
Tribbles Pseudokinase 3 Regulation and Contribution to Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
2. TRIB3 Gene and Transcripts
3. TRIB3 Protein Structure
4. Regulation of TRIB3 Expression
4.1. Transcriptional Regulation
4.2. Post-Translational Regulation
5. TRIB3 as A Scaffold for Diverse Signaling Proteins
5.1. TRIB3 Regulation of Kinase-Dependent Proteins
5.2. TRIB3 Regulation of Transcription Factors
5.3. TRIB3 Regulation of Pre-mRNA Splicing
6. TRIB3 and Cancer
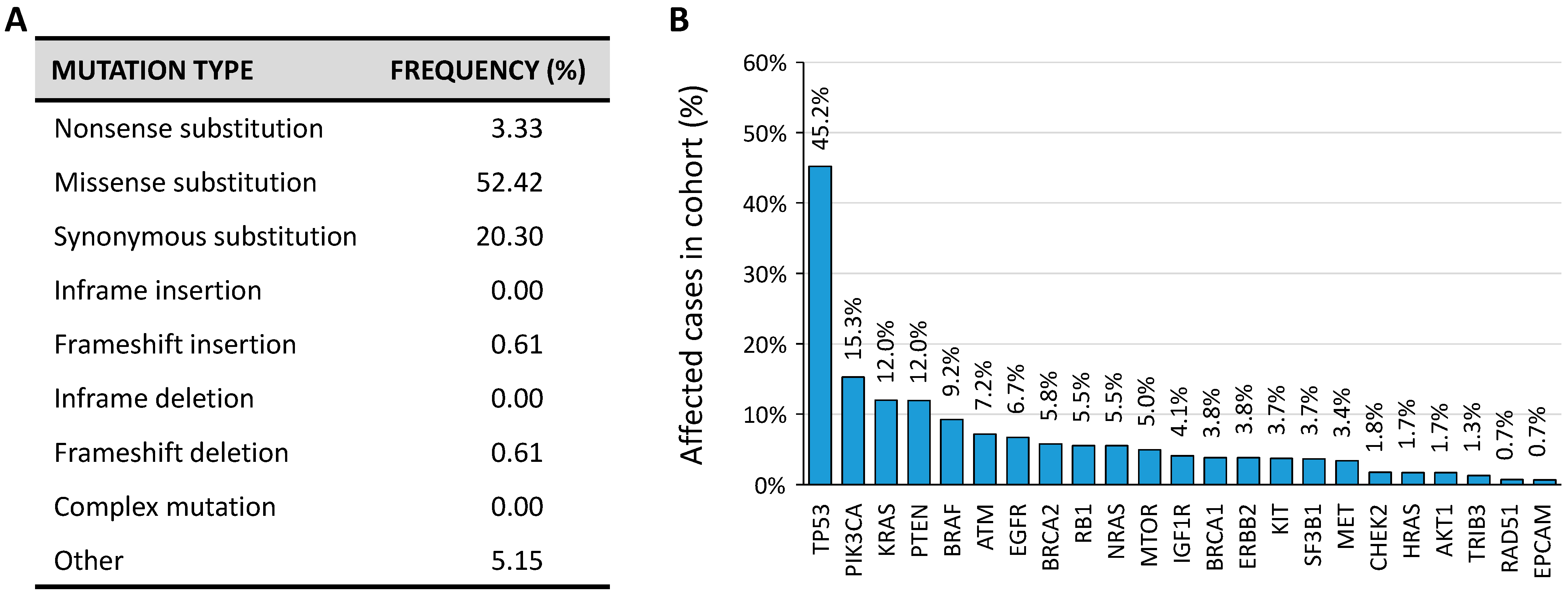
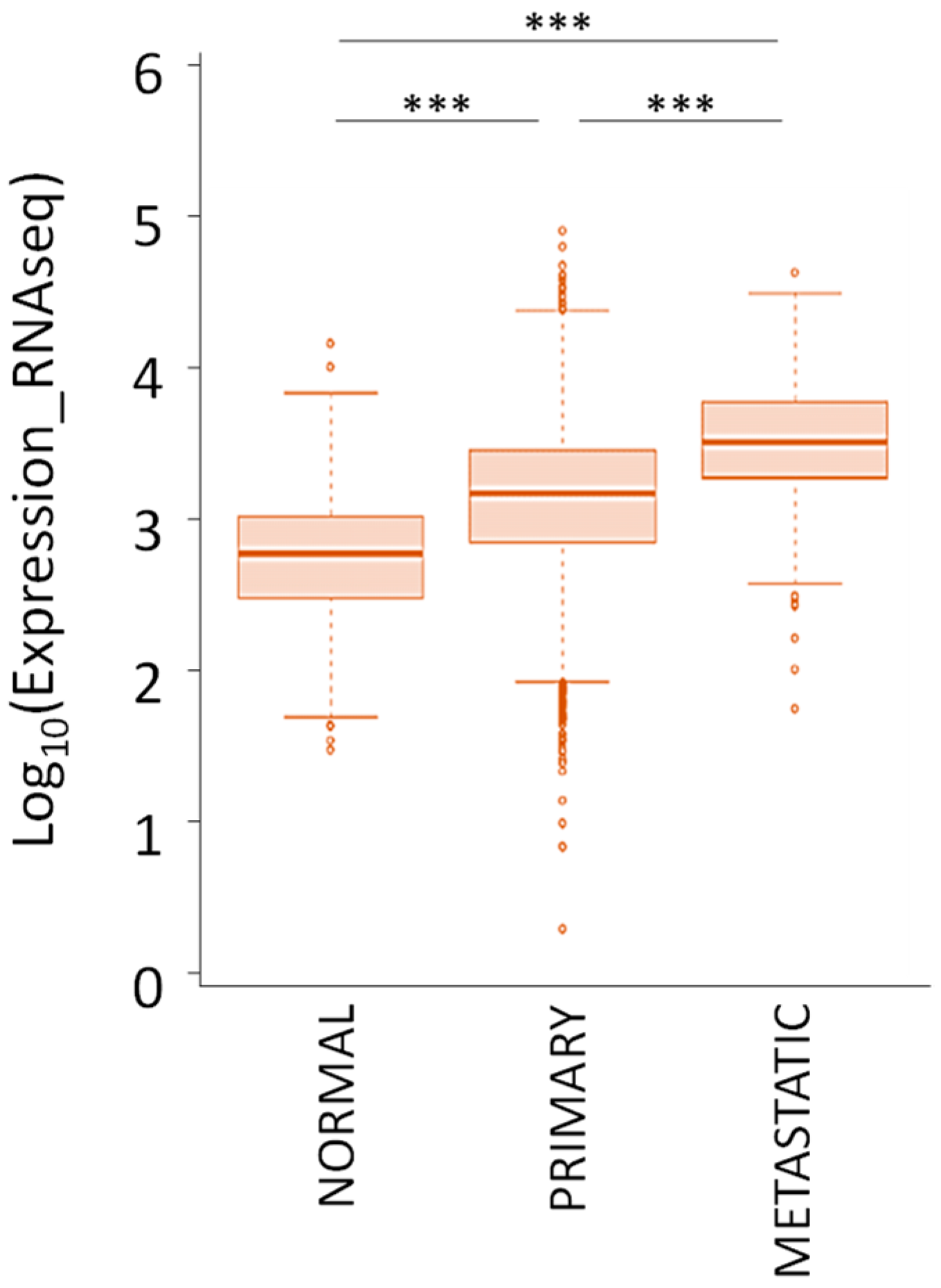
6.1. Genetic Alterations and Expression
6.2. TRIB3 Expression Levels and Patient’s Prognosis
6.3. Is TRIB3 A Tumor Suppressor?
6.4. Is TRIB3 An Oncogene?
7. Conclusions
Funding
Conflicts of Interest
References
- Großhans, J.; Wieschaus, E. A Genetic Link between Morphogenesis and Cell Division during Formation of the Ventral Furrow in Drosophila. Cell 2000, 101, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Seher, T.C.; Leptin, M. Tribbles, a cell-cycle brake that coordinates proliferation and morphogenesis during Drosophila gastrulation. Curr. Biol. 2000, 10, 623–629. [Google Scholar] [CrossRef] [Green Version]
- Mata, J.; Curado, S.; Ephrussi, A.; Rørth, P. Tribbles Coordinates Mitosis and Morphogenesis in Drosophila by Regulating String/CDC25 Proteolysis. Cell 2000, 101, 511–522. [Google Scholar] [CrossRef] [Green Version]
- Hegedus, Z.; Czibula, A.; Kiss-Toth, E. Tribbles: Novel regulators of cell function; evolutionary aspects. Cell. Mol. Life Sci. 2006, 63, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Eyers, P.A.; Keeshan, K.; Kannan, N. Tribbles in the 21st Century: The Evolving Roles of Tribbles Pseudokinases in Biology and Disease. Trends Cell Biol. 2017, 27, 284–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiss-Toth, E.; Bagstaff, S.M.; Sung, H.Y.; Jozsa, V.; Dempsey, C.; Caunt, J.C.; Oxley, K.M.; Wyllie, D.H.; Polgar, T.; Harte, M.; et al. Human Tribbles, a Protein Family Controlling Mitogen-activated Protein Kinase Cascades. J. Biol. Chem. 2004, 279, 42703–42708. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, N.; Ishii, H.; Mimori, K.; Takatsuno, Y.; Kim, H.; Hirose, H.; Sekimoto, M.; Doki, Y.; Mori, M. Abnormal expression of TRIB3 in colorectal cancer: A novel marker for prognosis. Br. J. Cancer 2009, 101, 1664–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, B.; Zhou, J.; Ma, K.; Zhang, J.; Xie, H.; Zhang, K.; Li, L.; Cai, L.; Zhang, N.; Zhang, Z.; et al. TRIB3 Promotes the Proliferation and Invasion of Renal Cell Carcinoma Cells via Activating MAPK Signaling Pathway. Int. J. Biol. Sci. 2019, 15, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Wu, I.-J.; Lin, R.-J.; Wang, H.-C.; Yuan, T.-M.; Chuang, S.-M. TRIB3 downregulation enhances doxorubicin-induced cytotoxicity in gastric cancer cells. Arch. Biochem. Biophys. 2017, 622, 26–35. [Google Scholar] [CrossRef]
- Zhang, J.; Wen, H.-J.; Guo, Z.-M.; Zeng, M.-S.; Li, M.-Z.; Jiang, Y.-E.; He, X.-G.; Sun, C.-Z. TRB3 overexpression due to endoplasmic reticulum stress inhibits AKT kinase activation of tongue squamous cell carcinoma. Oral Oncol. 2011, 47, 934–939. [Google Scholar] [CrossRef]
- Zhou, H.; Luo, Y.; Chen, J.-H.; Hu, J.; Luo, Y.-Z.; Wang, W.; Zeng, Y.; Xiao, L. Knockdown of TRB3 induces apoptosis in human lung adenocarcinoma cells through regulation of Notch 1 expression. Mol. Med. Rep. 2013, 8, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Stefanovska, B.; Vicier, C.E.; Dayris, T.; Ogryzko, V.; Scott, V.; Bouakka, I.; Delaloge, S.; Rocca, A.; Le Saux, O.; Trédan, O.; et al. Rapalog-mediated repression of Tribbles pseudokinase 3 regulates pre-mRNA splicing. Cancer Res. 2020, 80, 2190–2203. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.M.; Nakatani, Y.; Jamieson, S.A.; Dai, W.; Lucet, I.S.; Mace, P.D. Molecular Mechanism of CCAAT-Enhancer Binding Protein Recruitment by the TRIB1 Pseudokinase. Structure 2015, 23, 2111–2121. [Google Scholar] [CrossRef] [Green Version]
- Bowers, A.J.; Scully, S.; Boylan, J.F. SKIP3, a novel Drosophila tribbles ortholog, is overexpressed in human tumors and is regulated by hypoxia. Oncogene 2003, 22, 2823–2835. [Google Scholar] [CrossRef] [Green Version]
- Bailey, F.P.; Byrne, D.P.; Oruganty, K.; Eyers, C.E.; Novotny, C.J.; Shokat, K.M.; Kannan, N.; Eyers, P.A. The Tribbles 2 (TRB2) pseudokinase binds to ATP and autophosphorylates in a metal-independent manner. Biochem. J. 2015, 467, 47–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, S.; Wells, R.; Rechsteiner, M. Amino acid sequences common to rapidly degraded proteins: The PEST hypothesis. Science 1986, 234, 364–368. [Google Scholar] [CrossRef]
- Wennemers, M.; Bussink, J.; Beucken, T.V.D.; Sweep, F.C.G.J.; Span, P.N. Regulation of TRIB3 mRNA and Protein in Breast Cancer. PLoS ONE 2012, 7, e49439. [Google Scholar] [CrossRef] [PubMed]
- Örd, D.; Örd, T. Characterization of human NIPK (TRB3, SKIP3) gene activation in stressful conditions. Biochem. Biophys. Res. Commun. 2005, 330, 210–218. [Google Scholar] [CrossRef]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4–CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Jousse, C.; Deval, C.; Maurin, A.-C.; Parry, L.; Chérasse, Y.; Chaveroux, C.; Lefloch, R.; Lenormand, P.; Bruhat, A.; Fafournoux, P. TRB3 Inhibits the Transcriptional Activation of Stress-regulated Genes by a Negative Feedback on the ATF4 Pathway. J. Biol. Chem. 2007, 282, 15851–15861. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wu, X.; Franklin, J.L.; Messina, J.L.; Hill, H.S.; Moellering, D.R.; Walton, R.G.; Martin, M.; Garvey, W.T. Mammalian Tribbles homolog 3 impairs insulin action in skeletal muscle: Role in glucose-induced insulin resistance. Am. J. Physiol. Metab. 2010, 298, E565–E576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Örd, T.; Örd, D.; Adler, P.; Vilo, J.; Örd, T. TRIB3 enhances cell viability during glucose deprivation in HEK293-derived cells by upregulating IGFBP2, a novel nutrient deficiency survival factor. Biochim. Biophys. Acta (BBA) Bioenerg. 2015, 1853, 2492–2505. [Google Scholar] [CrossRef] [Green Version]
- Geng, T.; Hu, W.; Broadwater, M.H.; Snider, J.M.; Bielawski, J.; Russo, S.B.; Schwacke, J.H.; Ross, J.; Cowart, L.A. Fatty acids differentially regulate insulin resistance through endoplasm reticulum stress-mediated induction of tribbles homologue 3: A potential link between dietary fat composition and the pathophysiological outcomes of obesity. Diabetologia 2013, 56, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Örd, T.; Örd, T. Mammalian Pseudokinase TRIB3 in Normal Physiology and Disease: Charting the Progress in Old and New Avenues. Curr. Protein Pept. Sci. 2017, 18, 1. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.-O.; Aw, T.-Y.; Holbrook, N.J. Gadd153 Sensitizes Cells to Endoplasmic Reticulum Stress by Down-Regulating Bcl2 and Perturbing the Cellular Redox State. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mkrtchian, S. Targeting unfolded protein response in cancer and diabetes. Endocrine-Related Cancer 2015, 22, C1–C4. [Google Scholar] [CrossRef] [Green Version]
- Koo, S.-H.; Satoh, H.; Herzig, S.; Lee, C.-H.; Hedrick, S.; Kulkarni, R.N.; Evans, R.M.; Olefsky, J.M.; Montminy, M. PGC-1 promotes insulin resistance in liver through PPAR-α-dependent induction of TRB-3. Nat. Med. 2004, 10, 530–534. [Google Scholar] [CrossRef]
- Chan, M.C.; Weisman, A.S.; Kang, H.; Nguyen, P.H.; Hickman, T.; Mecker, S.V.; Hill, N.S.; Lagna, G.; Hata, A. The Amiloride Derivative Phenamil Attenuates Pulmonary Vascular Remodeling by Activating NFAT and the Bone Morphogenetic Protein Signaling Pathway. Mol. Cell. Biol. 2010, 31, 517–530. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Han, S.; Kitamura, T.; Accili, M. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J. Clin. Investig. 2006, 116, 2464–2472. [Google Scholar] [CrossRef] [Green Version]
- Zareen, N.; Biswas, S.C.; Greene, L. A feed-forward loop involving Trib3, Akt and FoxO mediates death of NGF-deprived neurons. Cell Death Differ. 2013, 20, 1719–1730. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Bjornsti, M.-A.; Houghton, P.J. Rapamycins: Mechanism of action and cellular resistance. Cancer Biol. Ther. 2003, 2, 222–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galat, A.; Thai, R. Rapamycin-binding FKBP25 associates with diverse proteins that form large intracellular entities. Biochem. Biophys. Res. Commun. 2014, 450, 1255–1260. [Google Scholar] [CrossRef]
- Rasmussen, T.L.; Ma, Y.; Park, C.Y.; Harriss, J.; Pierce, S.A.; Dekker, J.D.; Valenzuela, N.; Srivastava, D.; Schwartz, R.J.; Stewart, M.D.; et al. Smyd1 Facilitates Heart Development by Antagonizing Oxidative and ER Stress Responses. PLoS ONE 2015, 10, e0121765. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; De Vries, E.G.; et al. Everolimus for Advanced Pancreatic Neuroendocrine Tumors. New Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Li, L.; Liu, Q.; Xing, G.; Kuai, X.; Sun, J.; Yin, X.; Wang, J.; Zhang, L.; He, F. E3 ubiquitin ligase SIAH1 mediates ubiquitination and degradation of TRB3. Cell. Signal. 2008, 20, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Takahama, S.; Endo, Y.; Sawasaki, T. Stress-Inducible Caspase Substrate TRB3 Promotes Nuclear Translocation of Procaspase-3. PLoS ONE 2012, 7, e42721. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Du, K. TRB3: A tribbles Homolog That Inhibits Akt/PKB Activation by Insulin in Liver. Science 2003, 300, 1574–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsting, E.; Patel, S.V.; Declèves, A.-E.; Lee, S.J.; Rahman, Q.M.; Akira, S.; Satriano, J.; Sharma, K.; Vallon, V.; Cunard, R. Tribbles Homolog 3 Attenuates Mammalian Target of Rapamycin Complex-2 Signaling and Inflammation in the Diabetic Kidney. J. Am. Soc. Nephrol. 2014, 25, 2067–2078. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Fang, X.; Malik, W.S.; He, Y.; Li, X.; Xie, M.; Sun, W.; Xu, Y.; Liu, X. TRB3 interacts with ERK and JNK and contributes to the proliferation, apoptosis, and migration of lung adenocarcinoma cells. J. Cell. Physiol. 2020, 235, 538–547. [Google Scholar] [CrossRef]
- Izrailit, J.; Berman, H.K.; Datti, A.; Wrana, J.L.; Reedijk, M. High throughput kinase inhibitor screens reveal TRB3 and MAPK-ERK/TGF pathways as fundamental Notch regulators in breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 1714–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Ohoka, N.; Hayashi, H.; Sato, R. TRB3 suppresses adipocyte differentiation by negatively regulating PPARγ transcriptional activity. J. Lipid Res. 2008, 49, 880–892. [Google Scholar] [CrossRef] [Green Version]
- Dedhia, P.H.; Keeshan, K.; Uljon, S.; Xu, L.; Vega, M.E.; Shestova, O.; Zaks-Zilberman, M.; Romany, C.; Blacklow, S.C.; Pear, W.S. Differential ability of Tribbles family members to promote degradation of C/EBPα and induce acute myelogenous leukemia. Blood 2010, 116, 1321–1328. [Google Scholar] [CrossRef]
- Örd, D.; Meerits, K.; Örd, T. TRB3 protects cells against the growth inhibitory and cytotoxic effect of ATF4. Exp. Cell Res. 2007, 313, 3556–3567. [Google Scholar] [CrossRef]
- Luo, X.; Zhong, L.; Yu, L.; Xiong, L.; Dan, W.; Li, J.; Ye, J.; Chu, X.; Liu, C.; Liu, B. TRIB3 destabilizes tumor suppressor PPARα expression through ubiquitin-mediated proteasome degradation in acute myeloid leukemia. Life Sci. 2020, 257, 118021. [Google Scholar] [CrossRef] [PubMed]
- Fang, N.; Zhang, W.; Xu, S.; Lin, H.; Wang, Z.; Liu, H.; Fang, Q.; Li, C.; Peng, L.; Lou, J. TRIB3 alters endoplasmic reticulum stress-induced β-cell apoptosis via the NF-κB pathway. Metabolism 2014, 63, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Duggan, S.P.; Behan, F.M.; Kirca, M.; Smith, S.; Reynolds, J.V.; Long, A.; Kelleher, D. An integrative genomic approach in oesophageal cells identifies TRB3 as a bile acid responsive gene, downregulated in Barrett’s oesophagus, which regulates NF-κB activation and cytokine levels. Carcinogenesis 2010, 31, 936–945. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Xu, L.-G.; Zhai, Z.; Shu, H.-B. SINK Is a p65-interacting Negative Regulator of NF-κB-dependent Transcription. J. Biol. Chem. 2003, 278, 27072–27079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rzymski, T.; Paantjens, A.; Bod, J.; Harris, A.L. Multiple pathways are involved in the anoxia response of SKIP3 including HuR-regulated RNA stability, NF-κB and ATF4. Oncogene 2008, 27, 4532–4543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Örd, D.; Örd, T. Mouse NIPK interacts with ATF4 and affects its transcriptional activity. Exp. Cell Res. 2003, 286, 308–320. [Google Scholar] [CrossRef]
- Hua, F.; Shang, S.; Zhang, X.-W.; Liu, S.-S.; Yu, J.-M.; Wang, F.; Zhang, C.; Huang, B.; Hu, Z.-W.; Yang, Y.-W.; et al. TRIB3 Interacts With β-Catenin and TCF4 to Increase Stem Cell Features of Colorectal Cancer Stem Cells and Tumorigenesis. Gastroenterology 2019, 156, 708–721.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Z.; Yang, L.; Yang, Y.; Yang, J.; Niu, Z.; Zhang, X.; Song, Q.; Lei, Y.; Wu, H.; Guo, J. Activation of Wnt/β-catenin pathway causes insulin resistance and increases lipogenesis in HepG2 cells via regulation of endoplasmic reticulum stress. Biochem. Biophys. Res. Commun. 2020, 526, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.T.; Fink, G.R.; Bartel, D.P. Excised linear introns regulate growth in yeast. Nat. Cell Biol. 2019, 565, 606–611. [Google Scholar] [CrossRef]
- Edwards, S.R.; Johnson, T.L. Intron RNA sequences help yeast cells to survive starvation. Nat. Cell Biol. 2019, 565, 578–579. [Google Scholar] [CrossRef] [Green Version]
- Jacob, A.G.; Smith, C.W.J. Intron retention as a component of regulated gene expression programs. Qual. Life Res. 2017, 136, 1043–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.; Zheng, Y.; Cho, S.; Jang, C.; England, C.; Dempsey, J.M.; Yu, Y.; Liu, X.; He, L.; Cavaliere, P.M.; et al. Post-transcriptional Regulation of De Novo Lipogenesis by mTORC1-S6K1-SRPK2 Signaling. Cell 2017, 171, 1545–1558.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prudente, S.; Sesti, G.; Pandolfi, A.; Andreozzi, F.; Consoli, A.; Trischitta, V. The mammalian tribbles homolog TRIB3, glucose homeostasis, and cardiovascular diseases. Endocr. Rev. 2012, 33, 526–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, N.Z.A.; Mokhtar, N.M.; Sin, T.K.; Rose, I.M.; Sagap, I.; Harun, R.; Jamal, R. Integrated Analysis of Copy Number Variation and Genome-Wide Expression Profiling in Colorectal Cancer Tissues. PLoS ONE 2014, 9, e92553. [Google Scholar] [CrossRef]
- Loo, L.W.M.; Tiirikainen, M.; Cheng, I.; Lum-Jones, A.; Seifried, A.; Church, J.M.; Gryfe, R.; Weisenberger, D.J.; Lindor, N.M.; Gallinger, S.; et al. Integrated analysis of genome-wide copy number alterations and gene expression in microsatellite stable, CpG island methylator phenotype-negative colon cancer. Genes, Chromosom. Cancer 2013, 52, 450–466. [Google Scholar] [CrossRef] [Green Version]
- Salazar, M.; Lorente, M.; García-Taboada, E.; Gomez, E.P.; Davila, D.; Zúñiga-García, P.; Flores, J.M.; Rodríguez, A.; Hegedus, Z.; Mosén-Ansorena, D.; et al. TRIB3 suppresses tumorigenesis by controlling mTORC2/AKT/FOXO signaling. Mol. Cell. Oncol. 2015, 2, e980134. [Google Scholar]
- Salazar, M.; Lorente, M.; García-Taboada, E.; Pérez-Gómez, E.; Dávila, D.; Zúñiga-García, P.; Flores, J.M.; Rodríguez, A.; Hegedus, Z.; Mosén-Ansorena, D.; et al. Loss of Tribbles pseudokinase-3 promotes Akt-driven tumorigenesis via FOXO inactivation. Cell Death Differ. 2014, 22, 131–144. [Google Scholar] [CrossRef] [Green Version]
- Swanton, C.; McGranahan, N.; Starrett, G.J.; Harris, R.S. APOBEC Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discov. 2015, 5, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Aynaud, M.-M.; Suspène, R.; Vidalain, P.-O.; Mussil, B.; Guétard, D.; Tangy, F.; Wain-Hobson, S.; Vartanian, J.-P. Human Tribbles 3 Protects Nuclear DNA from Cytidine Deamination by APOBEC3A. J. Biol. Chem. 2012, 287, 39182–39192. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Hua, F.; Li, K.; Yu, J.-J.; Lv, X.; Yan, J.; Zhang, X.-W.; Sun, W.; Lin, H.; Shang, S.; Wang, F.; et al. TRB3 links insulin/IGF to tumour promotion by interacting with p62 and impeding autophagic/proteasomal degradations. Nat. Commun. 2015, 6, 7951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, F.; Li, K.; Yu, J.-J.; Hu, Z.-W. The TRIB3-SQSTM1 interaction mediates metabolic stress-promoted tumorigenesis and progression via suppressing autophagic and proteasomal degradation. Autophagy 2015, 11, 1929–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izrailit, J.; Jaiswal, A.; Zheng, W.; Moran, M.F.; Reedijk, M. Cellular stress induces TRB3/USP9x-dependent Notch activation in cancer. Oncogene 2017, 36, 1048–1057. [Google Scholar] [CrossRef]
- Hua, F.; Mu, R.; Liu, J.; Xue, J.; Wang, Z.; Lin, H.; Yang, H.; Chen, X.; Hu, Z. TRB3 interacts with SMAD3 promoting tumor cell migration and invasion. J. Cell Sci. 2011, 124, 3235–3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Transcript ID | bp | Protein | UniProt | Flags |
|---|---|---|---|---|---|
| TRIB3-201 | ENST00000217233.8 | 2095 | 358aa | Q96RU7 | TSL:1, APPRIS P3 |
| TRIB3-202 | ENST00000422053.3 | 1533 | 385aa | J3KR25 | TSL:2, APPRIS ALT2 |
| TRIB3-203 | ENST00000449710.5 | 1070 | 271aa | B0QYQ2 | CDS 3′ incomplete, TSL:5 |
| TRIB3-204 | ENST00000615226.4 | 877 | 130aa | A0A087WTX3 | CDS 3′ incomplete, TSL:3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stefanovska, B.; André, F.; Fromigué, O. Tribbles Pseudokinase 3 Regulation and Contribution to Cancer. Cancers 2021, 13, 1822. https://doi.org/10.3390/cancers13081822
Stefanovska B, André F, Fromigué O. Tribbles Pseudokinase 3 Regulation and Contribution to Cancer. Cancers. 2021; 13(8):1822. https://doi.org/10.3390/cancers13081822
Chicago/Turabian StyleStefanovska, Bojana, Fabrice André, and Olivia Fromigué. 2021. "Tribbles Pseudokinase 3 Regulation and Contribution to Cancer" Cancers 13, no. 8: 1822. https://doi.org/10.3390/cancers13081822
APA StyleStefanovska, B., André, F., & Fromigué, O. (2021). Tribbles Pseudokinase 3 Regulation and Contribution to Cancer. Cancers, 13(8), 1822. https://doi.org/10.3390/cancers13081822