
Cross-Resistance to Abiraterone and Enzalutamide in Castration Resistance Prostate Cancer Cellular Models Is Mediated by AR Transcriptional Reactivation

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Cell Culture
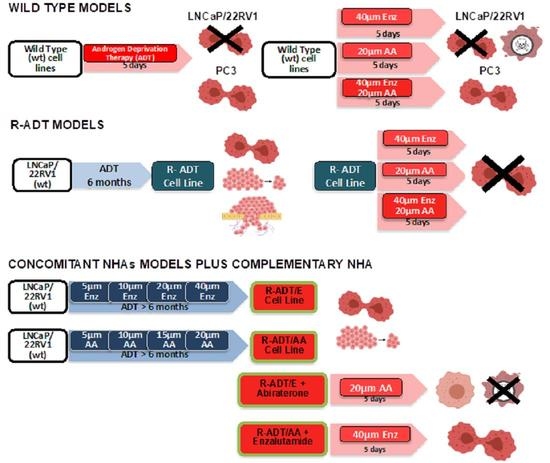
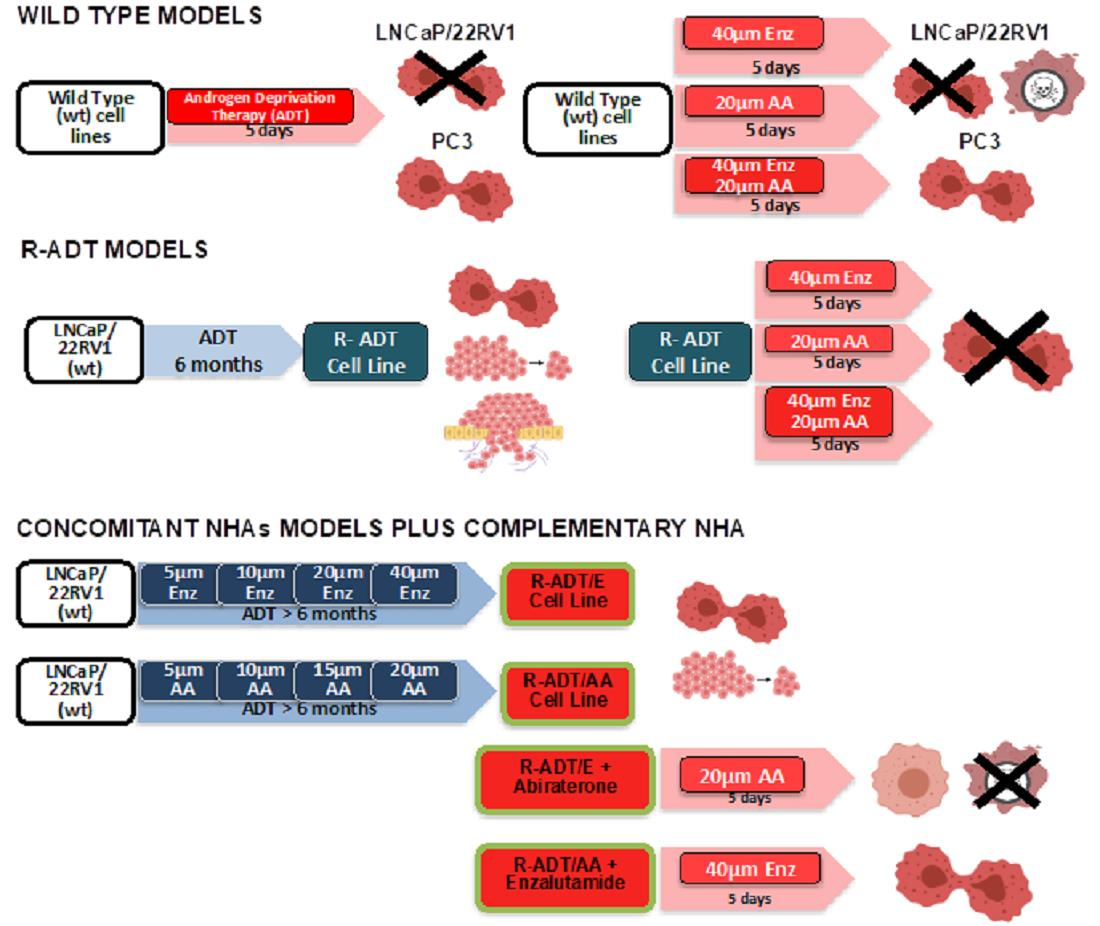
2.2. Generation of Androgen-Deprivation-Treatment-Resistant Cell Lines (R-ADT)
2.3. Generation of Cell Lines Resistant to ADT/NHAs (R-ADT/AA, R-ADT/Enz and R-ADT/AA + Enz) by a Concomitant Use of Treatments
2.4. Treatment with AA or Enz as Second-Line Treatment after Concomitant Therapy (R-ADT/NHAs)
2.5. Cell Proliferation Assays
2.6. Cell Cycle Experiments
2.7. Cell Migration and Invasion Assays
2.8. Quantification of AR Full-Length, AR-V7 and AR-V9 Expression and Isoform Sequencing
2.9. AR Transcriptional Activity
2.10. Statistical Analysis
3. Results
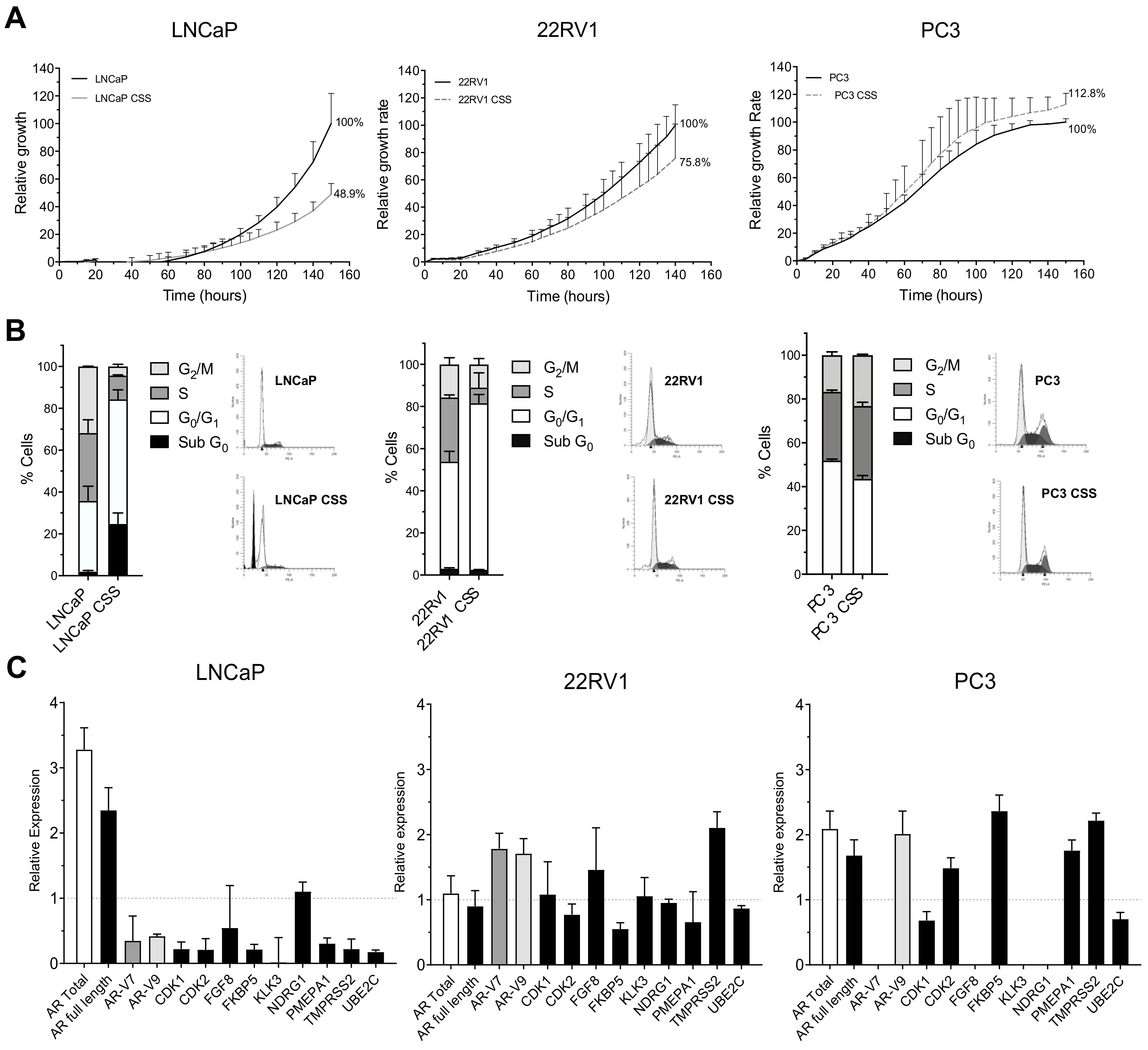
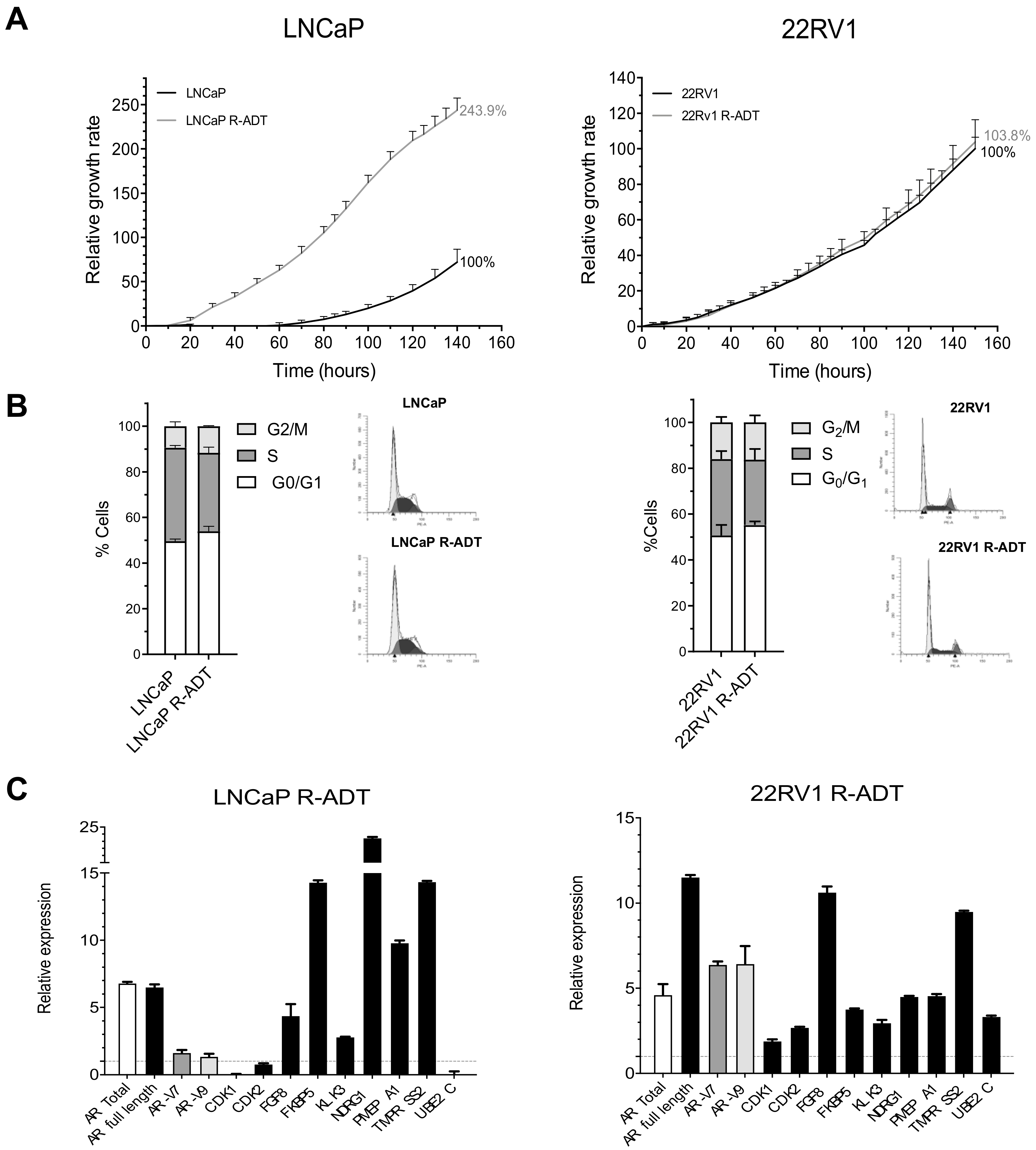
3.1. Functional and Genetic Analyses of the Response to ADT, AA and Enz as First-Line Therapy in PCa Cells Lines: Wild-Type Models
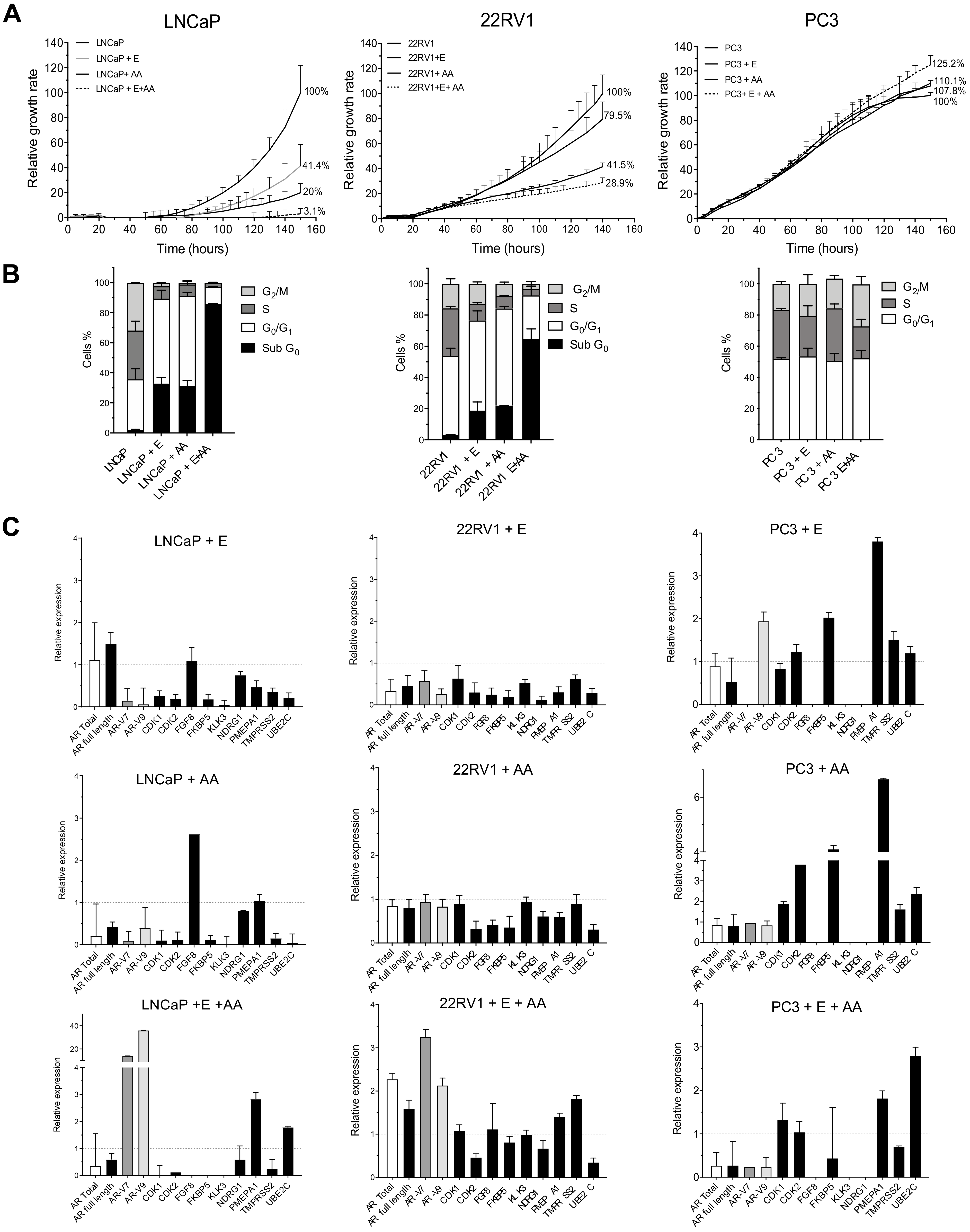
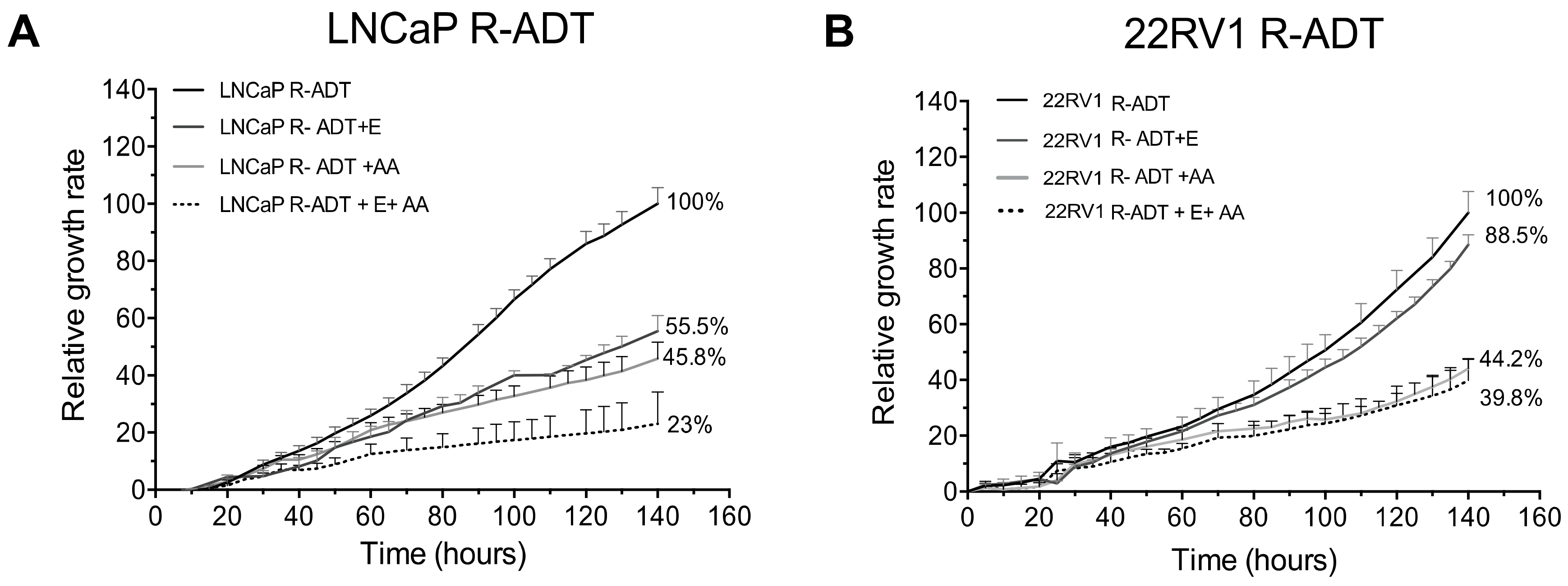
3.2. ADT Resistance Increases AR Transcriptional Activity and Confers Enzalutamide and/or Abiraterone Cross-Resistance (R-ADT Model)
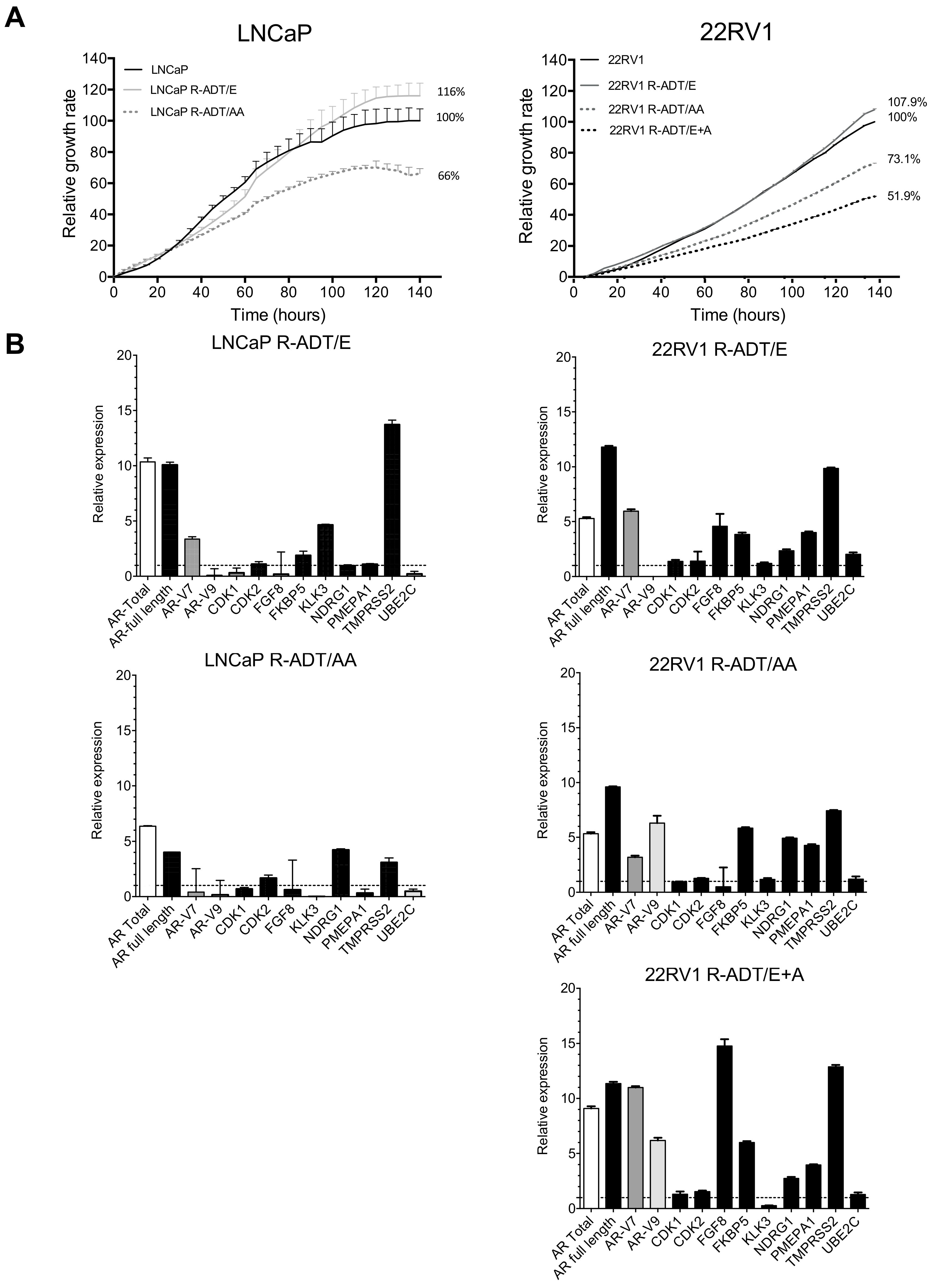
3.3. Resistance to ADT Combined with NHAs Increases AR Full Length Expression and AR Transcriptional Activity in Both PCa Cell Lines (Concomitant Model: R-ADT/NHAs)
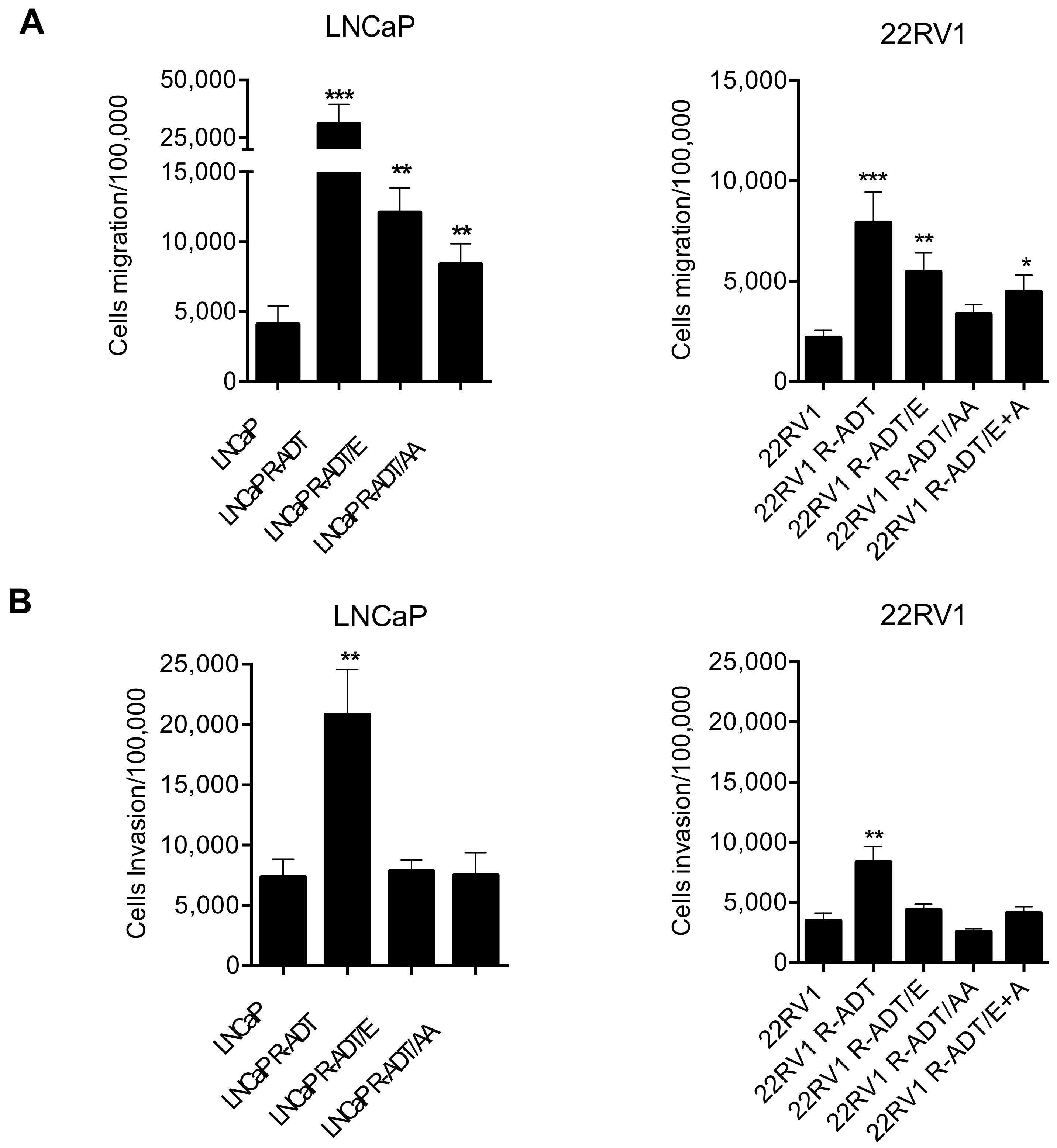
3.4. Acquisition of ADT Resistance Increases Migration and Invasion in Both PCa Cell Lines
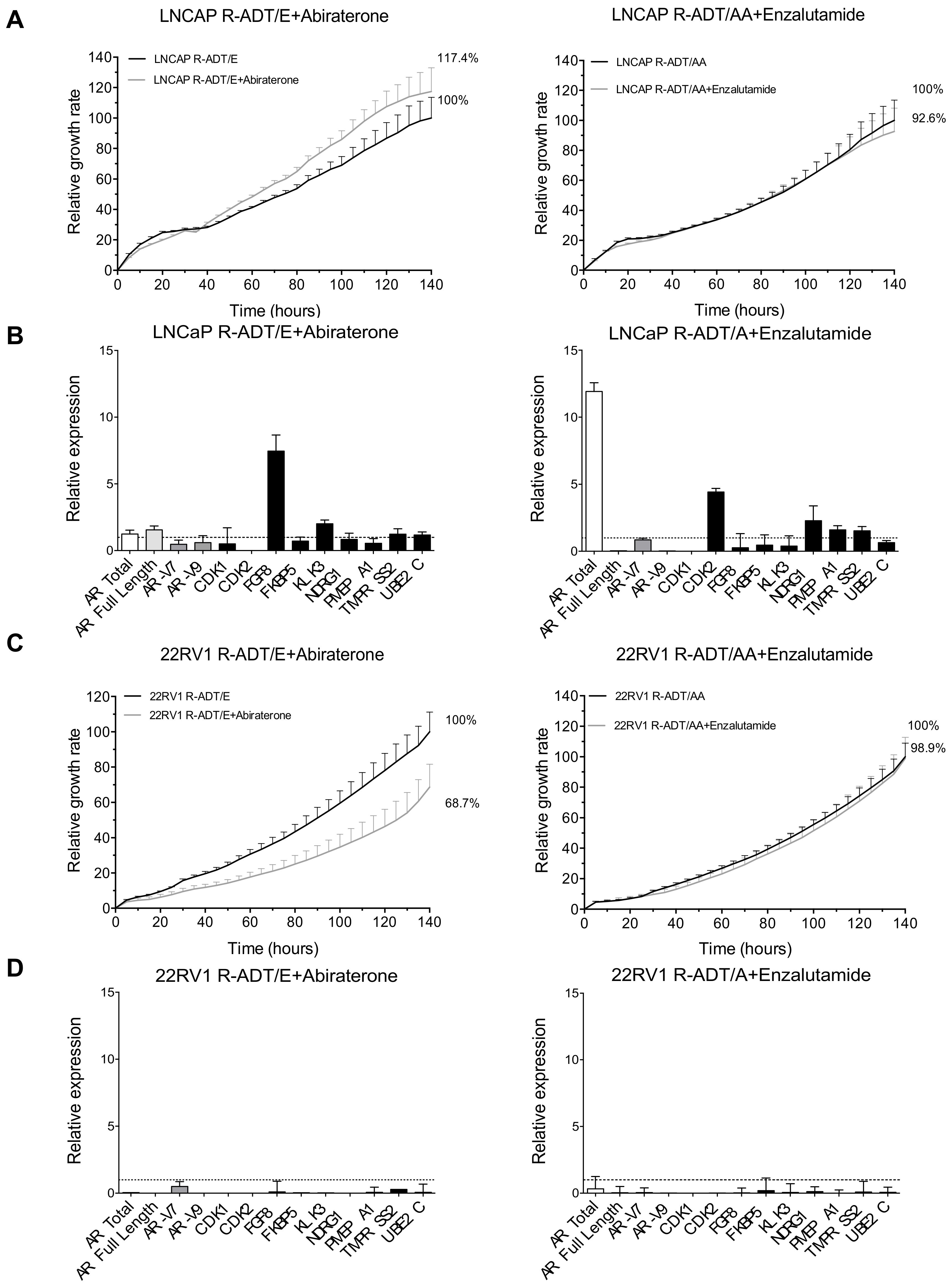
3.5. Most of the Concomitant PCa Models Developed Cross-Resistance to the Alternative NHA Used as a Second-Line Treatment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banerjee, P.P.; Banerjee, S.; Brown, T.R.; Zirkin, B.R. Androgen action in prostate function and disease. Am. J. Clin. Exp. Urol. 2018, 6, 62–77. [Google Scholar]
- Buttigliero, C.; Tucci, M.; Bertaglia, V.; Vignani, F.; Bironzo, P.; Di Maio, M.; Scagliotti, G.V. Understanding and overcoming the mechanisms of primary and acquired resistance to abiraterone and enzalutamide in castration resistant prostate cancer. Cancer Treat. Rev. 2015, 41, 884–892. [Google Scholar] [CrossRef]
- De Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B.; Saad, F.; et al. Abiraterone and Increased Survival in Metastatic Prostate Cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrie, S.E.; Potter, G.A.; Goddard, P.M.; Haynes, B.P.; Dowsett, M.; Jarman, M. Pharmacology of novel steroidal inhibitors of cytochrome P450(17) alpha (17 alpha-hydroxylase/C17-20 lyase). J. Steroid Biochem. Mol. Biol. 1994, 50, 267–273. [Google Scholar] [CrossRef]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a Second-Generation Antiandrogen for Treatment of Advanced Prostate Cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loriot, Y.; Bianchini, D.; Ileana, E.; Sandhu, S.; Patrikidou, A.; Pezaro, C.; Albiges, L.; Attard, G.; Fizazi, K.; De Bono, J.S.; et al. Antitumour activity of abiraterone acetate against metastatic castration-resistant prostate cancer progressing after docetaxel and enzalutamide (MDV3100). Ann. Oncol. 2013, 24, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Noonan, K.L.; North, S.; Bitting, R.L.; Armstrong, A.J.; Ellard, S.L.; Chi, K.N. Clinical activity of abiraterone acetate in patients with metastatic castration-resistant prostate cancer progressing after enzalutamide. Ann. Oncol. 2013, 24, 1802–1807. [Google Scholar] [CrossRef]
- Bianchini, D.; Lorente, D.; Rodriguez-Vida, A.; Omlin, A.; Pezaro, C.; Ferraldeschi, R.; Zivi, A.; Attard, G.; Chowdhury, S.; de Bono, J.S. Antitumour activity of enzalutamide (MDV3100) in patients with metastatic castration-resistant prostate cancer (CRPC) pre-treated with docetaxel and abiraterone. Eur. J. Cancer 2014, 50, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Nadal, R.; Schweizer, M.; Kryvenko, O.N.; Epstein, J.I.; Eisenberger, M.A. Small cell carcinoma of the prostate. Nat. Rev. Urol. 2014, 11, 213–219. [Google Scholar] [CrossRef]
- Badrising, S.; van der Noort, V.; van Oort, I.M.; van den Berg, H.P.; Los, M.; Hamberg, P.; Coenen, J.L.; van den Eertwegh, A.J.M.; de Jong, I.J.; Kerver, E.D.; et al. Clinical activity and tolerability of enzalutamide (MDV3100) in patients with metastatic, castration-resistant prostate cancer who progress after docetaxel and abiraterone treatment. Cancer 2014, 120, 968–975. [Google Scholar] [CrossRef]
- Waltering, K.K.; Urbanucci, A.; Visakorpi, T. Androgen receptor (AR) aberrations in castration-resistant prostate cancer. Mol. Cell. Endocrinol. 2012, 360, 38–43. [Google Scholar] [CrossRef]
- Grist, E.; de Bono, J.S.; Attard, G. Targeting extra-gonadal androgens in castration-resistant prostate cancer. J. Steroid Biochem. Mol. Biol. 2015, 145, 157–163. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, X.; Liang, X.; Jiang, G. Molecular and cellular mechanisms of castration resistant prostate cancer. Oncol. Lett. 2018, 15, 6063–6076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Ueda, T.; Ichikawa, T.; Ito, H. Androgen receptor involvement in the progression of prostate cancer. Endocr. Relat. Cancer 2003, 10, 209–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggero, K.; Farran-Matas, S.; Martinez-Tebar, A.; Aytes, A. Epigenetic Regulation in Prostate Cancer Progression. Curr. Mol. Biol. Rep. 2018, 4, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cato, L.; de Tribolet-Hardy, J.; Lee, I.; Rottenberg, J.T.; Coleman, I.; Melchers, D.; Houtman, R.; Xiao, T.; Li, W.; Uo, T.; et al. ARv7 Represses Tumor-Suppressor Genes in Castration-Resistant Prostate Cancer. Cancer Cell 2019, 35, 401–413.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, M.; Ho, Y.; Hillman, D.W.; Van Etten, J.L.; Henzler, C.; Yang, R.; Sperger, J.M.; Li, Y.; Tseng, E.; Hon, T.; et al. Androgen Receptor Variant AR-V9 Is Coexpressed with AR-V7 in Prostate Cancer Metastases and Predicts Abiraterone Resistance. Clin. Cancer Res. 2017, 23, 4704–4715. [Google Scholar] [CrossRef] [Green Version]
- Taplin, M.-E.; Bubley, G.J.; Shuster, T.D.; Frantz, M.E.; Spooner, A.E.; Ogata, G.K.; Keer, H.N.; Balk, S.P. Mutation of the Androgen-Receptor Gene in Metastatic Androgen-Independent Prostate Cancer. N. Engl. J. Med. 1995, 332, 1393–1398. [Google Scholar] [CrossRef]
- Gregory, C.W.; Johnson, R.T.; Mohler, J.L.; French, F.S.; Wilson, E.M. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001, 61, 2892–2898. [Google Scholar]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef]
- Stanbrough, M.; Bubley, G.J.; Ross, K.; Golub, T.R.; Rubin, M.A.; Penning, T.M.; Febbo, P.G.; Balk, S.P. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006, 66, 2815–2825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.; Wang, H.; Xu, Y.; Chen, S.; Balk, S.P. Reactivation of androgen receptor-regulated TMPRSS2:ERG gene expression in castration-resistant prostate cancer. Cancer Res. 2009, 69, 6027–6032. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; He, H.H.; Chen, S.; Coleman, I.; Wang, H.; Fang, Z.; Chen, S.; Nelson, P.S.; Liu, X.S.; Brown, M.; et al. Androgen Receptor Gene Expression in Prostate Cancer Is Directly Suppressed by the Androgen Receptor Through Recruitment of Lysine-Specific Demethylase 1. Cancer Cell 2011, 20, 457–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.; Balk, S.P. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr. Relat. Cancer 2011, 18, R175–R182. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.J.; Sowalsky, A.G.; Gao, S.; Cai, C.; Voznesensky, O.; Schaefer, R.; Loda, M.; True, L.D.; Ye, H.; Troncoso, P.; et al. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin. Cancer Res. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Hwang, T.H.; Oseth, L.A.; Hauge, A.; Vessella, R.L.; Schmechel, S.C.; Hirsch, B.; Beckman, K.B.; Silverstein, K.A.; Dehm, S.M. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene 2012, 31, 4759–4767. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.T.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.S.; Stockert, J.A.; Hackert, V.; Yadav, K.K.; Tewari, A.K. Intratumor heterogeneity in prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 349–360. [Google Scholar] [CrossRef]
- Katsuoka, Y.; Hoshino, H.; Shiramizu, M.; Sakabe, K.; Seiki, K. Autoradiographic and cytochemical localization of androgen in human prostatic cancer cell lines. Urology 1986, 28, 228–231. [Google Scholar] [CrossRef]
- Takayoshi, D.; Noboru, K.; Atsushi, O.; Hisakazu, F.; Teruo, I.; Tomohiko, K. Establishment and characterization of monoclonal antibody against androgen receptor. J. Steroid Biochem. 1989, 33, 845–851. [Google Scholar] [CrossRef]
- Ghashghaei, M.; Paliouras, M.; Heravi, M.; Bekerat, H.; Trifiro, M.; Niazi, T.M.; Muanza, T. Enhanced radiosensitization of enzalutamide via schedule dependent administration to androgen-sensitive prostate cancer cells. Prostate 2018, 78, 64–75. [Google Scholar] [CrossRef] [Green Version]
- Salami, J.; Alabi, S.; Willard, R.R.; Vitale, N.J.; Wang, J.; Dong, H.; Jin, M.; McDonnell, D.P.; Crew, A.P.; Neklesa, T.K.; et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol. 2018, 1, 100. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dehigaspitiya, D.C.; Levine, P.M.; Profit, A.A.; Haugbro, M.; Imberg-Kazdan, K.; Logan, S.K.; Kirshenbaum, K.; Garabedian, M.J. Multivalent peptoid conjugates which overcome enzalutamide resistance in prostate cancer cells. Cancer Res. 2016, 76, 5124–5132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.; Liu, M.; Liby, T.; Bayani, N.; Bucher, E.; Chiotti, K.; Derrick, D.; Chauchereau, A.; Heiser, L.; Alumkal, J.; et al. Enzalutamide response in a panel of prostate cancer cell lines reveals a role for glucocorticoid receptor in enzalutamide resistant disease. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Claessens, F.; Helsen, C.; Prekovic, S.; Van den Broeck, T.; Spans, L.; Van Poppel, H.; Joniau, S. Emerging mechanisms of enzalutamide resistance in prostate cancer. Nat. Rev. Urol. 2014, 11, 712–716. [Google Scholar] [CrossRef]
- Agus, D.B.; Cordon-Cardo, C.; William, F.; Drobnjak, M.; Koff, A.; Golde, D.W.; Scher, H.I. Prostate cancer cell cycle regulators: Response to androgen withdrawal and development of androgen independence. J. Natl. Cancer Inst. 1999, 91, 1869–1876. [Google Scholar] [CrossRef] [Green Version]
- Scher, H.I.; Sawyers, C.L. Biology of progressive, castration-resistant prostate cancer: Directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol. 2005, 23, 8253–8261. [Google Scholar] [CrossRef]
- Blute, M.L.; Damaschke, N.; Wagner, J.; Yang, B.; Gleave, M.; Fazli, L.; Shi, F.; Abel, E.J.; Downs, T.M.; Huang, W.; et al. Persistence of senescent prostate cancer cells following prolonged neoadjuvant androgen deprivation therapy. PLoS ONE 2017, 12, e0172048. [Google Scholar] [CrossRef]
- Nakazawa, M.; Antonarakis, E.S.; Luo, J. Androgen receptor splice variants in the era of enzalutamide and abiraterone. Horm. Cancer 2014, 5, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, C.A. AR-V7—Repress to impress. Nat. Rev. Urol. 2019, 16, 269. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Itsumi, M.; Takeuchi, A.; Imada, K.; Yokomizo, A.; Kuruma, H.; Inokuchi, J.; Tatsugami, K.; Uchiumi, T.; Oda, Y.; et al. Crosstalk between epithelial-mesenchymal transition and castration resistance mediated by Twist1/AR signaling in prostate cancer. Endocr. Relat. Cancer 2015, 22, 889–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, L.; Yang, L.; Li, R.; Rodrigues, D.N.; Crespo, M.; Hsieh, J.T.; Tilley, W.D.; De Bono, J.; Selth, L.A.; Raj, G.V. Disrupting androgen receptor signaling induces Snail-mediated epithelial-mesenchymal plasticity in prostate cancer. Cancer Res. 2017, 77, 3101–3112. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Nakazawa, M.; Nadal, R.; Paller, C.J.; Denmeade, S.R.; Carducci, M.A.; et al. Androgen receptor splice variant 7 and efficacy of taxane chemotherapy in patients with metastatic castration-resistant prostate cancer. JAMA Oncol. 2015, 1, 582–591. [Google Scholar] [CrossRef] [Green Version]
- Jeong, C.W.; Kang, M.; Il Jung, S.; Kim, T.H.; Park, S.W.; Joung, J.Y.; Jeon, S.S.; Hong, J.H.; Lee, J.Y.; Chung, B.H.; et al. Importance of androgen-deprivation therapy during enzalutamide treatment in men with metastatic castration-resistant prostate cancer following chemotherapy: Results from retrospective, multicenter data. Prostate Cancer Prostatic Dis. 2019, 22, 150–158. [Google Scholar] [CrossRef]
- Morris, M.J.; Heller, G.; Bryce, A.H.; Armstrong, A.J.; Beltran, H.; Hahn, O.M.; McGary, E.C.; Mehan, P.T.; Goldkorn, A.; Roth, B.J.; et al. Alliance A031201: A phase III trial of enzalutamide (ENZ) versus enzalutamide, abiraterone, and prednisone (ENZ/AAP) for metastatic castration resistant prostate cancer (mCRPC). J. Clin. Oncol. 2019, 37, 5008. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simon, I.; Perales, S.; Casado-Medina, L.; Rodríguez-Martínez, A.; Garrido-Navas, M.d.C.; Puche-Sanz, I.; Diaz-Mochon, J.J.; Alaminos, C.; Lupiañez, P.; Lorente, J.A.; et al. Cross-Resistance to Abiraterone and Enzalutamide in Castration Resistance Prostate Cancer Cellular Models Is Mediated by AR Transcriptional Reactivation. Cancers 2021, 13, 1483. https://doi.org/10.3390/cancers13061483
Simon I, Perales S, Casado-Medina L, Rodríguez-Martínez A, Garrido-Navas MdC, Puche-Sanz I, Diaz-Mochon JJ, Alaminos C, Lupiañez P, Lorente JA, et al. Cross-Resistance to Abiraterone and Enzalutamide in Castration Resistance Prostate Cancer Cellular Models Is Mediated by AR Transcriptional Reactivation. Cancers. 2021; 13(6):1483. https://doi.org/10.3390/cancers13061483
Chicago/Turabian StyleSimon, Iris, Sonia Perales, Laura Casado-Medina, Alba Rodríguez-Martínez, Maria del Carmen Garrido-Navas, Ignacio Puche-Sanz, Juan J. Diaz-Mochon, Clara Alaminos, Pablo Lupiañez, Jose A. Lorente, and et al. 2021. "Cross-Resistance to Abiraterone and Enzalutamide in Castration Resistance Prostate Cancer Cellular Models Is Mediated by AR Transcriptional Reactivation" Cancers 13, no. 6: 1483. https://doi.org/10.3390/cancers13061483
APA StyleSimon, I., Perales, S., Casado-Medina, L., Rodríguez-Martínez, A., Garrido-Navas, M. d. C., Puche-Sanz, I., Diaz-Mochon, J. J., Alaminos, C., Lupiañez, P., Lorente, J. A., Serrano, M. J., & Real, P. J. (2021). Cross-Resistance to Abiraterone and Enzalutamide in Castration Resistance Prostate Cancer Cellular Models Is Mediated by AR Transcriptional Reactivation. Cancers, 13(6), 1483. https://doi.org/10.3390/cancers13061483