
The Tumor Suppressor MTUS1/ATIP1 Modulates Tumor Promotion in Glioma: Association with Epigenetics and DNA Repair

 ,
,  and
and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Assessment of MTUS1/ATIP1-Expression in Glioma
2.2. Methylation Status of the 5′ upstream MTUS1-Transcription Start Site
2.3. ATIP1 Modulates Cell Proliferation, Motility and Survival
2.4. ATIP1 Modulates ERK and AKT Activity
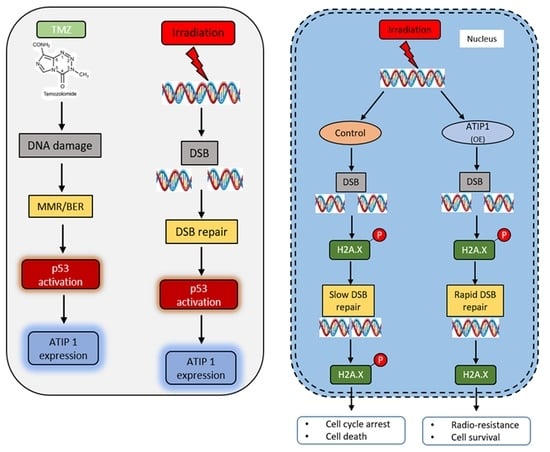
2.5. ATIP1 Expression Is p53 Dependent and Modulates Irradiation-Induced Growth Reduction
2.6. ATIP1 Promotes DNA Double-Strand Break (DSB) Repair
3. Discussion
4. Materials and Methods
4.1. Clinical Sample Collection and Processing
4.2. RNA Isolation, PCR and Quantitative Real-Time PCR (qRT-PCR)
4.3. Western Blotting
4.4. Histology and Immunohistochemistry
4.5. Cell Culture and Reagents
4.6. Plasmids and Transfection
4.7. CpG Island Identification, Bisulfite Conversion and Methylation Analysis
4.8. Cell Migration, Invasion and Adhesion Assay
4.9. siRNA Transfection
4.10. Animal Experiments
4.11. Radio-Sensitivity Analysis
4.12. Cell Cycle Analyses
4.13. Data Acquisition and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients with Glioblastoma. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beier, D.; Hau, P.; Proescholdt, M.A.; Lohmeier, A.; Wischhusen, J.; Oefner, P.J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C.P. CD133+ and CD133− Glioblastoma-Derived Cancer Stem Cells Show Differential Growth Characteristics and Molecular Profiles. Cancer Res. 2007, 67, 4010–4015. [Google Scholar] [CrossRef] [Green Version]
- Adamson, C.; O Kanu, O.; I Mehta, A.; Di, C.; Lin, N.; Mattox, A.K.; Bigner, D.D. Glioblastoma multiforme: A review of where we have been and where we are going. Expert Opin. Investig. Drugs 2009, 18, 1061–1083. [Google Scholar] [CrossRef]
- Klutstein, M.; Nejman, D.; Greenfield, R.; Cedar, H. DNA Methylation in Cancer and Aging. Cancer Res. 2016, 76, 3446–3450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabrowski, M.J.; Wojtas, B. Global DNA Methylation Patterns in Human Gliomas and Their Interplay with Other Epigenetic Modifications. Int. J. Mol. Sci. 2019, 20, 3478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nat. Cell Biol. 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R.; Pugh, J.E. DNA Modification Mechanisms and Gene Activity during Development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef]
- Kitange, G.J.; Carlson, B.L.; Mladek, A.C.; Decker, P.A.; Schroeder, M.A.; Wu, W.; Grogan, P.T.; Giannini, C.; Ballman, K.V.; Buckner, J.C.; et al. Evaluation of MGMT promoter methylation status and correlation with temozolomide response in orthotopic glioblastoma xenograft model. J. Neuro-Oncol. 2008, 92, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ippen, F.M.; Grosch, J.K.; Subramanian, M.; Kuter, B.M.; Liederer, B.M.; Plise, E.G.; Mora, J.L.; Nayyar, N.; Schmidt, S.P.; Giobbie-Hurder, A.; et al. Targeting the PI3K/Akt/mTOR pathway with the pan-Akt inhibitor GDC-0068 in PIK3CA-mutant breast cancer brain metastases. Neuro-Oncol. 2019, 21, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartos, M.; Jirik, R.; et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef] [Green Version]
- Parker, A.L.; Ekavallaris, M.; McCarroll, J.A. Microtubules and Their Role in Cellular Stress in Cancer. Front. Oncol. 2014, 4, 153. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dueva, R.; Iliakis, G. Alternative pathways of non-homologous end joining (NHEJ) in genomic instability and cancer. Transl. Cancer Res. 2013, 2, 163–177. [Google Scholar] [CrossRef]
- Osada, H.; Takahashi, T. Genetic alterations of multiple tumor suppressors and oncogenes in the carcinogenesis and progresion of lung cancer. Oncogene 2002, 21, 7421–7434. [Google Scholar] [CrossRef] [PubMed]
- Krug, U.; Ganser, A.; Koeffler, H.P. Tumor suppressor genes in normal and malignant hematopoiesis. Oncogene 2002, 21, 3475–3495. [Google Scholar] [CrossRef] [Green Version]
- Bozgeyik, I.; Yumrutas, O.; Bozgeyik, E. MTUS1, a gene encoding angiotensin-II type 2 (AT2) receptor-interacting proteins, in health and disease, with special emphasis on its role in carcinogenesis. Gene 2017, 626, 54–63. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, N.; Cai, Y.; Li, S.; Zheng, C.; Jin, Y.; Yu, T.; Wang, A.; Zhou, X. Down-regulation of tumor suppressor MTUS1/ATIP is associated with enhanced proliferation, poor differentiation and poor prognosis in oral tongue squamous cell carcinoma. Mol. Oncol. 2011, 6, 73–80. [Google Scholar] [CrossRef]
- Di Benedetto, M.; Bièche, I.; Deshayes, F.; Vacher, S.; Nouet, S.; Collura, V.; Seitz, I.; Louis, S.; Pineau, P.; Amsellem-Ouazana, D.; et al. Structural organization and expression of human MTUS1, a candidate 8p22 tumor suppressor gene encoding a family of angiotensin II AT2 receptor-interacting proteins, ATIP. Gene 2006, 380, 127–136. [Google Scholar] [CrossRef]
- Seibold, S.; Rudroff, C.; Weber, M.; Galle, J.; Wanner, C.; Marx, M. Identification of a new tumor suppressor gene located at chromosome 8p21.3. FASEB J. 2003, 17, 1180–1182. [Google Scholar] [CrossRef]
- Payet, G.N.; Guimond, M.O.; Bilodeau, L.; Wallinder, C.; Alterman, M.; Hallberg, A. Angiotensin II, a neuropeptide at the frontier between endocrinology and neuroscience: Is there a link between the angiotensin II type 2 receptor and Alzheimer’s disease? Front. Endocrinol. 2011, 2, 17. [Google Scholar] [CrossRef] [Green Version]
- Pinter, M.; Jain, R.K. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci. Transl. Med. 2017, 9, 5616. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Chen, Z.; Liu, X.; Wang, C.; Jin, Y.; Wang, Y.; Wang, A. p53 regulates the expression of human angiotensin II AT2 receptor interacting protein (ATIP1) gene. Oncol. Lett. 2011, 2, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Nouet, S.; Amzallag, N.; Li, J.M.; Louis, S.; Seitz, I.; Cui, T.X.; Alleaume, A.M.; Di Benedetto, M.; Boden, C.; Masson, M.; et al. Trans-inactivation of Receptor Tyrosine Kinases by Novel Angiotensin II AT2 Receptor-interacting Protein, ATIP. J. Biol. Chem. 2004, 279, 28989–28997. [Google Scholar] [CrossRef] [Green Version]
- Füchtbauer, L.; Hansen, T.H.; Khorooshi, R.; Owens, T. Expression of Astrocytic Type 2 Angiotensin Receptor in Central Nervous System Inflammation Correlates with Blood–Brain Barrier Breakdown. J. Mol. Neurosci. 2010, 42, 89–98. [Google Scholar] [CrossRef]
- Ferreira, R.S.; Le Rouzic, E.; Pawlowski, T.; Srivastava, A.; Goguet, M.F.; Nahmias, C. AT2 Receptor-Interacting Proteins ATIPs in the Brain. Int. J. Hypertens. 2013, 2013, 513047. [Google Scholar] [CrossRef] [Green Version]
- Broomfield, S.; Chow, B.L.; Xiao, W. MMS2, encoding a ubiquitin-conjugating-enzyme-like protein, is a member of the yeast error-free postreplication repair pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 5678–5683. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Belinsky, S.A.; Herman, J.G. Aberrant methylation of gene promoters in cancer—Concepts, misconcepts, and promise. J. Natl. Cancer Inst. 2000, 92, 1460–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brusky, J.; Zhu, Y.; Xiao, W. UBC13, a DNA-damage-inducible gene, is a member of the error-free postreplication repair pathway in Saccharomyces cerevisiae. Curr. Genet. 2000, 37, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Toss, A.; Tomasello, C.; Razzaboni, E.; Contu, G.; Grandi, G.; Cagnacci, A.; Schilder, R.J.; Cortesi, L. Hereditary Ovarian Cancer: Not OnlyBRCA1 and 2 Genes. Bio. Med. Res. Int. 2015, 2015, 341723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsopoulos, J. BRCA Mutations and Breast Cancer Prevention. Cancers 2018, 10, 524. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Guo, L.; Xi, J.; Wang, N.; Ping, H. Angiotensin II type 2 receptor–interacting protein 3a inhibits ovarian carcinoma metastasis via the extracellular HMGA2-mediated ERK/EMT pathway. Tumor Biol. 2017, 39, 3389. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Ding, X.; Chang, B.; Zhou, X.; Wang, A. MTUS1/ATIP3a down-regulation is associated with enhanced migration, invasion and poor prognosis in salivary adenoid cystic carcinoma. BMC Cancer 2015, 15, 203. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; He, Q.; Liu, Z.; Ding, X.; Zhou, X.; Wang, A. Angiotensin II type 2 receptor-interacting protein 3a suppresses proliferation, migration and invasion in tongue squamous cell carcinoma via the extracellular signal-regulated kinase-Snai2 pathway. Oncol. Lett. 2015, 11, 340–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuffin, L.L.J.; Rodriguez, F.; Giannini, C.; Scheithauer, B.; Necela, B.M.; Sarkaria, J.N.; Anastasiadis, P.Z. Misregulated E-Cadherin Expression Associated with an Aggressive Brain Tumor Phenotype. PLoS ONE 2010, 5, e13665. [Google Scholar] [CrossRef]
- Chen, H.; E Paradies, N.; Chaiken, F.M.; Brackenbury, R. E-cadherin mediates adhesion and suppresses cell motility via distinct mechanisms. J. Cell Sci. 1997, 110, 345–356. [Google Scholar] [PubMed]
- Gong, J.; Zhu, S.; Zhang, Y.; Wang, J. Interplay of VEGFa and MMP2 regulates invasion of glioblastoma. Tumor Biol. 2014, 35, 11879–11885. [Google Scholar] [CrossRef]
- Lau, M.T.; Klausen, C.; Leung, P.C.K. E-cadherin inhibits tumor cell growth by suppressing PI3K/Akt signaling via β-catenin-Egr1-mediated PTEN expression. Oncogene 2011, 30, 2753–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Guo, M.; Herman, J.G.; Clark, D.P. Aberrant Promoter Methylation Profiles of Tumor Suppressor Genes in Hepatocellular Carcinoma. Am. J. Pathol. 2003, 163, 1101–1107. [Google Scholar] [CrossRef] [Green Version]
- Razin, A.; Cedar, H. DNA Methylation and Gene Expression. Microbiol. Rev. 1991, 55, 451–458. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Varley, K.E.; Gertz, J.; Bowling, K.M.; Parker, S.L.; Reddy, T.E.; Behn, P.F.; Cross, M.K.; Williams, B.A.; Stamatoyannopoulos, J.A.; Crawford, G.E.; et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013, 23, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nat. Cell Biol. 2008, 454, 766–770. [Google Scholar] [CrossRef] [Green Version]
- Moarii, M.; Boeva, V.; Vert, J.P.; Reyal, F. Changes in correlation between promoter methylation and gene expression in cancer. BMC Genom. 2015, 16, 873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singkhorn, S.; Tantisira, M.H.; Tanasawet, S.; Hutamekalin, P.; Wongtawatchai, T.; Sukketsiri, W. Induction of keratinocyte migration by ECa 233 is mediated through FAK/Akt, ERK, and p38 MAPK signaling. Phytother. Res. 2018, 32, 1397–1403. [Google Scholar] [CrossRef] [PubMed]
- Wen, R.; Li, J.; Xu, X.; Cui, Z.; Xiao, W. Zebrafish Mms2 promotes K63-linked polyubiquitination and is involved in p53-mediated DNA-damage response. DNA Repair 2012, 11, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, P.; Capper, D. WHO 2016 Classification of gliomas. Neuropathol. Appl. Neurobiol. 2017, 44, 139–150. [Google Scholar] [CrossRef]
- Pandey, V.; Ranjan, N.; Narne, P.; Babu, P.P. Roscovitine effectively enhances antitumor activity of temozolomide in vitro and in vivo mediated by increased autophagy and Caspase-3 dependent apoptosis. Sci. Rep. 2019, 9, 5012. [Google Scholar] [CrossRef] [Green Version]
- Varghese, F.; Bukhari, A.B.; Malhotra, R.; De, A. IHC Profiler: An Open Source Plugin for the Quantitative Evaluation and Automated Scoring of Immunohistochemistry Images of Human Tissue Samples. PLoS ONE 2014, 9, e96801. [Google Scholar] [CrossRef] [Green Version]
- Pohl, U.; Wagenknecht, B.; Naumann, U.; Weller, M. p53 Enhances BAK and CD95 Expression in Human Malignant Glioma Cells but Does Not Enhance CD95L-Induced Apoptosis. Cell. Physiol. Biochem. 1999, 9, 29–37. [Google Scholar] [CrossRef]
- Van Meir, E.G.; Kikuchi, T.; Tada, M.; Li, H.; Diserens, A.C.; E Wojcik, B.; Huang, H.J.; Friedmann, T.; De Tribolet, N.; Cavenee, W.K. Analysis of the p53 gene and its expression in human glioblastoma cells. Cancer Res. 1994, 54, 649–652. [Google Scholar] [PubMed]
- Naumann, U.; Kügler, S.; Wolburg, H.; Wick, W.; Rascher, G.; Schulz, J.B.; Conseiller, E.; Bähr, M.; Weller, M. Chimeric tumor suppressor 1, a p53-derived chimeric tumor suppressor gene, kills p53 mutant and p53 wild-type glioma cells in synergy with irradiation and CD95 ligand. Cancer Res. 2001, 61, 5833–5842. [Google Scholar]
- Schindelin, J.; Carreras, A.I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filho, Z.A.; Braganhol, E.; Klafke, K.; Figueiró, F.; Terra, S.R.; Paludo, F.J.; Morrone, M.; Bristot, I.J.; Battastini, A.M.; Forcelini, C.M.; et al. Autophagy inhibition improves the efficacy of curcumin/temozolomide combination therapy in glioblastomas. Cancer Lett. 2015, 358, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Baumann, B.C.; Dorsey, J.F.; Benci, J.L.; Joh, D.Y.; Kao, G.D. Stereotactic intracranial implantation and in vivo biolumi-nescent imaging of tumor xenografts in a mouse model system of glioblastoma multiforme. J. Vis. Exp. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, S.N.; Chow, L.T.; Varghayee, N.; Rezmann, L.A.; Frauman, A.G.; Louis, W.J. The Expression of MTUS1/ATIP and Its Major Isoforms, ATIP1 and ATIP3, in Human Prostate Cancer. Cancers 2011, 3, 3824–3837. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranjan, N.; Pandey, V.; Panigrahi, M.K.; Klumpp, L.; Naumann, U.; Babu, P.P. The Tumor Suppressor MTUS1/ATIP1 Modulates Tumor Promotion in Glioma: Association with Epigenetics and DNA Repair. Cancers 2021, 13, 1245. https://doi.org/10.3390/cancers13061245
Ranjan N, Pandey V, Panigrahi MK, Klumpp L, Naumann U, Babu PP. The Tumor Suppressor MTUS1/ATIP1 Modulates Tumor Promotion in Glioma: Association with Epigenetics and DNA Repair. Cancers. 2021; 13(6):1245. https://doi.org/10.3390/cancers13061245
Chicago/Turabian StyleRanjan, Nikhil, Vimal Pandey, Manas Kumar Panigrahi, Lukas Klumpp, Ulrike Naumann, and Phanithi Prakash Babu. 2021. "The Tumor Suppressor MTUS1/ATIP1 Modulates Tumor Promotion in Glioma: Association with Epigenetics and DNA Repair" Cancers 13, no. 6: 1245. https://doi.org/10.3390/cancers13061245
APA StyleRanjan, N., Pandey, V., Panigrahi, M. K., Klumpp, L., Naumann, U., & Babu, P. P. (2021). The Tumor Suppressor MTUS1/ATIP1 Modulates Tumor Promotion in Glioma: Association with Epigenetics and DNA Repair. Cancers, 13(6), 1245. https://doi.org/10.3390/cancers13061245