E.; Lorsy, E.; et al. Multidimensional phenotyping of breast cancer cell lines to guide preclinical research. Breast Cancer Res. Treat. 2018, 167, 289–301. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [Green Version]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.R.; Paish, E.C.; Lee, A.H.; Ellis, I.O. Breast carcinoma with basal differentiation: A proposal for pathology definition based on basal cytokeratin expression. Histopathology 2007, 50, 434–438. [Google Scholar] [CrossRef]
- Lerma, E.; Barnadas, A.; Prat, J. Triple negative breast carcinomas: Similarities and differences with basal like carcinomas. Appl. Immunohistochem. Mol. Morphol. 2009, 17, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, C.N.; Smith, Y.E.; Cao, Y.; Burrows, A.D.; Cross, R.S.; Ling, X.; Redvers, R.P.; Doherty, J.P.; Eckhardt, B.L.; Natoli, A.L.; et al. Functional and molecular characterisation of EO771.LMB tumours, a new C57BL/6-mouse-derived model of spontaneously metastatic mammary cancer. Dis. Model. Mech. 2015, 8, 237–251. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, C.N.; Pattison, A.D.; Gorringe, K.L.; Harrison, P.F.; Powell, D.R.; Lock, P.; Baloyan, D.; Ernst, M.; Stewart, A.G.; Beilharz, T.H.; et al. Functional and genomic characterisation of a xenograft model system for the study of metastasis in triple-negative breast cancer. Dis. Model. Mech. 2018, 11, dmm032250. [Google Scholar] [CrossRef] [Green Version]
- Espinosa Fernandez, J.R.; Eckhardt, B.L.; Lee, J.; Lim, B.; Pearson, T.; Seitz, R.S.; Hout, D.R.; Schweitzer, B.L.; Nielsen, T.J.; Lawrence, O.R.; et al. Identification of triple-negative breast cancer cell lines classified under the same molecular subtype using different molecular characterization techniques: Implications for translational research. PLoS ONE 2020, 15, e0231953. [Google Scholar] [CrossRef] [PubMed]
- Fietz, E.R.; Keenan, C.R.; Lopez-Campos, G.; Tu, Y.; Johnstone, C.N.; Harris, T.; Stewart, A.G. Glucocorticoid resistance of migration and gene expression in a daughter MDA-MB-231 breast tumour cell line selected for high metastatic potential. Sci. Rep. 2017, 7, 43774. [Google Scholar] [CrossRef] [Green Version]
- Chang, X.Z.; Li, D.Q.; Hou, Y.F.; Wu, J.; Lu, J.S.; Di, G.H.; Jin, W.; Ou, Z.L.; Shen, Z.Z.; Shao, Z.M. Identification of the functional role of peroxiredoxin 6 in the progression of breast cancer. Breast Cancer Res. 2007, 9, R76. [Google Scholar] [CrossRef] [Green Version]
- Sampaio, N.G.; Kocan, M.; Schofield, L.; Pfleger, K.D.G.; Eriksson, E.M. Investigation of interactions between TLR2, MyD88 and TIRAP by bioluminescence resonance energy transfer is hampered by artefacts of protein overexpression. PLoS ONE 2018, 13, e0202408. [Google Scholar] [CrossRef] [PubMed]
- Haaksma, C.J.; Schwartz, R.J.; Tomasek, J.J. Myoepithelial cell contraction and milk ejection are impaired in mammary glands of mice lacking smooth muscle alpha-actin. Biol. Reprod. 2011, 85, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Moumen, M.; Chiche, A.; Cagnet, S.; Petit, V.; Raymond, K.; Faraldo, M.M.; Deugnier, M.A.; Glukhova, M.A. The mammary myoepithelial cell. Int. J. Dev. Biol. 2011, 55, 763–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, C.T.; Cardiff, R.D.; Muller, W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: A transgenic mouse model for metastatic disease. Mol. Cell. Biol. 1992, 12, 954–961. [Google Scholar] [CrossRef] [Green Version]
- Hollern, D.P.; Andrechek, E.R. A genomic analysis of mouse models of breast cancer reveals molecular features of mouse models and relationships to human breast cancer. Breast Cancer Res. 2014, 16, R59. [Google Scholar] [CrossRef] [Green Version]
- Rennhack, J.; To, B.; Wermuth, H.; Andrechek, E.R. Mouse Models of Breast Cancer Share Amplification and Deletion Events with Human Breast Cancer. J. Mammary Gland Biol. Neoplasia 2017, 22, 71–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, E.Y.; Jones, J.G.; Li, P.; Zhu, L.; Whitney, K.D.; Muller, W.J.; Pollard, J.W. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am. J. Pathol. 2003, 163, 2113–2126. [Google Scholar] [CrossRef] [Green Version]
- Goldman, J.P.; Blundell, M.P.; Lopes, L.; Kinnon, C.; Di Santo, J.P.; Thrasher, A.J. Enhanced human cell engraftment in mice deficient in RAG2 and the common cytokine receptor gamma chain. Br. J. Haematol. 1998, 103, 335–342. [Google Scholar] [CrossRef]
- Yi, M.; Schnitzer, J.E. Impaired tumor growth, metastasis, angiogenesis and wound healing in annexin A1-null mice. Proc. Natl. Acad. Sci. USA 2009, 106, 17886–17891. [Google Scholar] [CrossRef] [Green Version]
- Bumaschny, V.; Urtreger, A.; Diament, M.; Krasnapolski, M.; Fiszman, G.; Klein, S.; Joffe, E.B. Malignant myoepithelial cells are associated with the differentiated papillary structure and metastatic ability of a syngeneic murine mammary adenocarcinoma model. Breast Cancer Res. 2004, 6, R116–R129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savagner, P. Epithelial-mesenchymal transitions: From cell plasticity to concept elasticity. Curr. Top. Dev. Biol. 2015, 112, 273–300. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingthorsson, S.; Hilmarsdottir, B.; Kricker, J.; Magnusson, M.K.; Gudjonsson, T. Context-Dependent Function of Myoepithelial Cells in Breast Morphogenesis and Neoplasia. Curr. Mol. Biol. Rep. 2015, 1, 168–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inokuchi, J.; Lau, A.; Tyson, D.R.; Ornstein, D.K. Loss of annexin A1 disrupts normal prostate glandular structure by inducing autocrine IL-6 signaling. Carcinogenesis 2009, 30, 1082–1088. [Google Scholar] [CrossRef] [Green Version]
- Kendrick, H.; Regan, J.L.; Magnay, F.A.; Grigoriadis, A.; Mitsopoulos, C.; Zvelebil, M.; Smalley, M.J. Transcriptome analysis of mammary epithelial subpopulations identifies novel determinants of lineage commitment and cell fate. BMC Genom. 2008, 9, 591. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, T.O.; Hsu, F.D.; Jensen, K.; Cheang, M.; Karaca, G.; Hu, Z.; Hernandez-Boussard, T.; Livasy, C.; Cowan, D.; Dressler, L.; et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin. Cancer Res. 2004, 10, 5367–5374. [Google Scholar] [CrossRef] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillmore, C.M.; Kuperwasser, C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Louie, E.; Nik, S.; Chen, J.S.; Schmidt, M.; Song, B.; Pacson, C.; Chen, X.F.; Park, S.; Ju, J.; Chen, E.I. Identification of a stem-like cell population by exposing metastatic breast cancer cell lines to repetitive cycles of hypoxia and reoxygenation. Breast Cancer Res. 2010, 12, R94. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.C.; Deng, T.; Lehal, R.S.; Kim, J.; Zacksenhaus, E. Identification of tumorsphere- and tumor-initiating cells in HER2/Neu-induced mammary tumors. Cancer Res. 2007, 67, 8671–8681. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Knight, D.A.; Smyth, M.J.; Stewart, T.J. Sensitivity of a novel model of mammary cancer stem cell-like cells to TNF-related death pathways. Cancer Immunol. Immunother. 2012, 61, 1255–1268. [Google Scholar] [CrossRef] [Green Version]
- Shackleton, M.; Vaillant, F.; Simpson, K.J.; Stingl, J.; Smyth, G.K.; Asselin-Labat, M.L.; Wu, L.; Lindeman, G.J.; Visvader, J.E. Generation of a functional mammary gland from a single stem cell. Nature 2006, 439, 84–88. [Google Scholar] [CrossRef]
- Ma, J.; Lanza, D.G.; Guest, I.; Uk-Lim, C.; Glinskii, A.; Glinsky, G.; Sell, S. Characterization of mammary cancer stem cells in the MMTV-PyMT mouse model. Tumour Biol. 2012, 33, 1983–1996. [Google Scholar] [CrossRef]
- Cho, R.W.; Wang, X.; Diehn, M.; Shedden, K.; Chen, G.Y.; Sherlock, G.; Gurney, A.; Lewicki, J.; Clarke, M.F. Isolation and molecular characterization of cancer stem cells in MMTV-Wnt-1 murine breast tumors. Stem Cells 2008, 26, 364–371. [Google Scholar] [CrossRef]
- Lanza, D.G.; Ma, J.; Guest, I.; Uk-Lim, C.; Glinskii, A.; Glinsky, G.; Sell, S. Tumor-derived mesenchymal stem cells and orthotopic site increase the tumor initiation potential of putative mouse mammary cancer stem cells derived from MMTV-PyMT mice. Tumour Biol. 2012, 33, 1997–2005. [Google Scholar] [CrossRef] [PubMed]
- Sleeman, K.E.; Kendrick, H.; Ashworth, A.; Isacke, C.M.; Smalley, M.J. CD24 staining of mouse mammary gland cells defines luminal epithelial, myoepithelial/basal and non-epithelial cells. Breast Cancer Res. 2006, 8, R7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafee, N.; Smith, C.R.; Wei, S.; Kim, Y.; Mills, G.B.; Hortobagyi, G.N.; Stanbridge, E.J.; Lee, E.Y. Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer Res. 2008, 68, 3243–3250. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Behbod, F.; Atkinson, R.L.; Landis, M.D.; Kittrell, F.; Edwards, D.; Medina, D.; Tsimelzon, A.; Hilsenbeck, S.; Green, J.E.; et al. Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res. 2008, 68, 4674–4682. [Google Scholar] [CrossRef] [Green Version]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Swift, S.; Lorens, J.; Achacoso, P.; Nolan, G.P. Rapid Production of Retroviruses for Efficient Gene Delivery to Mammalian Cells Using 293T Cell-Based Systems. Curr. Protoc. Immunol. 1999, 31, 10.17.14–10.17.29. [Google Scholar] [CrossRef]
- Eckhardt, B.L.; Parker, B.S.; van Laar, R.K.; Restall, C.M.; Natoli, A.L.; Tavaria, M.D.; Stanley, K.L.; Sloan, E.K.; Moseley, J.M.; Anderson, R.L. Genomic analysis of a spontaneous model of breast cancer metastasis to bone reveals a role for the extracellular matrix. Mol. Cancer Res. 2005, 3, 1–13. [Google Scholar] [PubMed]
- Stewart, T.J.; Abrams, S.I. Altered immune function during long-term host-tumor interactions can be modulated to retard autochthonous neoplastic growth. J. Immunol. 2007, 179, 2851–2859. [Google Scholar] [CrossRef]
- Tan, E.M.; Ryhanen, L.; Uitto, J. Proline analogues inhibit human skin fibroblast growth and collagen production in culture. J. Investig. Dermatol. 1983, 80, 261–267. [Google Scholar] [CrossRef] [Green Version]
- Owens, R.B.; Smith, H.S.; Hackett, A.J. Epithelial cell cultures from normal glandular tissue of mice. J. Natl. Cancer Inst. 1974, 53, 261–269. [Google Scholar] [CrossRef]
- Okawa, T.; Michaylira, C.Z.; Kalabis, J.; Stairs, D.B.; Nakagawa, H.; Andl, C.D.; Johnstone, C.N.; Klein-Szanto, A.J.; El-Deiry, W.S.; Cukierman, E.; et al. The functional interplay between EGFR overexpression, hTERT activation, and p53 mutation in esophageal epithelial cells with activation of stromal fibroblasts induces tumor development, invasion, and differentiation. Genes Dev. 2007, 21, 2788–2803. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, C.N.; Pattison, A.D.; Harrison, P.F.; Powell, D.R.; Lock, P.; Ernst, M.; Anderson, R.L.; Beilharz, T.H. FGF13 promotes metastasis of triple-negative breast cancer. Int. J. Cancer 2020, 147, 230–243. [Google Scholar] [CrossRef]
- Johnstone, C.N.; Mongroo, P.S.; Rich, A.S.; Schupp, M.; Bowser, M.J.; Delemos, A.S.; Tobias, J.W.; Liu, Y.; Hannigan, G.E.; Rustgi, A.K. Parvin-beta inhibits breast cancer tumorigenicity and promotes CDK9-mediated peroxisome proliferator-activated receptor gamma 1 phosphorylation. Mol. Cell. Biol. 2008, 28, 687–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TNBC Subtype | Abbreviation | Sample Size (n) | Hazard Ratio & (95% CI) | Logrank p |
|---|---|---|---|---|
| Basal-like-1 | BL-1 | 171 | 1.57 (0.96–2.55) | 0.0687 |
| Basal-like-2 * | BL-2 | 76 | 3.12 (1.09–8.92) | 0.0257 |
| Mesenchymal | M | 177 | 1.25 (0.80–1.96) | 0.3215 |
| Luminal androgen receptor positive | LAR | 203 | 0.87 (0.54–1.42) | 0.5791 |
| Basal intrinsic subtype | Basal | 618 | 1.77 (1.37–2.28) | <0.0001 |
| ER− | ER− | 801 | 0.88 (0.52–1.50) | 0.6380 |
| HER2+ | HER+ | 251 | 0.85 (0.56–1.28) | 0.4342 |
| Luminal A | LumA | 1933 | 0.97 (0.81–1.17) | 0.7736 |
| Luminal B | LumB | 1149 | 0.95 (0.77–1.17) | 0.6305 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnstone, C.N.; Tu, Y.; Langenbach, S.; Baloyan, D.; Pattison, A.D.; Lock, P.; Britt, K.L.; Lehmann, B.D.; Beilharz, T.H.; Ernst, M.; et al. Annexin A1 Is Required for Efficient Tumor Initiation and Cancer Stem Cell Maintenance in a Model of Human Breast Cancer. Cancers 2021, 13, 1154. https://doi.org/10.3390/cancers13051154
Johnstone CN, Tu Y, Langenbach S, Baloyan D, Pattison AD, Lock P, Britt KL, Lehmann BD, Beilharz TH, Ernst M, et al. Annexin A1 Is Required for Efficient Tumor Initiation and Cancer Stem Cell Maintenance in a Model of Human Breast Cancer. Cancers. 2021; 13(5):1154. https://doi.org/10.3390/cancers13051154
Chicago/Turabian StyleJohnstone, Cameron N., Yan Tu, Shenna Langenbach, David Baloyan, Andrew D. Pattison, Peter Lock, Kara L. Britt, Brian D. Lehmann, Traude H. Beilharz, Matthias Ernst, and et al. 2021. "Annexin A1 Is Required for Efficient Tumor Initiation and Cancer Stem Cell Maintenance in a Model of Human Breast Cancer" Cancers 13, no. 5: 1154. https://doi.org/10.3390/cancers13051154
APA StyleJohnstone, C. N., Tu, Y., Langenbach, S., Baloyan, D., Pattison, A. D., Lock, P., Britt, K. L., Lehmann, B. D., Beilharz, T. H., Ernst, M., Anderson, R. L., & Stewart, A. G. (2021). Annexin A1 Is Required for Efficient Tumor Initiation and Cancer Stem Cell Maintenance in a Model of Human Breast Cancer. Cancers, 13(5), 1154. https://doi.org/10.3390/cancers13051154