
Arming Immune Cells for Battle: A Brief Journey through the Advancements of T and NK Cell Immunotherapy

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
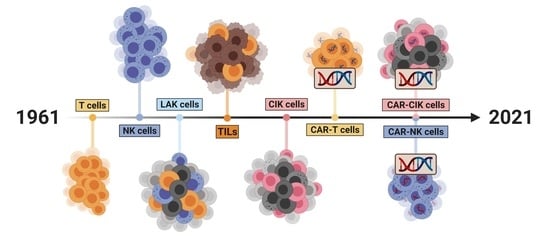
{kind=link}
{kind=link}
{kind=link}
1. Characteristics, Target Recognition and Antitumor Functionality of T and NK Cells
2. Cell Therapeutics: The Past, the Present and the Future
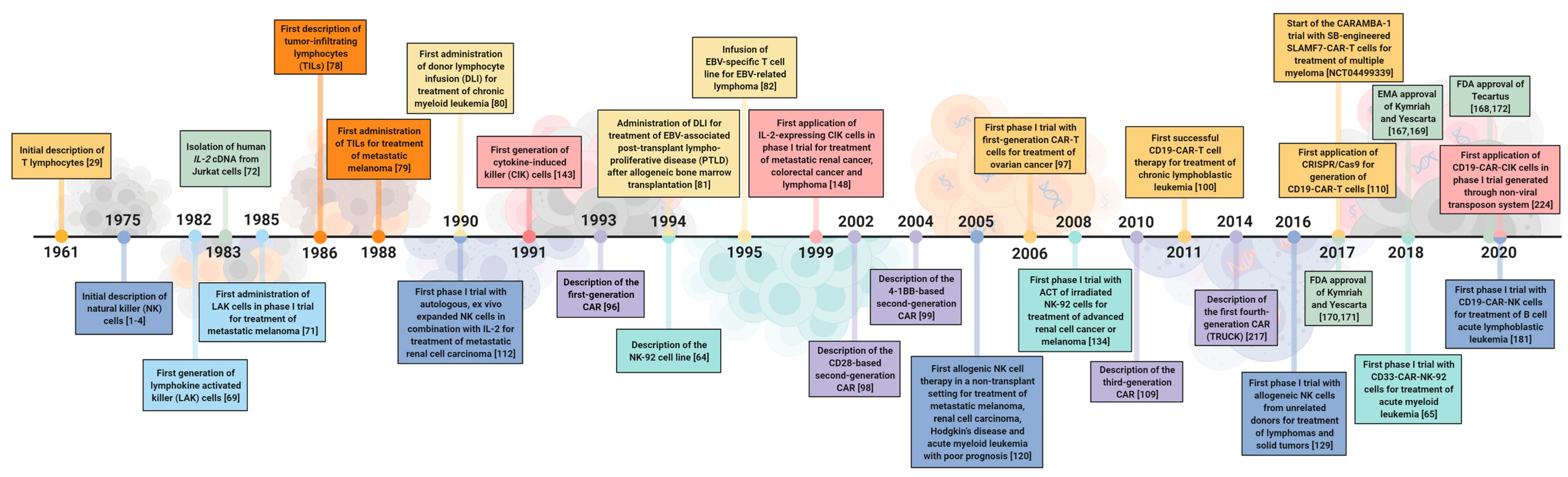
2.1. A Historical Review of the Development of Cell Therapeutics
2.1.1. Lymphokine-Activated Killer (LAK) Cells
2.1.2. T Cells
2.1.3. Natural Killer Cells
2.1.4. Cytokine-Induced Killer (CIK) Cells
2.2. Recent Clinical Studies of Immune Cell Therapy
2.3. Hurdles and Improvements for Immune Cell Therapy
3. Summary and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kiessling, R.; Klein, E.; Wigzell, H. ”Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur. J. Immunol. 1975, 5, 112–117. [Google Scholar] [CrossRef]
- Kiessling, R.; Klein, E.; Pross, H.F.; Wigzell, H. ”Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur. J. Immunol. 1975, 5, 117–121. [Google Scholar] [CrossRef]
- Herberman, R.B.; Nunn, M.E.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. I. Distribution of reactivity and specificity. Int. J. Cancer 1975, 16, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Herberman, R.B.; Nunn, M.E.; Holden, H.T.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int. J. Cancer 1975, 16, 230–239. [Google Scholar] [CrossRef]
- Cerottini, J.-C.; Brunner, K.T. Cell-Mediated Cytotoxicity, Allograft Rejection, and Tumor Immunity. In Advances in Immunology; Elsevier: Amsterdam, The Netherlands, 1974; Volume 18, pp. 67–132. [Google Scholar] [CrossRef]
- Lanier, L.L.; Phillips, J.H.; Hackett, J.; Tutt, M.; Kumar, V. Natural killer cells: Definition of a cell type rather than a function. J. Immunol. 1986, 137, 2735–2739. [Google Scholar]
- Kärre, K.; Ljunggren, H.G.; Piontek, G.; Kiessling, R. Selective rejection of H–2-deficient lymphoma variants suggests alternative immune defence strategy. Nat. Cell Biol. 1986, 319, 675–678. [Google Scholar] [CrossRef]
- Reindl, L.M.; Albinger, N.; Bexte, T.; Müller, S.; Hartmann, J.; Ullrich, E. Immunotherapy with NK cells: Recent developments in gene modification open up new avenues. OncoImmunology 2020, 9, 1777651. [Google Scholar] [CrossRef]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK Cells and T Cells by NKG2D, a Receptor for Stress-Inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef]
- Lazarova, M.; Wels, W.S.; Steinle, A. Arming cytotoxic lymphocytes for cancer immunotherapy by means of the NKG2D/NKG2D-ligand system. Expert Opin. Biol. Ther. 2020, 20, 1491–1501. [Google Scholar] [CrossRef]
- Mehta, R.S.; Rezvani, K. Chimeric Antigen Receptor Expressing Natural Killer Cells for the Immunotherapy of Cancer. Front. Immunol. 2018, 9, 283. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [Green Version]
- Raulet, D.H. Missing self recognition and self tolerance of natural killer (NK) cells. Semin. Immunol. 2006, 18, 145–150. [Google Scholar] [CrossRef]
- Raulet, D.H.; Guerra, N. Oncogenic stress sensed by the immune system: Role of natural killer cell receptors. Nat. Rev. Immunol. 2009, 9, 568–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullrich, E.; Koch, J.; Cerwenka, A.; Steinle, A. New prospects on the NKG2D/NKG2DL system for oncology. OncoImmunology 2013, 2, e26097. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Arnon, T.I.; Lev, M.; Katz, G.; Chernobrov, Y.; Porgador, A.; Mandelboim, O. Recognition of viral hemagglutinins by NKp44 but not by NKp30. Eur. J. Immunol. 2001, 31, 2680–2689. [Google Scholar] [CrossRef]
- Iwaszko, M.; Bogunia-Kubik, K. Clinical Significance of the HLA-E and CD94/NKG2 Interaction. Arch. Immunol. Ther. Exp. 2011, 59, 353–367. [Google Scholar] [CrossRef]
- Smyth, M.J.; Cretney, E.; Kelly, J.M.; Westwood, J.A.; Street, S.E.; Yagita, H.; Takeda, K.; van Dommelen, S.L.; Degli-Esposti, M.A.; Hayakawa, Y. Activation of NK cell cytotoxicity. Mol. Immunol. 2005, 42, 501–510. [Google Scholar] [CrossRef]
- Vivier, E.; Nunès, J.A.; Vély, F. Natural Killer Cell Signaling Pathways. Science 2004, 306, 1517–1519. [Google Scholar] [CrossRef]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Robertson, M.J.; Ritz, J. Biology and Clinical Relevance of Human Natural Killer Cells. Blood 1990, 76, 2421–2438. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L.; Le, A.M.; Civin, C.I.; Loken, M.R.; Phillips, J.H. The relationship of CD16 (Leu-11) and Leu-19 (NKH-1) antigen expression on human peripheral blood NK cells and cytotoxic T lymphocytes. J. Immunol. 1986, 136, 4480–4486. [Google Scholar]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activating Receptors and Coreceptors Involved in Human Natural Killer Cell-Mediated Cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Smyth, M.J.; Trapani, J.A. Perforin-mediated target-cell death and immune homeostasis. Nat. Rev. Immunol. 2006, 6, 940–952. [Google Scholar] [CrossRef]
- Terme, M.; Chaput, N.; Combadiere, B.; Ma, A.; Ohteki, T.; Zitvogel, L. Regulatory T cells control dendritic cell/NK cell cross-talk in lymph nodes at the steady state by inhibiting CD4+ self-reactive T cells. J. Immunol. 2008, 180, 4679–4686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.F. Immunological Function of the Thymus. Lancet 1961, 278, 748–749. [Google Scholar] [CrossRef]
- Miller, J.F.A.P. Analysis of the Thymus Influence in Leukæmogenesis. Nat. Cell Biol. 1961, 191, 248–249. [Google Scholar] [CrossRef]
- Miller, J.F.A.P. Role of the Thymus in Murine Leukæmia. Nat. Cell Biol. 1959, 183, 1069. [Google Scholar] [CrossRef]
- Miller, J.F.A.P. Fate of Subcutaneous Thymus Grafts in Thymectomized Mice inoculated with Leukæmic Filtrate. Nat. Cell Biol. 1959, 184, 1809–1810. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.F.A.P. Effect of neonatal thymectomy on the immunological responsiveness of the mouse. Proc. R. Soc. Lond. Ser. B Boil. Sci. 1962, 156, 415–428. [Google Scholar] [CrossRef]
- Miller, J.F.A.P. The function of the thymus and its impact on modern medicine. Science 2020, 369, eaba2429. [Google Scholar] [CrossRef]
- Vos, O.; De Vries, M.J.; Collenteur, J.C.; Van Bekkum, D.W. Transplantation of Homologous and Heterologous Lymphoid Cells in X-Irradiated and Non-irradiated Mice. J. Natl. Cancer Inst. 1959, 23, 53–73. [Google Scholar] [CrossRef]
- Miller, J.F.A.P.; Mitchell, G.F. The Thymus and the Precursors of Antigen Reactive Cells. Nat. Cell Biol. 1967, 216, 659–663. [Google Scholar] [CrossRef]
- Katz, D.H.; Benacerraf, B. The Regulatory Influence of Activated T Cells on B Cell Responses to Antigen. Adv. Immunol. 1972, 15, 1–94. [Google Scholar] [CrossRef]
- Miller, J.F.A.P.; Mitchell, G.F. CELL TO CELL INTERACTION IN THE IMMUNE RESPONSE. I. Hemolysin-forming cells in neonatally thymectomized mice reconstituted with thymus or thoracic duct lymphocytes. J. Exp. Med. 1968, 128, 801–820. [Google Scholar] [CrossRef]
- Mitchell, G.F.; Miller, J.F.A.P. CELL TO CELL INTERACTION IN THE IMMUNE RESPONSE. II. The source of hemolysin-forming cells in irradiated mice given bone marrow and thymus or thoracic duct lymphocytes. J. Exp. Med. 1968, 128, 821–837. [Google Scholar] [CrossRef] [Green Version]
- Nossal, G.J.V.; Cunningham, A.; Mitchell, G.F.; Miller, J.F.A.P. Cell to Cell Interaction in the Immune Response. 3. Chromosomal Marker Analysis of Single Antibody-Forming Cells in Reconstituted, Irradiated, or Thymectomized Mice. J. Exp. Med. 1968, 128, 839–853. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.K.; Weiss, A. Insights into the initiation of TCR signaling. Nat. Immunol. 2014, 15, 798–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clambey, E.T.; Davenport, B.; Kappler, J.W.; Marrack, P.; Homann, D. Molecules in medicine mini review: The αβ T cell receptor. J. Mol. Med. 2014, 92, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Mousset, C.M.; Hobo, W.; Woestenenk, R.; Preijers, F.; Dolstra, H.; Van Der Waart, A.B. Comprehensive Phenotyping of T Cells Using Flow Cytometry. Cytom. Part. A 2019, 95, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Hadeiba, H.; Lahl, K.; Edalati, A.; Oderup, C.; Habtezion, A.; Pachynski, R.; Nguyen, L.; Ghodsi, A.; Adler, S.; Butcher, E.C. Plasmacytoid Dendritic Cells Transport Peripheral Antigens to the Thymus to Promote Central Tolerance. Immunity 2012, 36, 438–450. [Google Scholar] [CrossRef] [Green Version]
- Kwan, J.; Killeen, N. CCR7 Directs the Migration of Thymocytes into the Thymic Medulla. J. Immunol. 2004, 172, 3999–4007. [Google Scholar] [CrossRef] [PubMed]
- Levelt, C.N.; Carsetti, R.; Eichmann, K. Regulation of thymocyte development through CDII. Expression of T cell receptor beta CD3 epsilon and maturation to the CD4+8+ stage are highly correlated in individual thymocytes. J. Exp. Med. 1993, 178, 1867–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lind, E.F.; Prockop, S.E.; Porritt, H.E.; Petrie, H.T. Mapping Precursor Movement through the Postnatal Thymus Reveals Specific Microenvironments Supporting Defined Stages of Early Lymphoid Development. J. Exp. Med. 2001, 194, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, B.A.; Bhandoola, A. Trafficking from the bone marrow to the thymus: A prerequisite for thymopoiesis. Immunol. Rev. 2006, 209, 47–57. [Google Scholar] [CrossRef]
- Ulges, A.; Schmitt, E.; Becker, C.; Bopp, T. Chapter One—Context- and Tissue-Specific Regulation of Immunity and Tolerance by Regulatory T Cells. In Advances in Immunology; Alt, F.W., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 132, pp. 1–46. [Google Scholar]
- Appay, V.; Van Lier, R.A.W.; Sallusto, F.; Roederer, M. Phenotype and function of human T lymphocyte subsets: Consensus and issues. Cytom. Part A 2008, 73, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Bassing, C.H.; Swat, W.; Alt, F.W. The Mechanism and Regulation of Chromosomal V(D)J Recombination. Cell 2002, 109, S45–S55. [Google Scholar] [CrossRef] [Green Version]
- Bluestone, J.A.; Tang, Q. Treg cells—the next frontier of cell therapy. Science 2018, 362, 154–155. [Google Scholar] [CrossRef]
- Janssen, E.M.; Droin, N.M.; Lemmens, E.E.; Pinkoski, M.J.; Bensinger, S.J.; Ehst, B.D.; Griffith, T.S.; Green, D.R.; Schoenberger, S.P. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nat. Cell Biol. 2005, 434, 88–93. [Google Scholar] [CrossRef]
- Russell, J.H.; Ley, T.J. Lymphocyte-Mediated Cytotoxicity. Annu. Rev. Immunol. 2002, 20, 323–370. [Google Scholar] [CrossRef]
- Cowan, J.E.; Jenkinson, W.E.; Anderson, G. Thymus medulla fosters generation of natural Treg cells, invariant γδ T cells, and invariant NKT cells: What we learn from intrathymic migration. Eur. J. Immunol. 2015, 45, 652–660. [Google Scholar] [CrossRef] [Green Version]
- Gapin, L. Development of invariant natural killer T cells. Curr. Opin. Immunol. 2016, 39, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerud, E.S.; Bricard, G.; Porcelli, S.A. CD1d-Restricted Natural Killer T Cells: Roles in Tumor Immunosurveillance and Tolerance. Transfus. Med. Hemotherapy 2006, 33, 18–36. [Google Scholar] [CrossRef]
- Vivier, E.; Anfossi, N. Inhibitory NK-cell receptors on T cells: Witness of the past, actors of the future. Nat. Rev. Immunol. 2004, 4, 190–198. [Google Scholar] [CrossRef]
- Van Der Vliet, H.J.; Nishi, N.; Koezuka, Y.; Peyrat, M.A.; Von Blomberg, B.M.E.; Eertwegh, A.J.M.V.D.; Pinedo, H.M.; Giaccone, G.; Scheper, R.J. Effects of α-galactosylceramide (KRN7000), interleukin-12 and interleukin-7 on phenotype and cytokine profile of human Vα24+ Vβ11+ T cells. Immunology 1999, 98, 557–563. [Google Scholar] [CrossRef]
- Kriegsmann, K.; Kriegsmann, M.; Von Bergwelt-Baildon, M.; Cremer, M.; Witzens-Harig, M. NKT cells—New players in CAR cell immunotherapy? Eur. J. Haematol. 2018, 101, 750–757. [Google Scholar] [CrossRef] [Green Version]
- Simonetta, F.; Alvarez, M.; Negrin, R.S. Natural Killer Cells in Graft-versus-Host-Disease after Allogeneic Hematopoietic Cell Transplantation. Front. Immunol. 2017, 8, 465. [Google Scholar] [CrossRef]
- Terme, M.; Ullrich, E.; Delahaye, N.F.; Chaput, N.; Zitvogel, L. Natural killer cell–directed therapies: Moving from unexpected results to successful strategies. Nat. Immunol. 2008, 9, 486–494. [Google Scholar] [CrossRef]
- Cheng, M.; Zhang, J.; Jiang, W.; Chen, Y.; Tian, Z. Natural killer cell lines in tumor immunotherapy. Front. Med. 2012, 6, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.H.; Maki, G.; Klingemann, H.G. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 1994, 8, 652–658. [Google Scholar]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar]
- Tonn, T.; Becker, S.; Esser, R.; Schwabe, D.; Seifried, E. Cellular Immunotherapy of Malignancies Using the Clonal Natural Killer Cell Line NK-J. Hematotherapy 2001, 10, 535–544. [Google Scholar] [CrossRef]
- Fang, F.; Xiao, W.; Tian, Z. NK cell-based immunotherapy for cancer. Semin. Immunol. 2017, 31, 37–54. [Google Scholar] [CrossRef]
- Montagner, I.M.; Penna, A.; Fracasso, G.; Carpanese, D.; Pietà, A.D.; Barbieri, V.; Zuccolotto, G.; Rosato, A. Anti-PSMA CAR-Engineered NK-92 Cells: An Off-the-Shelf Cell Therapy for Prostate Cancer. Cells 2020, 9, 1382. [Google Scholar] [CrossRef]
- Grimm, E.A.; Mazumder, A.; Zhang, H.Z.; A Rosenberg, S. Lymphokine-activated killer cell phenomenon. Lysis of natural killer-resistant fresh solid tumor cells by interleukin 2-activated autologous human peripheral blood lymphocytes. J. Exp. Med. 1982, 155, 1823–1841. [Google Scholar] [CrossRef] [Green Version]
- Grimm, E.A.; Ramsey, K.M.; Mazumder, A.; Wilson, D.J.; Djeu, J.Y.; Rosenberg, S.A. Lymphokine-activated killer cell phenomenon. II. Precursor phenotype is serologically distinct from peripheral T lymphocytes, memory cytotoxic thymus-derived lymphocytes, and natural killer cells. J. Exp. Med. 1983, 157, 884–897. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S. Lymphokine-activated killer cells: A new approach to immunotherapy of cancer. J. Natl. Cancer Inst. 1985, 75, 595–603. [Google Scholar] [CrossRef]
- Taniguchi, T.; Matsui, H.; Fujita, T.; Takaoka, C.; Kashima, N.; Yoshimoto, R.; Hamuro, J. Structure and expression of a cloned cDNA for human interleukin-2. Nat. Cell Biol. 1983, 302, 305–310. [Google Scholar] [CrossRef]
- Rosenberg, S.A. Immunotherapy of cancer by systemic administration of lymphoid cells plus interleukin-J. Boil. Response Modif. 1984, 3, 501–511. [Google Scholar]
- Lotze, M.T.; Matory, Y.L.; Ettinghausen, S.E.; Rayner, A.A.; O Sharrow, S.; Seipp, C.A.; Custer, M.C.; Rosenberg, S.A. In vivo administration of purified human interleukin II. Half life, immunologic effects, and expansion of peripheral lymphoid cells in vivo with recombinant IL. J. Immunol. 1985, 135, 2865–2875. [Google Scholar]
- Mule, J.; Shu, S.; Schwarz, S.; Rosenberg, S. Adoptive immunotherapy of established pulmonary metastases with LAK cells and recombinant interleukin-2. Science 1984, 225, 1487–1489. [Google Scholar] [CrossRef]
- Law, T.M.; Motzer, R.J.; Mazumdar, M.; Sell, K.W.; Walther, P.; O’Connell, M.; Khan, A.; Vlamis, V.; Vogelzang, N.J.; Bajorin, D.F. Phase iii randomized trial of interleukin-2 with or without lymphokine-activated killer cells in the treatment of patients with advanced renal cell carcinoma. Cancer 1995, 76, 824–832. [Google Scholar] [CrossRef]
- Margolin, K.A.; Aronson, F.R.; Sznol, M.; Atkins, M.B.; Ciobanu, N.; Fisher, R.I.; Weiss, G.R.; Doroshow, J.H.; Bar, M.H.; Hawkins, M.J.; et al. Phase II Trial of High-Dose Interleukin-2 and Lymphokine-Activated Killer Cells in Hodgkin’s Disease and Non-Hodgkin’s Lymphom. J. Immunother. 1991, 10, 214–220. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Spiess, P.; LaFreniere, R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 1986, 233, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.-H.; Seipp, C.A.; et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Kolb, H.; Mittermuller, J.; Clemm, C.; Holler, E.; Ledderose, G.; Brehm, G.; Heim, M.; Wilmanns, W. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood 1990, 76, 2462–2465. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, E.B.; Ladanyi, M.; Emanuel, D.; MacKinnon, S.; Boulad, F.; Carabasi, M.H.; Castro-Malaspina, H.; Childs, B.H.; Gillio, A.P.; Small, T.N.; et al. Infusions of Donor Leukocytes to Treat Epstein-Barr Virus-Associated Lymphoproliferative Disorders after Allogeneic Bone Marrow Transplantation. N. Engl. J. Med. 1994, 330, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Rooney, C.; Ng, C.; Loftin, S.; Smith, C.; Li, C.; Krance, R.; Brenner, M.; Heslop, H.; Rooney, C.M.; Brenner, M.K.; et al. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet 1995, 345, 9–13. [Google Scholar] [CrossRef]
- Rooney, C.M.; Smith, C.A.; Ng, C.Y.; Loftin, S.K.; Sixbey, J.W.; Gan, Y.; Srivastava, D.-K.; Bowman, L.C.; Krance, R.A.; Brenner, M.K.; et al. Infusion of Cytotoxic T Cells for the Prevention and Treatment of Epstein-Barr Virus–Induced Lymphoma in Allogeneic Transplant Recipients. Blood 1998, 92, 1549–1555. [Google Scholar] [CrossRef]
- Clay, T.M.; Custer, M.C.; Sachs, J.; Hwu, P.; Rosenberg, S.A.; I Nishimura, M. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J. Immunol. 1999, 163, 507–513. [Google Scholar] [PubMed]
- Cole, D.J.; Weil, D.P.; Shilyansky, J.; Custer, M.; Kawakami, Y.; Rosenberg, S.A.; Nishimura, M.I. Characterization of the functional specificity of a cloned T-cell receptor heterodimer recognizing the MART-1 melanoma antigen. Cancer Res. 1995, 55, 748–752. [Google Scholar]
- Hughes, M.S.; Yu, Y.Y.; Dudley, M.E.; Zheng, Z.; Robbins, P.F.; Li, Y.; Wunderlich, J.; Hawley, R.G.; Moayeri, M.; Rosenberg, S.A.; et al. Transfer of a TCR Gene Derived from a Patient with a Marked Antitumor Response Conveys Highly Active T-Cell Effector Functions. Hum. Gene Ther. 2005, 16, 457–472. [Google Scholar] [CrossRef]
- Morgan, R.A.; Dudley, M.E.; Yu, Y.Y.L.; Zheng, Z.; Robbins, P.F.; Theoret, M.R.; Wunderlich, J.R.; Hughes, M.S.; Restifo, N.P.; Rosenberg, S.A. High Efficiency TCR Gene Transfer into Primary Human Lymphocytes Affords Avid Recognition of Melanoma Tumor Antigen Glycoprotein 100 and Does Not Alter the Recognition of Autologous Melanoma Antigens. J. Immunol. 2003, 171, 3287–3295. [Google Scholar] [CrossRef] [Green Version]
- Stancovski, I.; Schindler, D.G.; Waks, T.; Yarden, Y.; Sela, M.; Eshhar, Z. Targeting of T lymphocytes to Neu/HER2-expressing cells using chimeric single chain Fv receptors. J. Immunol. 1993, 151, 6577–6582. [Google Scholar] [PubMed]
- Sadelain, M.; Rivière, I.; Brentjens, R.J. Targeting tumours with genetically enhanced T lymphocytes. Nat. Rev. Cancer 2003, 3, 35–45. [Google Scholar] [CrossRef]
- Dembić, Z.; Haas, W.; Weiss, S.; McCubrey, J.A.; Kiefer, H.; Von Boehmer, H.; Steinmetz, M. Transfer of specificity by murine α and β T-cell receptor genes. Nat. Cell Biol. 1986, 320, 232–238. [Google Scholar] [CrossRef]
- Stanislawski, T.; Voss, R.-H.; Lotz, C.; Sadovnikova, E.; Willemsen, R.A.; Kuball, J.; Ruppert, T.; Bolhuis, R.L.H.; Melief, C.J.; Huber, C.; et al. Circumventing tolerance to a human MDM2-derived tumor antigen by TCR gene transfer. Nat. Immunol. 2001, 2, 962–970. [Google Scholar] [CrossRef]
- Zoete, V.; Irving, M.; Ferber, M.; Cuendet, M.; Michielin, O. Structure-Based, Rational Design of T Cell Receptors. Front. Immunol. 2013, 4, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seliger, B.; Cabrerab, T.; Garridob, F.; Ferronec, S. HLA class I antigen abnormalities and immune escape by malignant cells. Semin. Cancer Biol. 2002, 12, 3–13. [Google Scholar] [CrossRef]
- González-Galarza, F.F.; Takeshita, L.Y.; Santos, E.J.; Kempson, F.; Maia, M.H.T.; Da Silva, A.L.S.; Silva, A.L.T.E.; Ghattaoraya, G.S.; Alfirevic, A.; Jones, A.R.; et al. Allele frequency net 2015 update: New features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res. 2015, 43, D784–D788. [Google Scholar] [CrossRef] [PubMed]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A Phase I Study on Adoptive Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [Green Version]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.-H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [Green Version]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Van Der Stegen, S.J.C.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.-S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric Antigen Receptors Combining 4-1BB and CD28 Signaling Domains Augment PI3kinase/AKT/Bcl-XL Activation and CD8+ T Cell–mediated Tumor Eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; Van Der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, K.M.C.M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nat. Cell Biol. 2017, 543, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Leon, E.; Ranganathan, R.; Savoldo, B. Adoptive T cell therapy: Boosting the immune system to fight cancer. Semin. Immunol. 2020, 49, 101437. [Google Scholar] [CrossRef]
- Hercend, T.; Farace, F.; Baume, D.; Charpentier, F.; Droz, J.P.; Triebel, F.; Escudier, B. Immunotherapy with lymphokine-activated natural killer cells and recombinant interleukin-2: A feasibility trial in metastatic renal cell carcinoma. J. Boil. Response Modif. 1990, 9, 546–555. [Google Scholar]
- Miller, J.S.; Tessmer-Tuck, J.; Pierson, B.A.; Weisdorf, D.; McGlave, P.; Blazar, B.R.; Katsanis, E.; Verfaillie, C.; Lebkowski, J.; Radford, J.; et al. Low dose subcutaneous interleukin-2 after autologous transplantation generates sustained in vivo natural killer cell activity. Biol. Blood Marrow Transpl. 1997, 3, 34–44. [Google Scholar]
- Burns, L.J.; Weisdorf, D.J.; DeFor, T.E.; Vesole, D.H.; Repka, T.L.; Blazar, B.R.; Burger, S.R.; Panoskaltsismortari, A.; Keevertaylor, C.A.; Zhang, M.-J.; et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: A phase I/II trial. Bone Marrow Transpl. 2003, 32, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, G.; Hemmers, S.; Firth, M.A.; Le Floc’H, A.; Huse, M.; Sun, J.C.; Rudensky, A.Y. IL-2–dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. J. Exp. Med. 2013, 210, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Ménard, C.; Terme, M.; Flament, C.; Taieb, J.; Chaput, N.; Puig, P.E.; Novault, S.; Escudier, B.; Vivier, E.; et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor–β–dependent manner. J. Exp. Med. 2005, 202, 1075–1085. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggeri, L.; Capanni, M.; Casucci, M.; Volpi, I.; Tosti, A.; Perruccio, K.; Urbani, E.; Negrin, R.S.; Martelli, M.F.; Velardi, A. Role of Natural Killer Cell Alloreactivity in HLA-Mismatched Hematopoietic Stem Cell Transplantation. Blood 1999, 94, 333–339. [Google Scholar] [CrossRef]
- Farag, S.S.; Bacigalupo, A.; Eapen, M.; Hurley, C.; Dupont, B.; Caligiuri, M.A.; Boudreau, C.; Nelson, G.; Oudshoorn, M.; Van Rood, J.; et al. The Effect of KIR Ligand Incompatibility on the Outcome of Unrelated Donor Transplantation: A Report from the Center for International Blood and Marrow Transplant Research, the European Blood and Marrow Transplant Registry, and the Dutch Registry. Biol. Blood Marrow Transpl. 2006, 12, 876–884. [Google Scholar] [CrossRef] [Green Version]
- Passweg, J.R.; Tichelli, A.; Meyer-Monard, S.; Heim, D.; Stern, M.; Kühne, T.; Favre, G.; Gratwohl, A. Purified donor NK-lymphocyte infusion to consolidate engraftment after haploidentical stem cell transplantation. Leukemia 2004, 18, 1835–1838. [Google Scholar] [CrossRef]
- Koehl, U.; Esser, R.; Zimmermann, S.; Tonn, T.; Kotchetkov, R.; Bartling, T.; Sörensen, J.; Grüttner, H.-P.; Bader, P.; Seifried, E.; et al. Ex vivo Expansion of Highly Purified NK Cells for Immunotherapy after Haploidentical Stem Cell Transplantation in Children. Klin. Pädiatrie 2005, 217, 345–350. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.R.; Lee, Y.S.; Yang, S.H.; Ahn, K.H.; Lee, J.-H.; Lee, J.-H.; Kim, D.Y.; Kang, Y.A.; Jeon, M.; Seol, M.; et al. Generation of donor natural killer cells from CD34+ progenitor cells and subsequent infusion after HLA-mismatched allogeneic hematopoietic cell transplantation: A feasibility study. Bone Marrow Transpl. 2009, 45, 1038–1046. [Google Scholar] [CrossRef]
- Shi, J.; Tricot, G.; Szmania, S.; Rosen, N.; Garg, T.K.; Malaviarachchi, P.A.; Moreno, A.; Dupont, B.; Hsu, K.C.; Baxter-Lowe, L.A.; et al. Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br. J. Haematol. 2008, 143, 641–653. [Google Scholar] [CrossRef] [Green Version]
- Brehm, C.; Huenecke, S.; Quaiser, A.; Esser, R.; Bremm, M.; Kloess, S.; Soerensen, J.; Kreyenberg, H.; Seidl, C.; Becker, P.S.A.; et al. IL-2 Stimulated but Not Unstimulated NK Cells Induce Selective Disappearance of Peripheral Blood Cells: Concomitant Results to a Phase I/II Study. PLoS ONE 2011, 6, e27351. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; DeFor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [Green Version]
- Bachanova, V.; Cooley, S.; DeFor, T.E.; Verneris, M.R.; Zhang, B.; McKenna, D.H.; Curtsinger, J.; Panoskaltsis-Mortari, A.; Lewis, D.; Hippen, K.; et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood 2014, 123, 3855–3863. [Google Scholar] [CrossRef]
- Granzin, M.; Wagner, J.; Köhl, U.; Cerwenka, A.; Huppert, V.; Ullrich, E. Shaping of Natural Killer Cell Antitumor Activity by Ex Vivo Cultivation. Front. Immunol. 2017, 8, 458. [Google Scholar] [CrossRef] [Green Version]
- Lim, O.; Lee, Y.; Chung, H.; Her, J.H.; Kang, S.M.; Jung, M.-Y.; Min, B.; Shin, H.; Kim, T.M.; Heo, D.S.; et al. GMP-Compliant, Large-Scale Expanded Allogeneic Natural Killer Cells Have Potent Cytolytic Activity against Cancer Cells In Vitro and In Vivo. PLoS ONE 2013, 8, e53611. [Google Scholar] [CrossRef]
- Yang, Y.; Lim, O.; Kim, T.M.; Ahn, Y.-O.; Choi, H.; Chung, H.; Min, B.; Her, J.H.; Cho, S.Y.; Keam, B.; et al. Phase I Study of Random Healthy Donor–Derived Allogeneic Natural Killer Cell Therapy in Patients with Malignant Lymphoma or Advanced Solid Tumors. Cancer Immunol. Res. 2016, 4, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Iliopoulou, E.G.; Kountourakis, P.; Karamouzis, M.V.; Doufexis, D.; Ardavanis, A.; Baxevanis, C.N.; Rigatos, G.; Papamichail, M.; Perez, S.A. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol. Immunother. 2010, 59, 1781–1789. [Google Scholar] [CrossRef]
- Rubnitz, J.E.; Inaba, H.; Ribeiro, R.C.; Pounds, S.; Rooney, B.; Bell, T.; Pui, C.-H.; Leung, W. NKAML: A Pilot Study to Determine the Safety and Feasibility of Haploidentical Natural Killer Cell Transplantation in Childhood Acute Myeloid Leukemia. J. Clin. Oncol. 2010, 28, 955–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curti, A.; Ruggeri, L.; D’Addio, A.; Bontadini, A.; Dan, E.; Motta, M.R.; Trabanelli, S.; Giudice, V.; Urbani, E.; Martinelli, G.; et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood 2011, 118, 3273–3279. [Google Scholar] [CrossRef]
- Geller, M.A.; Cooley, S.; Judson, P.L.; Ghebre, R.; Carson, L.F.; Argenta, P.A.; Jonson, A.L.; Panoskaltsis-Mortari, A.; Curtsinger, J.; McKenna, D.; et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011, 13, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Boyiadzis, M.; Agha, M.; Redner, R.L.; Sehgal, A.; Im, A.; Hou, J.-Z.; Farah, R.; Dorritie, K.A.; Raptis, A.; Lim, S.H.; et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy 2017, 19, 1225–1232. [Google Scholar] [CrossRef]
- Klingemann, H.-G.; Martinson, J. Ex vivo expansion of natural killer cells for clinical applications. Cytotherapy 2004, 6, 15–22. [Google Scholar] [CrossRef]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef]
- Williams, B.A.; Law, A.D.; Routy, B.; Denhollander, N.; Gupta, V.; Wang, X.-H.; Chaboureau, A.; Viswanathan, S.; Keating, A. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget 2017, 8, 89256–89268. [Google Scholar] [CrossRef] [Green Version]
- Burger, M.C.; Zhang, C.; Harter, P.N.; Romanski, A.; Strassheimer, F.; Senft, C.; Tonn, T.; Steinbach, J.P.; Wels, W.S. CAR-Engineered NK Cells for the Treatment of Glioblastoma: Turning Innate Effectors Into Precision Tools for Cancer Immunotherapy. Front. Immunol. 2019, 10, 2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suck, G.; Odendahl, M.; Nowakowska, P.; Seidl, C.; Wels, W.S.; Klingemann, H.G.; Tonn, T. NK-92: An ‘off-the-shelf therapeutic’ for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol. Immunother. 2016, 65, 485–492. [Google Scholar] [CrossRef]
- Kottaridis, P.D.; North, J.; Tsirogianni, M.; Marden, C.; Samuel, E.R.; Jide-Banwo, S.; Grace, S.; Lowdell, M.W. Two-Stage Priming of Allogeneic Natural Killer Cells for the Treatment of Patients with Acute Myeloid Leukemia: A Phase I Trial. PLoS ONE 2015, 10, e0123416. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Wolf, I.G.; Negrin, R.S.; Kiem, H.P.; Blume, K.G.; Weissman, I.L. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J. Exp. Med. 1991, 174, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Introna, M.; Correnti, F. Innovative Clinical Perspectives for CIK Cells in Cancer Patients. Int. J. Mol. Sci. 2018, 19, 358. [Google Scholar] [CrossRef] [Green Version]
- Pievani, A.; Borleri, G.; Pende, D.; Moretta, L.; Rambaldi, A.; Golay, J.; Introna, M. Dual-functional capability of CD3+CD56+ CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood 2011, 118, 3301–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolin, K.A.; Negrin, R.S.; Wong, K.K.; Chatterjee, S.; Wright, C.; Forman, S.J. Cellular immunotherapy and autologous transplantation for hematologic malignancy. Immunol. Rev. 1997, 157, 231–240. [Google Scholar] [CrossRef]
- Lu, P.H.; Negrin, R.S. A novel population of expanded human CD3+CD56+ cells derived from T cells with potent in vivo antitumor activity in mice with severe combined immunodeficiency. J. Immunol. 1994, 153, 1687–1696. [Google Scholar]
- Schmidt-Wolf, I.G.H.; Finke, S.; Trojaneck, B.; Denkena, A.; Lefterova, P.; Schwella, N.; Heuft, H.-G.; Prange, G.; Korte, M.; Takeya, M.; et al. Phase I clinical study applying autologous immunological effector cells transfected with the interleukin-2 gene in patients with metastatic renal cancer, colorectal cancer and lymphoma. Br. J. Cancer 1999, 81, 1009–1016. [Google Scholar] [CrossRef]
- Leemhuis, T.; Wells, S.; Scheffold, C.; Edinger, M.; Negrin, R.S. A phase I trial of autologous cytokine-induced killer cells for the treatment of relapsed Hodgkin disease and non-Hodgkin lymphoma. Biol. Blood Marrow Transpl. 2005, 11, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Xu, N.; Wu, C.; Deng, H.; Lu, M.; Li, M.; Xu, B.; Wu, J.; Wang, R.; Xu, J.; et al. Treatment of advanced gastric cancer by chemotherapy combined with autologous cytokine-induced killer cells. Anticancer. Res. 2006, 26, 2237–2242. [Google Scholar]
- Li, R.; Wang, C.; Liu, L.; Du, C.; Cao, S.; Yu, J.; Wang, S.E.; Hao, X.; Ren, X.; Li, H. Autologous cytokine-induced killer cell immunotherapy in lung cancer: A phase II clinical study. Cancer Immunol. Immunother. 2012, 61, 2125–2133. [Google Scholar] [CrossRef]
- Niu, Q.; Wang, W.; Li, Y.; Qin, S.; Wang, Y.; Wan, G.; Guan, J.; Zhu, W. Cord blood-derived cytokine-induced killer cells biotherapy combined with second-line chemotherapy in the treatment of advanced solid malignancies. Int. Immunopharmacol. 2011, 11, 449–456. [Google Scholar] [CrossRef]
- Kong, D.-S.; Nam, D.-H.; Kang, S.-H.; Lee, J.W.; Chang, J.-H.; Kim, J.-H.; Lim, Y.-J.; Koh, Y.-C.; Chung, Y.-G.; Kim, J.-M.; et al. Phase III randomized trial of autologous cytokine-induced killer cell immunotherapy for newly diagnosed glioblastoma in korea. Oncotarget 2017, 8, 7003–7013. [Google Scholar] [CrossRef] [Green Version]
- Rettinger, E.; Kuçi, S.; Naumann, I.; Becker, P.; Kreyenberg, H.; Anzaghe, M.; Willasch, A.; Koehl, U.; Bug, G.; Ruthardt, M.; et al. The cytotoxic potential of interleukin-15-stimulated cytokine-induced killer cells against leukemia cells. Cytotherapy 2012, 14, 91–103. [Google Scholar] [CrossRef]
- Merker, M.; Salzmann-Manrique, E.; Katzki, V.; Huenecke, S.; Bremm, M.; Bakhtiar, S.; Willasch, A.; Jarisch, A.; Soerensen, J.; Schulz, A.; et al. Clearance of Hematologic Malignancies by Allogeneic Cytokine-Induced Killer Cell or Donor Lymphocyte Infusions. Biol. Blood Marrow Transpl. 2019, 25, 1281–1292. [Google Scholar] [CrossRef]
- Zhang, Y.; Schmidt-Wolf, I.G.H. Ten-year update of the international registry on cytokine-induced killer cells in cancer immunotherapy. J. Cell. Physiol. 2020, 235, 9291–9303. [Google Scholar] [CrossRef]
- Introna, M.; Lussana, F.; Algarotti, A.; Gotti, E.; Valgardsdottir, R.; Micò, C.; Grassi, A.; Pavoni, C.; Ferrari, M.L.; Delaini, F.; et al. Phase II Study of Sequential Infusion of Donor Lymphocyte Infusion and Cytokine-Induced Killer Cells for Patients Relapsed after Allogeneic Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transpl. 2017, 23, 2070–2078. [Google Scholar] [CrossRef] [Green Version]
- Rettinger, E.; Huenecke, S.; Bönig, H.; Merker, M.; Jarisch, A.; Soerensen, J.; Willasch, A.; Bug, G.; Schulz, A.; Klingebiel, T.; et al. Interleukin-15-activated cytokine-induced killer cells may sustain remission in leukemia patients after allogeneic stem cell transplantation: Feasibility, safety and first insights on efficacy. Haematology 2016, 101, e153–e156. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef]
- Benson, D.M.; Cohen, A.D.; Jagannath, S.; Munshi, N.C.; Spitzer, G.; Hofmeister, C.C.; Efebera, Y.A.; Andre, P.; Zerbib, R.; Caligiuri, M.A. A Phase I Trial of the Anti-KIR Antibody IPH2101 and Lenalidomide in Patients with Relapsed/Refractory Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4055–4061. [Google Scholar] [CrossRef] [Green Version]
- Tinker, A.V.; Hirte, H.W.; Provencher, D.; Butler, M.; Ritter, H.; Tu, D.; Azim, H.A.; Paralejas, P.; Grenier, N.; Hahn, S.-A.; et al. Dose-Ranging and Cohort-Expansion Study of Monalizumab (IPH2201) in Patients with Advanced Gynecologic Malignancies: A Trial of the Canadian Cancer Trials Group (CCTG): IND221. Clin. Cancer Res. 2019, 25, 6052–6060. [Google Scholar] [CrossRef] [Green Version]
- Vey, N.; Karlin, L.; Sadot-Lebouvier, S.; Broussais, F.; Berton-Rigaud, D.; Rey, J.; Charbonnier, A.; Marie, D.; André, P.; Paturel, C.; et al. A phase 1 study of lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D.; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget 2018, 9, 17675–17688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franks, S.E.; Wolfson, B.; Hodge, J.W. Natural Born Killers: NK Cells in Cancer Therapy. Cancers 2020, 12, 2131. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Arooj, S.; Wang, H. NK Cell-Based Immune Checkpoint Inhibition. Front. Immunol. 2020, 11, 167. [Google Scholar] [CrossRef]
- Pei, X.-Y.; Zhao, X.-Y.; Chang, Y.-J.; Liu, J.; Xu, L.-P.; Wang, Y.; Zhang, X.-H.; Han, W.; Chen, Y.-H.; Huang, X.-J. Cytomegalovirus-Specific T-Cell Transfer for Refractory Cytomegalovirus Infection After Haploidentical Stem Cell Transplantation: The Quantitative and Qualitative Immune Recovery for Cytomegalovirus. J. Infect. Dis. 2017, 216, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Beagley, L.; Rehan, S.; Neller, M.A.; Crooks, P.; Solomon, M.; Holmes-Liew, C.-L.; Holmes, M.; McKenzie, S.C.; Hopkins, P.; et al. Autologous Adoptive T-cell Therapy for Recurrent or Drug-resistant Cytomegalovirus Complications in Solid Organ Transplant Recipients: A Single-arm Open-label Phase I Clinical Trial. Clin. Infect. Dis. 2019, 68, 632–640. [Google Scholar] [CrossRef]
- Ali, S.; Kjeken, R.; Niederlaender, C.; Markey, G.; Saunders, T.S.; Opsata, M.; Moltu, K.; Bremnes, B.; Grønevik, E.; Muusse, M.; et al. The European Medicines Agency Review of Kymriah (Tisagenlecleucel) for the Treatment of Acute Lymphoblastic Leukemia and Diffuse Large B-Cell Lymphoma. Oncologist 2019, 25, e321–e327. [Google Scholar] [CrossRef] [Green Version]
- Gilead Sciences, Inc. U.S. FDA Approves Kite’s TecartusTM, the First and Only CAR T Treatment for Relapsed or Refractory Mantle Cell Lymphoma. Available online: https://www.gilead.com/news-and-press (accessed on 7 October 2020).
- European Medicines Agency. YESCARTA: Axicabtagene Ciloleucel. Available online: https://www.ema.europa.eu/ (accessed on 7 October 2020).
- U.S. Food and Drug Administration. KYMRIAH (Tisagenlecleucel). Available online: http://www.fda.gov/ (accessed on 7 October 2020).
- U.S. Food & Drug Administration. YESCARTA (Axicabtagene Ciloleucel). Available online: https://www.fda.gov (accessed on 7 October 2020).
- U.S. Food & Drug Administration. TECARTUS (Brexucabtagene Autoleucel). Available online: https://www.fda.gov (accessed on 7 October 2020).
- Marin, V.; Kakuda, H.; Dander, E.; Imai, C.; Campana, D.; Biondi, A.; D’Amico, G. Enhancement of the anti-leukemic activity of cytokine induced killer cells with an anti-CD19 chimeric receptor delivering a 4-1BB-ζ activating signal. Exp. Hematol. 2007, 35, 1388–1397. [Google Scholar] [CrossRef]
- Leuci, V.; Casucci, G.M.; Grignani, G.; Rotolo, R.; Rossotti, U.; Vigna, E.; Gammaitoni, L.; Mesiano, G.; Fiorino, E.; Donini, C.; et al. CD44v6 as innovative sarcoma target for CAR-redirected CIK cells. OncoImmunology 2018, 7, e1423167. [Google Scholar] [CrossRef] [Green Version]
- Market, M.; Angka, L.; Martel, A.B.; Bastin, D.; Olanubi, O.; Tennakoon, G.; Boucher, D.M.; Ng, J.; Ardolino, M.; Auer, R.C. Flattening the COVID-19 Curve With Natural Killer Cell Based Immunotherapies. Front. Immunol. 2020, 11, 1512. [Google Scholar] [CrossRef]
- Modak, S.; Le Luduec, J.-B.; Cheung, I.Y.; Goldman, D.A.; Ostrovnaya, I.; Doubrovina, E.; Basu, E.; Kushner, B.H.; Kramer, K.; Roberts, S.S.; et al. Adoptive immunotherapy with haploidentical natural killer cells and Anti-GD2 monoclonal antibody m3F8 for resistant neuroblastoma: Results of a phase I study. OncoImmunology 2018, 7, e1461305. [Google Scholar] [CrossRef] [Green Version]
- Ahmadzadeh, M.; Rosenberg, S.A. IL-2 administration increases CD4+CD25hi Foxp3+ regulatory T cells in cancer patients. Blood 2006, 107, 2409–2414. [Google Scholar] [CrossRef] [Green Version]
- Buchbinder, E.I.; Dutcher, J.P.; Daniels, G.A.; Curti, B.D.; Patel, S.P.; Holtan, S.G.; Miletello, G.P.; Fishman, M.N.; Gonzalez, R.; Clark, J.I.; et al. Therapy with high-dose Interleukin-2 (HD IL-2) in metastatic melanoma and renal cell carcinoma following PD1 or PDL1 inhibition. J. Immunother. Cancer 2019, 7, 49. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.; Rosenblum, J.; Jeng, E.K.; Alter, S.; Rhode, P.R.; Lee, J.H.; Lee, D.; Wong, H.C.; Cairo, M.S. Efficiently Targeting Metastatic Osteosarcoma, Neuroblastoma and Glioblastoma with Ex-Vivo Expanded Natural Killer Cells Combined with N-803 (ALT-803, IL-15 Superagonist) and TIM-3 Blockage. Biol. Blood Marrow Transpl. 2019, 25, S336. [Google Scholar] [CrossRef]
- Kim, P.S.; Kwilas, A.R.; Xu, W.; Alter, S.; Jeng, E.K.; Wong, H.C.; Schlom, J.; Hodge, J.W. IL-15 superagonist/IL-15RαSushi-Fc fusion complex (IL-15SA/IL-15RαSu-Fc; ALT-803) markedly enhances specific subpopulations of NK and memory CD8+ T cells, and mediates potent anti-tumor activity against murine breast and colon carcinomas. Oncotarget 2016, 7, 16130–16145. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Jochems, C.; Hodge, J.W.; Fantini, M.; Fujii, R.; Ii, Y.M.M.; Greiner, J.W.; Padget, M.R.; Tritsch, S.R.; Tsang, K.Y.; Campbell, K.S.; et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget 2016, 7, 86359–86373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romee, R.; Cooley, S.; Berrien-Elliott, M.M.; Westervelt, P.; Verneris, M.R.; Wagner, J.E.; Weisdorf, D.J.; Blazar, B.R.; Ustun, C.; DeFor, T.E.; et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood 2018, 131, 2515–2527. [Google Scholar] [CrossRef]
- Hellström, I.; Hellström, K.E.; Pierce, G.E.; Yang, J.P.S. Cellular and Humoral immunity to Different Types of Human Neoplasms. Nat. Cell Biol. 1968, 220, 1352–1354. [Google Scholar] [CrossRef]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef] [PubMed]
- Macmillan, M.L.; Robin, M.; Harris, A.C.; DeFor, T.E.; Martin, P.J.; Alousi, A.M.; Ho, V.T.; Bolaños-Meade, J.; Ferrara, J.L.; Jones, R.; et al. A Refined Risk Score for Acute Graft-versus-Host Disease that Predicts Response to Initial Therapy, Survival, and Transplant-Related Mortality. Biol. Blood Marrow Transpl. 2015, 21, 761–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor–Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef] [Green Version]
- Kambhampati, S.; Gray, L.; Fakhri, B.; Lo, M.; Vu, K.; Arora, S.; Kaplan, L.; Ai, W.Z.; Andreadis, C. Immune-related Adverse Events Associated With Checkpoint Inhibition in the Setting of CAR T Cell Therapy: A Case Series. Clin. Lymphoma Myeloma Leuk. 2020, 20, e118–e123. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol. Immunother. 2019, 68, 365–377. [Google Scholar] [CrossRef]
- Nakazawa, T.; Natsume, A.; Nishimura, F.; Morimoto, T.; Matsuda, R.; Nakamura, M.; Yamada, S.; Nakagawa, I.; Motoyama, Y.; Park, Y.-S.; et al. Effect of CRISPR/Cas9-Mediated PD-1-Disrupted Primary Human Third-Generation CAR-T Cells Targeting EGFRvIII on In Vitro Human Glioblastoma Cell Growth. Cells 2020, 9, 998. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef]
- Restifo, N.P.; Esquivel, F.; Kawakami, Y.; Yewdell, J.W.; Mulé, J.J.; Rosenberg, S.A.; Bennink, J.R. Identification of human cancers deficient in antigen processing. J. Exp. Med. 1993, 177, 265–272. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Contribution of regulatory T cells to cancer: A review. J. Cell. Physiol. 2019, 234, 7983–7993. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P.; Chornoguz, O.; Ecker, C. Regulating the suppressors: Apoptosis and inflammation govern the survival of tumor-induced myeloid-derived suppressor cells (MDSC). Cancer Immunol. Immunother. 2012, 61, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Smyth, M.J. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell. Mol. Immunol. 2020, 17, 1–12. [Google Scholar] [CrossRef]
- Neo, S.Y.; Yang, Y.; Record, J.; Ma, R.; Chen, X.; Chen, Z.; Tobin, N.P.; Blake, E.; Seitz, C.; Thomas, R.; et al. CD73 immune checkpoint defines regulatory NK cells within the tumor microenvironment. J. Clin. Investig. 2020, 130, 1185–1198. [Google Scholar] [CrossRef] [Green Version]
- Waldhauer, I.; Steinle, A. NK cells and cancer immunosurveillance. Oncogene 2008, 27, 5932–5943. [Google Scholar] [CrossRef] [Green Version]
- De Andrade, L.F.; Kumar, S.; Luoma, A.M.; Ito, Y.; Da Silva, P.H.A.; Pan, D.; Pyrdol, J.W.; Yoon, C.H.; Wucherpfennig, K.W. Inhibition of MICA and MICB Shedding Elicits NK-Cell–Mediated Immunity against Tumors Resistant to Cytotoxic T Cells. Cancer Immunol. Res. 2020, 8, 769–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imura, Y.; Ando, M.; Kondo, T.; Ito, M.; Yoshimura, A. CD19-targeted CAR regulatory T cells suppress B cell pathology without GvHD. JCI Insight 2020, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Vassaux, G.; Martin-Duque, P. Use of suicide genes for cancer gene therapy: Study of the different approaches. Expert Opin. Biol. Ther. 2004, 4, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Moolten, F.L.; Wells, J.M. Curability of Tumors Bearing Herpes Thymidine Kinase Genes Transfered by Retroviral Vectors. J. Natl. Cancer Inst. 1990, 82, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Sangro, B.; Mazzolini, G.; Ruiz, M.; Ruiz, J.; Quiroga, J.; Herrero, I.; Qian, C.; Benito, A.; Larrache, J.; Olagüe, C.; et al. A phase I clinical trial of thymidine kinase-based gene therapy in advanced hepatocellular carcinoma. Cancer Gene Ther. 2010, 17, 837–843. [Google Scholar] [CrossRef]
- Bonini, C.; Ferrari, G.; Verzeletti, S.; Servida, P.; Zappone, E.; Ruggieri, L.; Ponzoni, M.; Rossini, S.; Mavilio, F.; Traversari, C.; et al. HSV-TK Gene Transfer into Donor Lymphocytes for Control of Allogeneic Graft-Versus-Leukemia. Science 1997, 276, 1719–1724. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, F.; Bonini, C.; Marktel, S.; Zappone, E.; Servida, P.; Bernardi, M.; Pescarollo, A.; Bondanza, A.; Peccatori, J.; Rossini, S.; et al. Antitumor effects of HSV-TK–engineered donor lymphocytes after allogeneic stem-cell transplantation. Blood 2007, 109, 4698–4707. [Google Scholar] [CrossRef]
- Casucci, M.; Falcone, L.; Camisa, B.; Norelli, M.; Porcellini, S.; Stornaiuolo, A.; Ciceri, F.; Traversari, C.; Bordignon, C.; Bonini, C.; et al. Extracellular NGFR Spacers Allow Efficient Tracking and Enrichment of Fully Functional CAR-T Cells Co-Expressing a Suicide Gene. Front. Immunol. 2018, 9, 507. [Google Scholar] [CrossRef] [Green Version]
- Tiberghien, P. Administration of herpes simplex-thymidine kinase-expressing donor T cells with a T-cell-depleted allogeneic marrow graft. Blood 2001, 97, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Di Stasi, A.; Tey, S.-K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Dotti, G.; Krance, R.A.; Martinez, C.A.; Naik, S.; Kamble, R.T.; Durett, A.G.; Dakhova, O.; Savoldo, B.; Di Stasi, A.; et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood 2015, 125, 4103–4113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffioen, M.; Van Egmond, E.H.; Kester, M.G.; Willemze, R.; Falkenburg, J.F.; Heemskerk, M.H. Retroviral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. Haematologica 2009, 94, 1316–1320. [Google Scholar] [CrossRef] [Green Version]
- Paszkiewicz, P.J.; Fräßle, S.P.; Srivastava, S.; Sommermeyer, D.; Hudecek, M.; Drexler, I.; Sadelain, M.; Liu, L.; Jensen, M.C.; Riddell, S.R.; et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J. Clin. Investig. 2016, 126, 4262–4272. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Yi, M.; Qin, S.; Wu, K. Next generation chimeric antigen receptor T cells: Safety strategies to overcome toxicity. Mol. Cancer 2019, 18, 1–13. [Google Scholar] [CrossRef]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schönfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. Nucleic Acids 2013, 2, e105. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Ramakrishna, S.; Nguyen, S.; Fountaine, T.J.; Ponduri, A.; Stetler-Stevenson, M.; Yuan, C.M.; Haso, W.; Shern, J.F.; Shah, N.N.; et al. Preclinical Development of Bivalent Chimeric Antigen Receptors Targeting Both CD19 and CD. Mol. Ther. Oncolytics 2018, 11, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.; Xiong, Y.; Wu, D.; Nölle, V.; Schmitz, S.; Haso, W.; Kaiser, A.; Dropulic, B.; Orentas, R.J. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J. Immunother. Cancer 2017, 5, 42. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Grote, S.; Chan, C.-H.; Baden, C.; Huber, S.M.; Eckert, F.; Mittelstaet, J.; Kaiser, A.; Seitz, C.; Schlegel, P.; Handgretinger, R.; et al. Abstract B70: Universal adapter CAR-engineered NK-92 cells target patient-derived glioblastoma cancer stem cells. Poster Present. Proffered Abstr. 2020, 8, B70. [Google Scholar] [CrossRef]
- Seitz, C.M.; Kieble, C.V.; Illi, C.C.; Reiter, C.S.; Grote, M.S.; Mittelstaet, J.; Lock, M.D.; Kaiser, A.; Schleicher, S.; Handgretinger, R.; et al. Combinatorial Targeting of Multiple Shared Antigens By Adapter-CAR-T Cells (aCAR-Ts) Allows Target Cell Discrimination and Specific Lysis Based on Differential Expression Profiles. Blood 2018, 132, 4543. [Google Scholar] [CrossRef]
- Jamali, A.; Hadjati, J.; Madjd, Z.; Mirzaei, H.R.; Thalheimer, F.B.; Agarwal, S.; Bonig, H.; Ullrich, E.; Hartmann, J. Highly Efficient Generation of Transgenically Augmented CAR NK Cells Overexpressing CXCR. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Kebriaei, P.; Singh, H.; Huls, M.H.; Figliola, M.J.; Bassett, R.; Olivares, S.; Jena, B.; Dawson, M.J.; Kumaresan, P.R.; Su, S.; et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J. Clin. Investig. 2016, 126, 3363–3376. [Google Scholar] [CrossRef]
- Hudecek, M.; Gogishvili, T.; Monjezi, R.; Wegner, J.; Shankar, R.; Kruesemann, C.; Miskey, C.; Ivics, Z.; Schmeer, M.; Schleef, M.; et al. Minicircle-Based Engineering of Chimeric Antigen Receptor (CAR) T Cells. Recent Results Cancer Res. 2016, 209, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Hudecek, M.; Ivics, Z. Non-viral therapeutic cell engineering with the Sleeping Beauty transposon system. Curr. Opin. Genet. Dev. 2018, 52, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Magnani, C.F.; Gaipa, G.; Belotti, D.; Matera, G.; Tettamanti, S.; Cabiati, B.; Buracchi, C.; Fazio, G.; Zaninelli, S.; Rigamonti, S.; et al. Donor-Derived CD19 CAR Cytokine Induced Killer (CIK) Cells Engineered with Sleeping Beauty Transposon for Relapsed B-Cell Acute Lymphoblastic Leukemia (B-ALL). Blood 2019, 134, 200. [Google Scholar] [CrossRef]
- Cappuzzello, E.; Tosi, A.; Zanovello, P.; Sommaggio, R.; Rosato, A. Retargeting cytokine-induced killer cell activity by CD16 engagement with clinical-grade antibodies. OncoImmunology 2016, 5, e1199311. [Google Scholar] [CrossRef] [Green Version]
- Sommaggio, R.; Cappuzzello, E.; Pietà, A.D.; Tosi, A.; Palmerini, P.; Carpanese, D.; Nicolè, L.; Rosato, A. Adoptive cell therapy of triple negative breast cancer with redirected cytokine-induced killer cells. OncoImmunology 2020, 9, 1777046. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef] [Green Version]
- Tarn, Y.; Martinson, J.; Doligosa, K.; Klingernann, H.-G. Ex vivo expansion of the highly cytotoxic human natural killer cell line NK-92 under current good manufacturing practice conditions for clinical adoptive cellular immunotherapy. Cytotherapy 2003, 5, 259–272. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wendel, P.; Reindl, L.M.; Bexte, T.; Künnemeyer, L.; Särchen, V.; Albinger, N.; Mackensen, A.; Rettinger, E.; Bopp, T.; Ullrich, E. Arming Immune Cells for Battle: A Brief Journey through the Advancements of T and NK Cell Immunotherapy. Cancers 2021, 13, 1481. https://doi.org/10.3390/cancers13061481
Wendel P, Reindl LM, Bexte T, Künnemeyer L, Särchen V, Albinger N, Mackensen A, Rettinger E, Bopp T, Ullrich E. Arming Immune Cells for Battle: A Brief Journey through the Advancements of T and NK Cell Immunotherapy. Cancers. 2021; 13(6):1481. https://doi.org/10.3390/cancers13061481
Chicago/Turabian StyleWendel, Philipp, Lisa Marie Reindl, Tobias Bexte, Leander Künnemeyer, Vinzenz Särchen, Nawid Albinger, Andreas Mackensen, Eva Rettinger, Tobias Bopp, and Evelyn Ullrich. 2021. "Arming Immune Cells for Battle: A Brief Journey through the Advancements of T and NK Cell Immunotherapy" Cancers 13, no. 6: 1481. https://doi.org/10.3390/cancers13061481
APA StyleWendel, P., Reindl, L. M., Bexte, T., Künnemeyer, L., Särchen, V., Albinger, N., Mackensen, A., Rettinger, E., Bopp, T., & Ullrich, E. (2021). Arming Immune Cells for Battle: A Brief Journey through the Advancements of T and NK Cell Immunotherapy. Cancers, 13(6), 1481. https://doi.org/10.3390/cancers13061481