
Multiple Myeloma: Heterogeneous in Every Way

 ,
,  ,
, 
{kind=link}
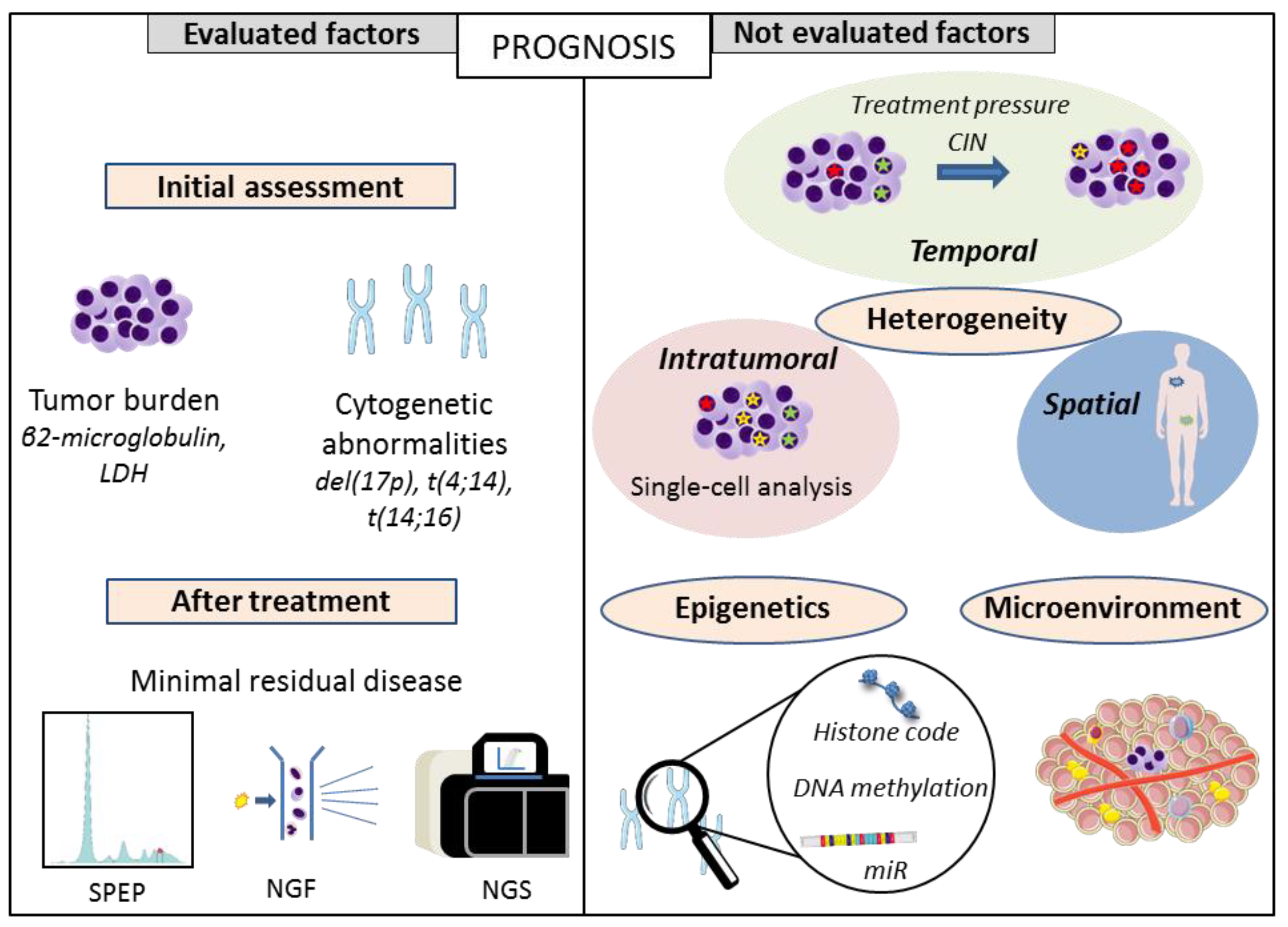
Abstract
:Simple Summary
Abstract
1. Introduction
2. Patient Prognosis: What We Know
2.1. Cytogenetic Abnormalities
2.2. Treatment Response and Minimal Residual Disease (MRD)
3. MM Heterogeneity as a Powerful Lever for Treatment Escape
3.1. Intratumoral Heterogeneity
3.2. Temporal Heterogeneity
3.3. Spatial Heterogeneity
3.4. Epigenetic, an Encouraging Line of Research
3.5. The Influence of the Microenvironment
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pawlyn, C.; Morgan, G.J. Evolutionary biology of high-risk multiple myeloma. Nat. Rev. Cancer 2017, 17, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Perrot, A.; Lauwers-Cances, V.; Tournay, E.; Hulin, C.; Chretien, M.-L.; Royer, B.; Dib, M.; Decaux, O.; Jaccard, A.; Belhadj, K.; et al. Development and Validation of a Cytogenetic Prognostic Index Predicting Survival in Multiple Myeloma. J. Clin. Oncol. 2019, 37, 1657–1665. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.G.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report from International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef]
- Corre, J.; Perrot, A.; Caillot, D.; Belhadj, K.; Hulin, C.; Leleu, X.; Mohty, M.; Facon, T.; Buisson, L.; Souto, L.D.; et al. del(17p) without TP53 mutation confers a poor prognosis in intensively treated newly diagnosed patients with multiple myeloma. Blood 2021, 137, 1192–1195. [Google Scholar] [CrossRef]
- Thakurta, A.; Ortiz, M.; Blecua, P.; Towfic, F.; Corre, J.; Serbina, N.V.; Flynt, E.; Yu, Z.; Yang, Z.; Palumbo, A.; et al. High subclonal fraction of 17p deletion is associated with poor prognosis in multiple myeloma. Blood 2019, 133, 1217–1221. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2019, 33, 159–170. [Google Scholar] [CrossRef]
- Hebraud, B.; Magrangeas, F.; Cleynen, A.; Lauwers-Cances, V.; Chretien, M.-L.; Hulin, C.; Leleu, X.; Yon, E.; Marit, G.; Karlin, L.; et al. Role of additional chromosomal changes in the prognostic value of t(4;14) and del(17p) in multiple myeloma: The IFM experience. Blood 2015, 125, 2095–2100. [Google Scholar] [CrossRef]
- Li, F.; Zhai, Y.-P.; Lai, T.; Zhao, Q.; Zhang, H.; Tang, Y.-M.; Hou, J. MB4-2/MB4-3 transcripts of IGH-MMSET fusion gene in t(4;14)pos multiple myeloma indicate poor prognosis. Oncotarget 2017, 8, 51608–51620. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Leleu, X.; Roussel, M.; Moreau, P.; Guerin-Charbonnel, C.; Caillot, D.; Marit, G.; Benboubker, L.; Voillat, L.; Mathiot, C.; et al. Bortezomib Plus Dexamethasone Induction Improves Outcome of Patients With t(4;14) Myeloma but Not Outcome of Patients With del(17p). J. Clin. Oncol. 2010, 28, 4630–4634. [Google Scholar] [CrossRef]
- Fonseca, R.; Blood, E.; Rue, M.; Harrington, D.; Oken, M.M.; Kyle, R.A.; Dewald, G.W.; Van Ness, B.; Van Wier, S.A.; Henderson, K.J.; et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood 2003, 101, 4569–4575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avet-Loiseau, H.; Malard, F.; Campion, L.; Magrangeas, F.; Sebban, C.; Lioure, B.; Decaux, O.; Lamy, T.; Legros, L.; Fuzibet, J.-G.; et al. Translocation t(14;16) and multiple myeloma: Is it really an independent prognostic factor? Blood 2011, 117, 2009–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mina, R.; Joseph, N.S.; Gay, F.; Kastritis, E.; Petrucci, M.T.; Kaufman, J.L.; Montefusco, V.; Gavriatopoulou, M.; Patriarca, F.; Omedé, P.; et al. Clinical features and survival of multiple myeloma patients harboring t(14;16) in the era of novel agents. Blood Cancer J. 2020, 10, 1–4. [Google Scholar] [CrossRef]
- Goldman-Mazur, S.; Jurczyszyn, A.; Castillo, J.J.; Waszczuk-Gajda, A.; Grząśko, N.; Radocha, J.; Bittrich, M.; Kortüm, K.M.; Gozzetti, A.; Usnarska-Zubkiewicz, L.; et al. A multicenter retrospective study of 223 patients with t(14;16) in multiple myeloma. Am. J. Hematol. 2020, 95, 503–509. [Google Scholar] [CrossRef]
- Fonseca, R.; Van Wier, S.A.; Chng, W.J.; Ketterling, R.P.; Lacy, M.Q.; Dispenzieri, A.; Bergsagel, P.L.; Rajkumar, S.V.; Greipp, P.R.; Litzow, M.R.; et al. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia 2006, 20, 2034–2040. [Google Scholar] [CrossRef] [Green Version]
- Hebraud, B.; Leleu, X.; Lauwers-Cances, V.; Roussel, M.; Caillot, D.; Marit, G.; Karlin, L.; Hulin, C.; Gentil, C.; Guilhot, F.; et al. Deletion of the 1p32 region is a major independent prognostic factor in young patients with myeloma: The IFM experience on 1195 patients. Leukemia 2013, 28, 675–679. [Google Scholar] [CrossRef]
- Chang, H.; Qi, X.; Jiang, A.; Xu, W.; Young, T.; Reece, D. 1p21 deletions are strongly associated with 1q21 gains and are an independent adverse prognostic factor for the outcome of high-dose chemotherapy in patients with multiple myeloma. Bone Marrow Transplant. 2009, 45, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Chretien, M.-L.; Corre, J.; Lauwers-Cances, V.; Magrangeas, F.; Cleynen, A.; Yon, E.; Hulin, C.; Leleu, X.; Orsini-Piocelle, F.; Blade, J.-S.; et al. Understanding the role of hyperdiploidy in myeloma prognosis: Which trisomies really matter? Blood 2015, 126, 2713–2719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuiper, R.; Zweegman, S.; Van Duin, M.; Van Vliet, M.H.; Van Beers, E.H.; Dumee, B.; Vermeulen, M.; Koenders, J.; Van Der Holt, B.; Visser-Wisselaar, H.; et al. Prognostic and predictive performance of R-ISS with SKY92 in older patients with multiple myeloma: The HOVON-87/NMSG-18 trial. Blood Adv. 2020, 4, 6298–6309. [Google Scholar] [CrossRef]
- Landgren, O.; Iskander, K. Modern multiple myeloma therapy: Deep, sustained treatment response and good clinical outcomes. J. Intern. Med. 2017, 281, 365–382. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Avet-Loiseau, H.; Rawstron, A.C.; Owen, R.G.; Child, J.A.; Thakurta, A.; Sherrington, P.; Samur, M.K.; Georgieva, A.; Anderson, K.C.; et al. Association of Minimal Residual Disease with Superior Survival Outcomes in Patients With Multiple Myeloma. JAMA Oncol. 2017, 3, 28–35. [Google Scholar] [CrossRef]
- Landgren, O.; Devlin, S.; Boulad, M.; Mailankody, S. Role of MRD status in relation to clinical outcomes in newly diagnosed multiple myeloma patients: A meta-analysis. Bone Marrow Transpl. 2016, 51, 1565–1568. [Google Scholar] [CrossRef] [Green Version]
- Avet-Loiseau, H.; Ludwig, H.; Landgren, O.; Paiva, B.; Morris, C.; Yang, H.; Zhou, K.; Ro, S.; Mateos, M.-V. Minimal Residual Disease Status as a Surrogate Endpoint for Progression-free Survival in Newly Diagnosed Multiple Myeloma Studies: A Meta-analysis. Clin. Lymphoma Myeloma Leuk. 2020, 20, e30–e37. [Google Scholar] [CrossRef] [Green Version]
- Goicoechea, I.; Puig, N.; Cedena, M.-T.; Burgos, L.; Cordón, L.; Vidriales, M.-B.; Flores-Montero, J.; Gutierrez, N.C.; Calasanz, M.-J.; Ramos, M.-L.M.; et al. Deep MRD profiling defines outcome and unveils different modes of treatment resistance in standard- and high-risk myeloma. Blood 2021, 137, 49–60. [Google Scholar] [CrossRef]
- Corre, J.; Montes, L.; Martin, E.; Perrot, A.; Caillot, D.; Leleu, X.; Belhadj, K.; Facon, T.; Hulin, C.; Mohty, M.; et al. Early relapse after autologous transplant for myeloma is associated with poor survival regardless of cytogenetic risk. Haematologica 2020, 105, e480–e483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread Genetic Heterogeneity in Multiple Myeloma: Implications for Targeted Therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, M.; Sive, J.I.; Allen, C.; Roddie, C.; Chavda, S.J.; Smith, D.; Blombery, P.; Jones, K.; Ryland, G.L.; Popat, R.; et al. Prevalence and timing of TP53 mutations in del(17p) myeloma and effect on survival. Blood Cancer J. 2017, 7, e610. [Google Scholar] [CrossRef] [Green Version]
- Ledergor, G.; Weiner, A.; Zada, M.; Wang, S.-Y.; Cohen, Y.C.; Gatt, M.E.; Snir, N.; Magen, H.; Koren-Michowitz, M.; Herzog-Tzarfati, K.; et al. Single cell dissection of plasma cell heterogeneity in symptomatic and asymptomatic myeloma. Nat. Med. 2018, 24, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fan, J.; Francis, J.M.; Georghiou, G.; Hergert, S.; Li, S.; Gambe, R.; Zhou, C.W.; Yang, C.; Chunxiao, Y.; et al. Integrated single-cell genetic and transcriptional analysis suggests novel drivers of chronic lymphocytic leukemia. Genome Res. 2017, 27, 1300–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, E.M.; Neja, S.A.; Fidan, K.; Chua, J.Y.H.; Chung, T.-H.; Bertin, N.; Tergaonkar, V.; Chng, W.-J.; Ooi, M.G.-M. Lymphocyte cytosolic protein 1 (LCP1) is a novel TRAF3 dysregulation biomarker with potential prognostic value in multiple myeloma. Genome Instab. Dis. 2020, 1, 1–14. [Google Scholar] [CrossRef]
- Amirouchene-Angelozzi, N.; Swanton, C.; Bardelli, A. Tumor Evolution as a Therapeutic Target. Cancer Discov. 2017, 7, 805–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corre, J.; Cleynen, A.; Du Pont, S.R.; Buisson, L.; Bolli, N.; Attal, M.; Munshi, N.; Avet-Loiseau, H. Multiple myeloma clonal evolution in homogeneously treated patients. Leukemia 2018, 32, 2636–2647. [Google Scholar] [CrossRef]
- Neuse, C.J.; Lomas, O.C.; Schliemann, C.; Shen, Y.J.; Manier, S.; Bustoros, M.; Ghobrial, I.M. Genome instability in multiple myeloma. Leukemia 2020, 34, 2887–2897. [Google Scholar] [CrossRef]
- Oliva, S.; De Paoli, L.; Ruggeri, M.; Caltagirone, S.; Troia, R.; Oddolo, D.; D’Agostino, M.; Gilestro, M.; Mina, R.; Saraci, E.; et al. A longitudinal analysis of chromosomal abnormalities in disease progression from MGUS/SMM to newly diagnosed and relapsed multiple myeloma. Ann. Hematol. 2021, 100, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Magrangeas, F.; Avet-Loiseau, H.; Munshi, N.C.; Minvielle, S. Chromothripsis identifies a rare and aggressive entity among newly diagnosed multiple myeloma patients. Blood 2011, 118, 675–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashby, C.; Bauer, M.A.; Wang, Y.; Wardell, M.C.P.; Tytarenko, M.R.G.; Patel, P.; Flynt, E.; Ortiz, M.; Dervan, A.; Gockley, J.; et al. Chromothripsis and Chromoplexy Are Associated with DNA Instability and Adverse Clinical Outcome in Multiple Myeloma. Blood 2018, 132, 408. [Google Scholar] [CrossRef]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Rasche, L.; Kortüm, K.M.; Raab, M.S.; Weinhold, N. The Impact of Tumor Heterogeneity on Diagnostics and Novel Therapeutic Strategies in Multiple Myeloma. Int. J. Mol. Sci. 2019, 20, 1248. [Google Scholar] [CrossRef] [Green Version]
- Zamagni, E.; Tacchetti, P.; Barbato, S.; Cavo, M. Role of Imaging in the Evaluation of Minimal Residual Disease in Multiple Myeloma Patients. J. Clin. Med. 2020, 9, 3519. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Khong, T.; Ramachandran, M.; Chow, A.; Klarica, D.; Mai, L.; Walsh, S.; Broemeling, D.; Marziali, A.; Wiggin, M.; et al. Circulating tumour DNA analysis demonstrates spatial mutational heterogeneity that coincides with disease relapse in myeloma. Leukemia 2017, 31, 1695–1705. [Google Scholar] [CrossRef]
- Kis, O.; Kaedbey, R.; Chow, S.; Danesh, A.; Dowar, M.; Li, T.; Li, Z.; Liu, J.; Mansour, M.; Masih-Khan, E.; et al. Circulating tumour DNA sequence analysis as an alternative to multiple myeloma bone marrow aspirates. Nat. Commun. 2017, 8, 15086. [Google Scholar] [CrossRef] [PubMed]
- Mazzotti, C.; Buisson, L.; Maheo, S.; Perrot, A.; Chretien, M.-L.; Leleu, X.; Hulin, C.; Manier, S.; Hébraud, B.; Roussel, M.; et al. Myeloma MRD by deep sequencing from circulating tumor DNA does not correlate with results obtained in the bone marrow. Blood Adv. 2018, 2, 2811–2813. [Google Scholar] [CrossRef] [Green Version]
- Dimopoulos, K.; Gimsing, P.; Grønbæk, K. The role of epigenetics in the biology of multiple myeloma. Blood Cancer J. 2014, 4, e207. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.F.; Johnson, D.C.; Wu, P.; Walker, B.A.; Brioli, A.; Mirabella, F.; Wardell, C.P.; Melchor, L.; Davies, F.E.; Morgan, G.J. Global methylation analysis identifies prognostically important epigenetically inactivated tumor suppressor genes in multiple myeloma. Blood 2013, 122, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Moreaux, J.; Bruyer, A.; Veyrune, J.-L.; Goldschmidt, H.; Hose, D.; Klein, B. DNA methylation score is predictive of myeloma cell sensitivity to 5-azacitidine. Br. J. Haematol. 2013, 164, 613–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalff, A.; Khong, T.; Mithraprabhu, S.; Bergin, K.; Reynolds, J.; Bowen, K.M.; Thakurta, A.; Guzman, R.; Wang, M.; Couto, S.; et al. Oral azacitidine (CC-486) in combination with lenalidomide and dexamethasone in advanced, lenalidomide-refractory multiple myeloma (ROAR study). Leuk. Lymphoma 2019, 60, 2143–2151. [Google Scholar] [CrossRef] [PubMed]
- Toor, A.A.; Payne, K.K.; Chung, H.M.; Sabo, R.T.; Hazlett, A.F.; Kmieciak, M.; Sanford, K.; Williams, D.C.; Clark, W.B.; Roberts, C.H.; et al. Epigenetic induction of adaptive immune response in multiple myeloma: Sequential azacitidine and lenalidomide generate cancer testis antigen-specific cellular immunity. Br. J. Haematol. 2012, 158, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Mithraprabhu, S.; Kalff, A.; Chow, A.; Khong, T.; Spencer, A. Dysregulated Class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics 2014, 9, 1511–1520. [Google Scholar] [CrossRef] [Green Version]
- Pawlyn, C.; Bright, M.D.; Buros, A.F.; Stein, C.K.; Walters, Z.; Aronson, L.I.; Mirabella, F.; Jones, J.R.; Kaiser, M.F.; Walker, B.A.; et al. Overexpression of EZH2 in multiple myeloma is associated with poor prognosis and dysregulation of cell cycle control. Blood Cancer J. 2017, 7, e549. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, K.; Gimsing, P.; Grønbaek, K.; Grønbæk, K. Aberrant microRNA expression in multiple myeloma. Eur. J. Haematol. 2013, 91, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Manier, S.; Liu, C.-J.; Avet-Loiseau, H.; Park, J.; Shi, J.; Campigotto, F.; Salem, K.Z.; Huynh, D.; Glavey, S.V.; Rivotto, B.; et al. Prognostic role of circulating exosomal miRNAs in multiple myeloma. Blood 2017, 129, 2429–2436. [Google Scholar] [CrossRef] [PubMed]
- Lomas, O.C.; Tahri, S.; Ghobrial, I.M. The microenvironment in myeloma. Curr. Opin. Oncol. 2020, 32, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M.; Liu, C.-J.; Redd, R.A.; Perez, R.P.; Baz, R.; Zavidij, O.; Sklavenitis-Pistofidis, R.; Richardson, P.G.; Anderson, K.C.; Laubach, J.P.; et al. A Phase Ib/II Trial of the First-in-Class Anti-CXCR4 Antibody Ulocuplumab in Combination with Lenalidomide or Bortezomib Plus Dexamethasone in Relapsed Multiple Myeloma. Clin. Cancer Res. 2020, 26, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Giannopoulos, K.; Kaminska, W.; Hus, I.; Dmoszynska, A. The frequency of T regulatory cells modulates the survival of multiple myeloma patients: Detailed characterisation of immune status in multiple myeloma. Br. J. Cancer 2012, 106, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Raja, K.R.M.; Rihova, L.; Zahradova, L.; Klincova, M.; Penka, M.; Hajek, R. Increased T Regulatory Cells Are Associated with Adverse Clinical Features and Predict Progression in Multiple Myeloma. PLoS ONE 2012, 7, e47077. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schavgoulidze, A.; Cazaubiel, T.; Perrot, A.; Avet-Loiseau, H.; Corre, J. Multiple Myeloma: Heterogeneous in Every Way. Cancers 2021, 13, 1285. https://doi.org/10.3390/cancers13061285
Schavgoulidze A, Cazaubiel T, Perrot A, Avet-Loiseau H, Corre J. Multiple Myeloma: Heterogeneous in Every Way. Cancers. 2021; 13(6):1285. https://doi.org/10.3390/cancers13061285
Chicago/Turabian StyleSchavgoulidze, Anaïs, Titouan Cazaubiel, Aurore Perrot, Hervé Avet-Loiseau, and Jill Corre. 2021. "Multiple Myeloma: Heterogeneous in Every Way" Cancers 13, no. 6: 1285. https://doi.org/10.3390/cancers13061285
APA StyleSchavgoulidze, A., Cazaubiel, T., Perrot, A., Avet-Loiseau, H., & Corre, J. (2021). Multiple Myeloma: Heterogeneous in Every Way. Cancers, 13(6), 1285. https://doi.org/10.3390/cancers13061285