
Sensitivity and Resistance of Oncogenic RAS-Driven Tumors to Dual MEK and ERK Inhibition

 , , , , and
, , , , and 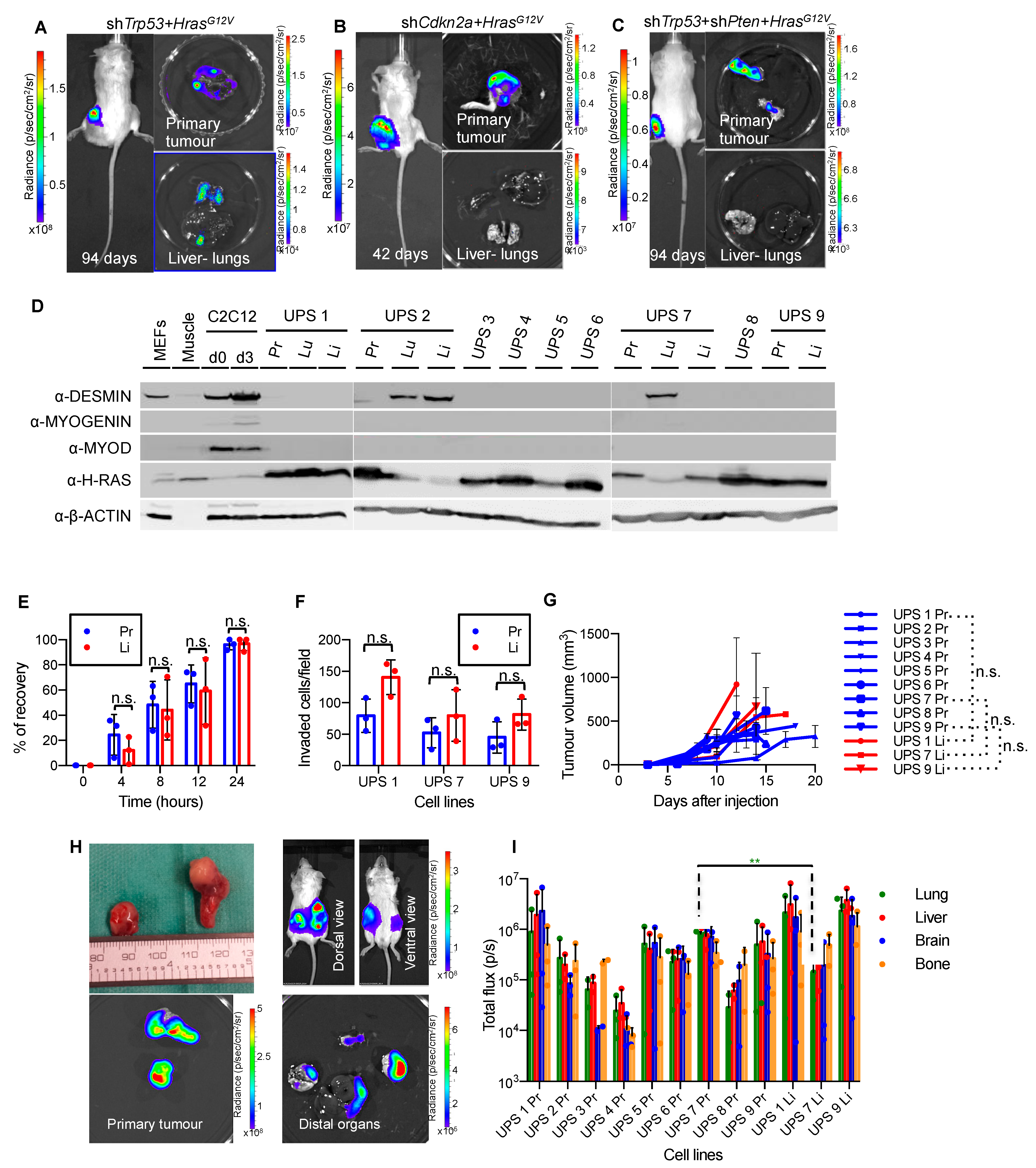{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of MuLE Vectors
2.2. Mouse Strains
2.3. In Vivo Tumour Formation and Metastases Study
2.4. Allograft Studies
2.5. In Vivo Imaging Studies
2.6. Preclinical Therapeutic Study
2.7. Cells
2.8. Scratch Assay
2.9. Transwell Migration Assay
2.10. Proliferation and Viability Assay of Inhibitor-Treated Cells
2.11. Short-Term Drug Screening
2.12. Drug Interaction Assay
2.13. Drug Resistance Assay
2.14. Colony Formation Assay
2.15. EGF Stimulation of KPC Cells
2.16. Western Blotting
2.17. PCR Analysis of Genetic Kras Recombination
2.18. Flow Cytometry
2.19. PamChip® Cell-Based Kinase Assay
2.20. Immunohistochemistry
2.21. Statistics and Data Evaluation
2.22. Study Approval
3. Results
3.1. Development of an HrasG12V-Driven Mouse Model of Metastatic Undifferentiated Pleomorphic Sarcoma
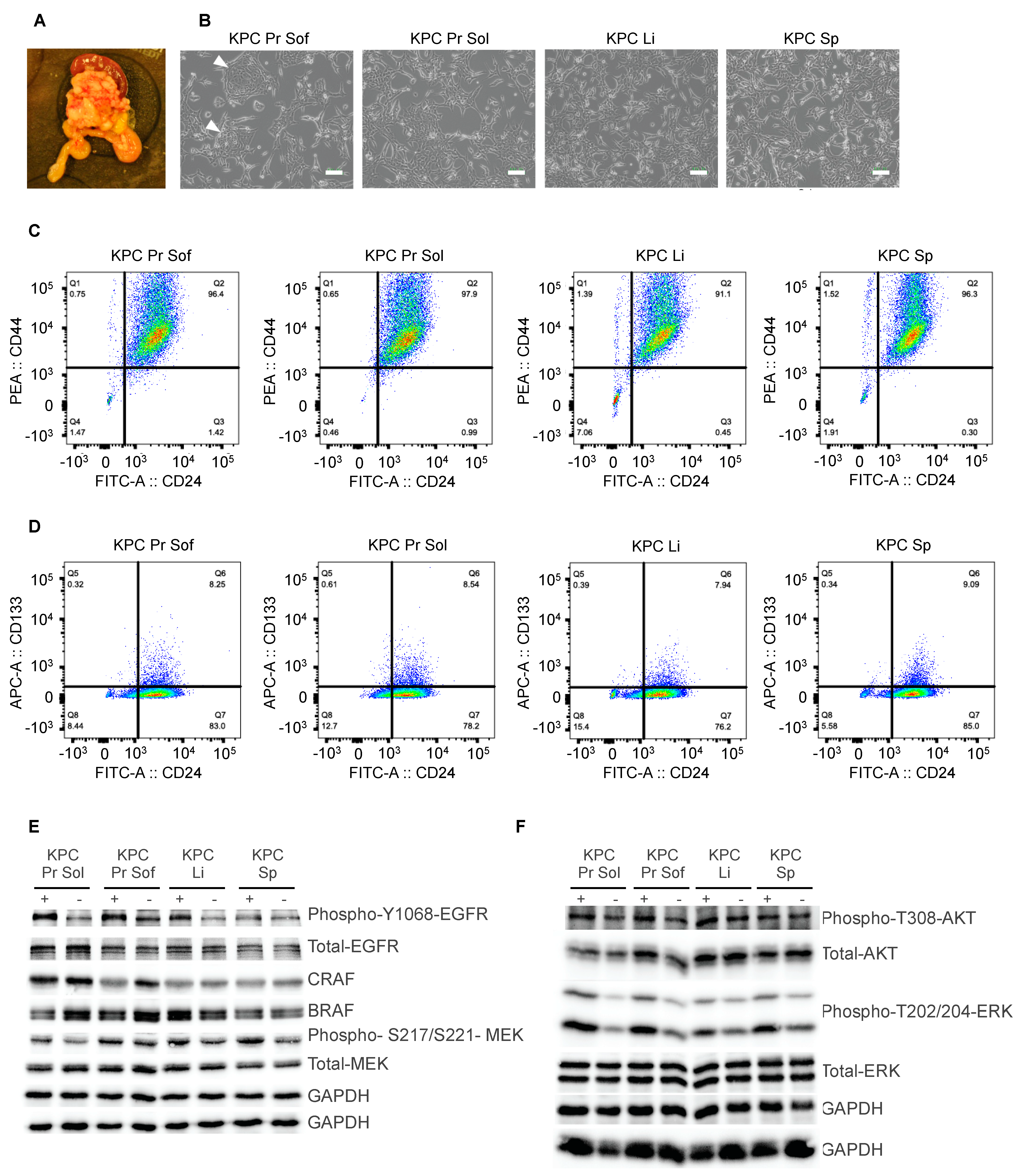
3.2. Development of a KrasG12D-Driven Mouse Model of Metastatic Pancreatic Ductal Adenocarcinoma
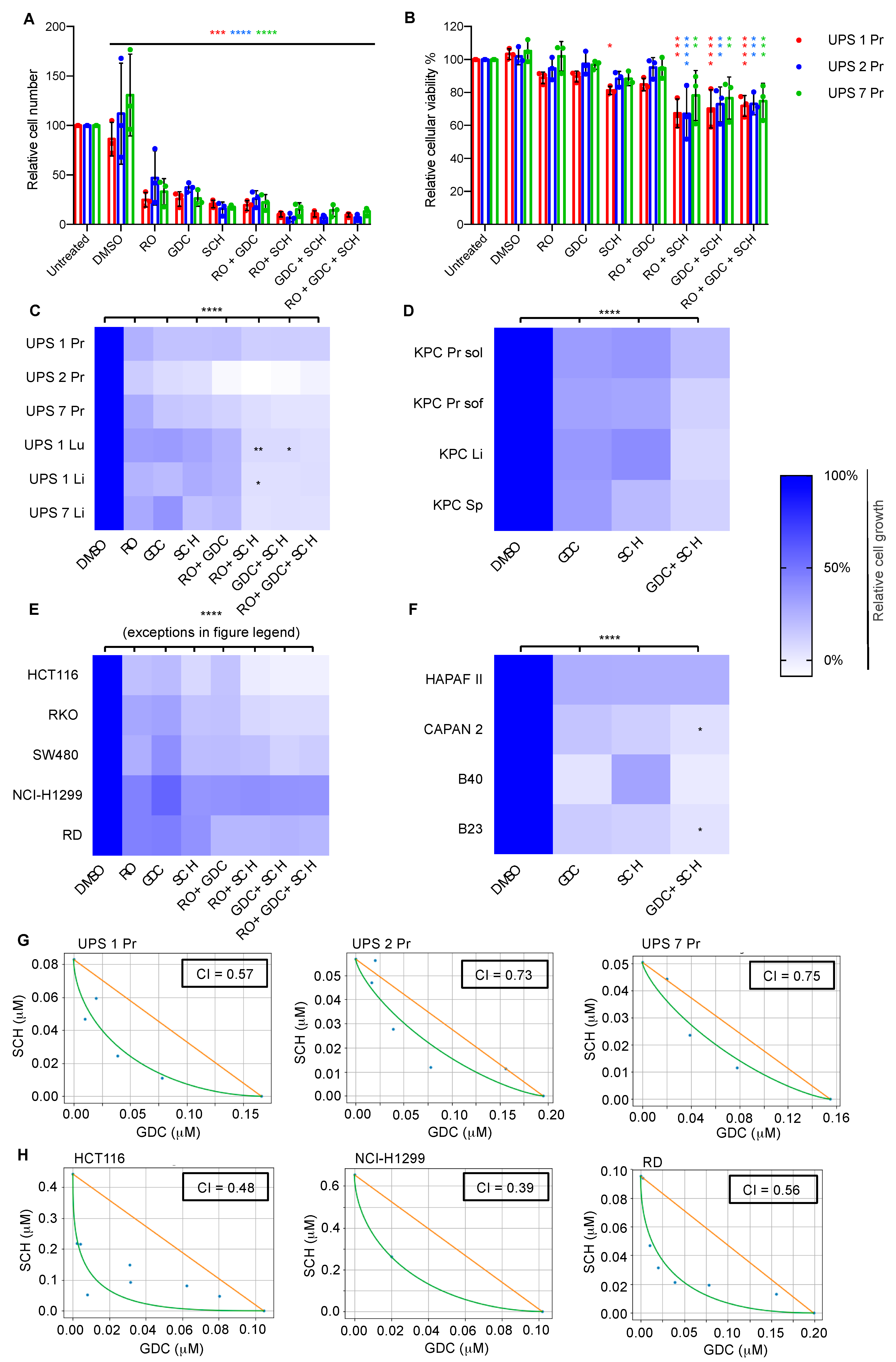
3.3. Diverse Oncogenic RAS-Driven Mouse and Human Cancer Cell Lines Are Sensitive to Dual MEKi and ERKi Treatment
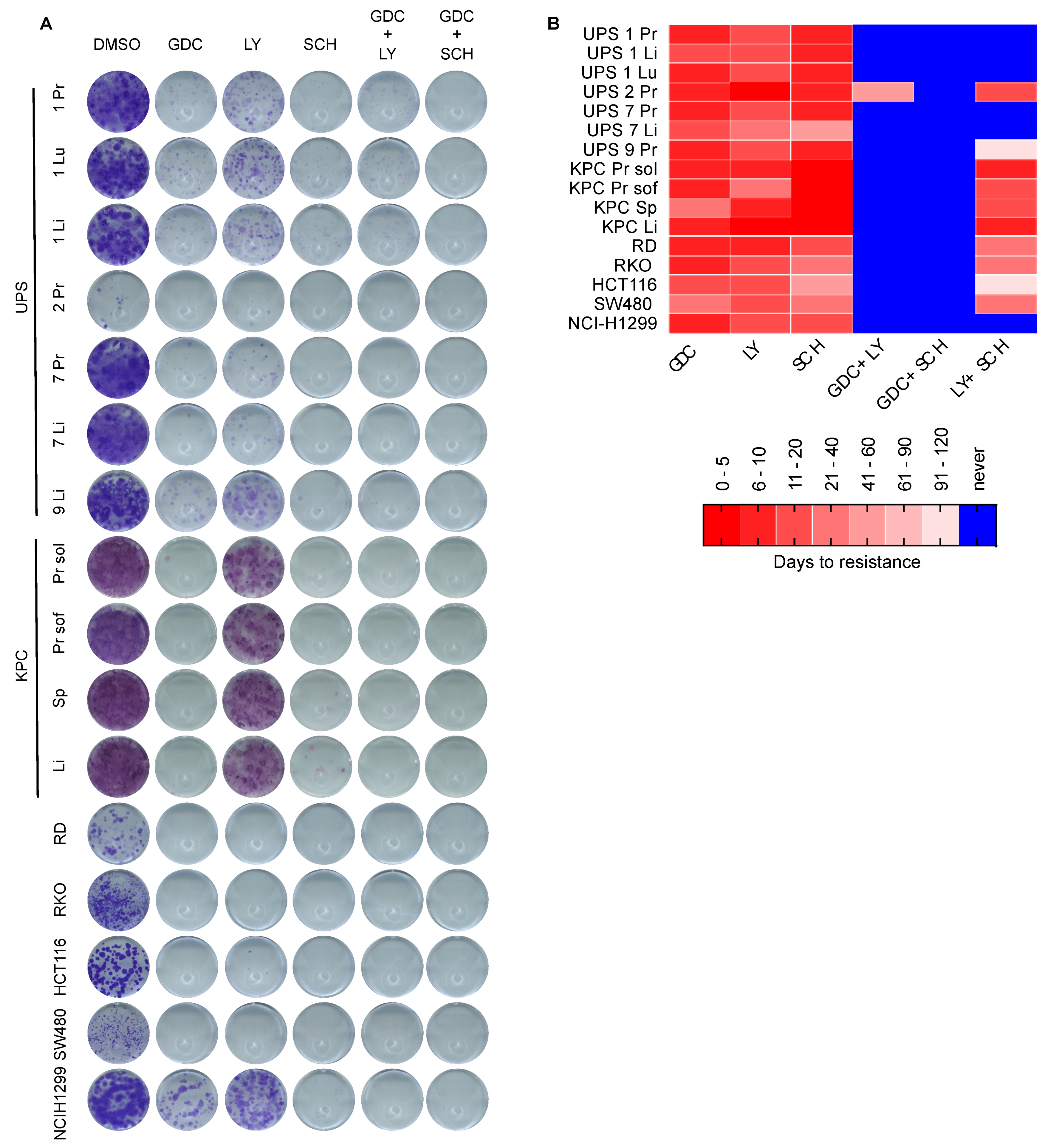
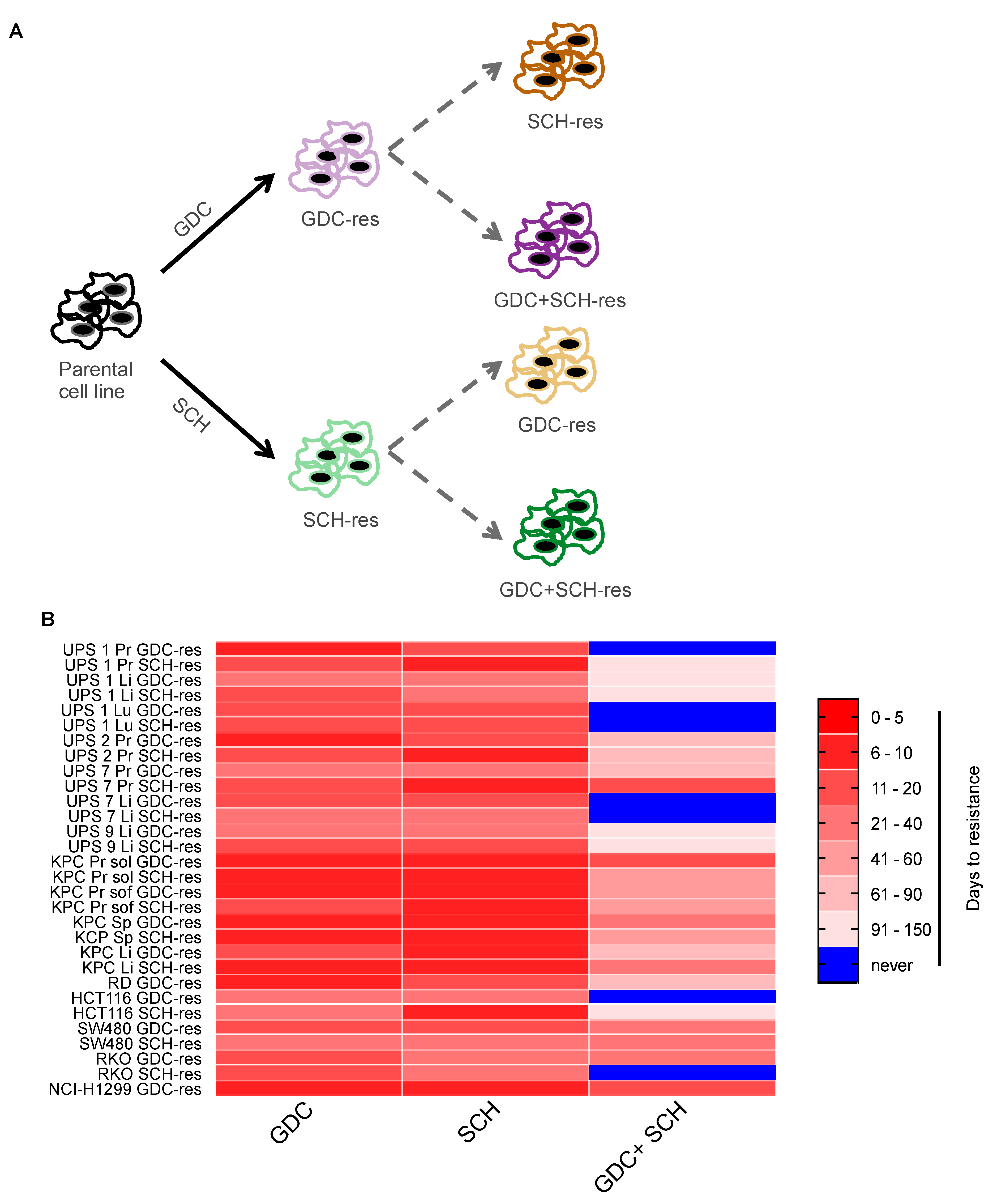
3.4. Dual MEKi Plus ERKi Prevents the Emergence of Drug Resistance
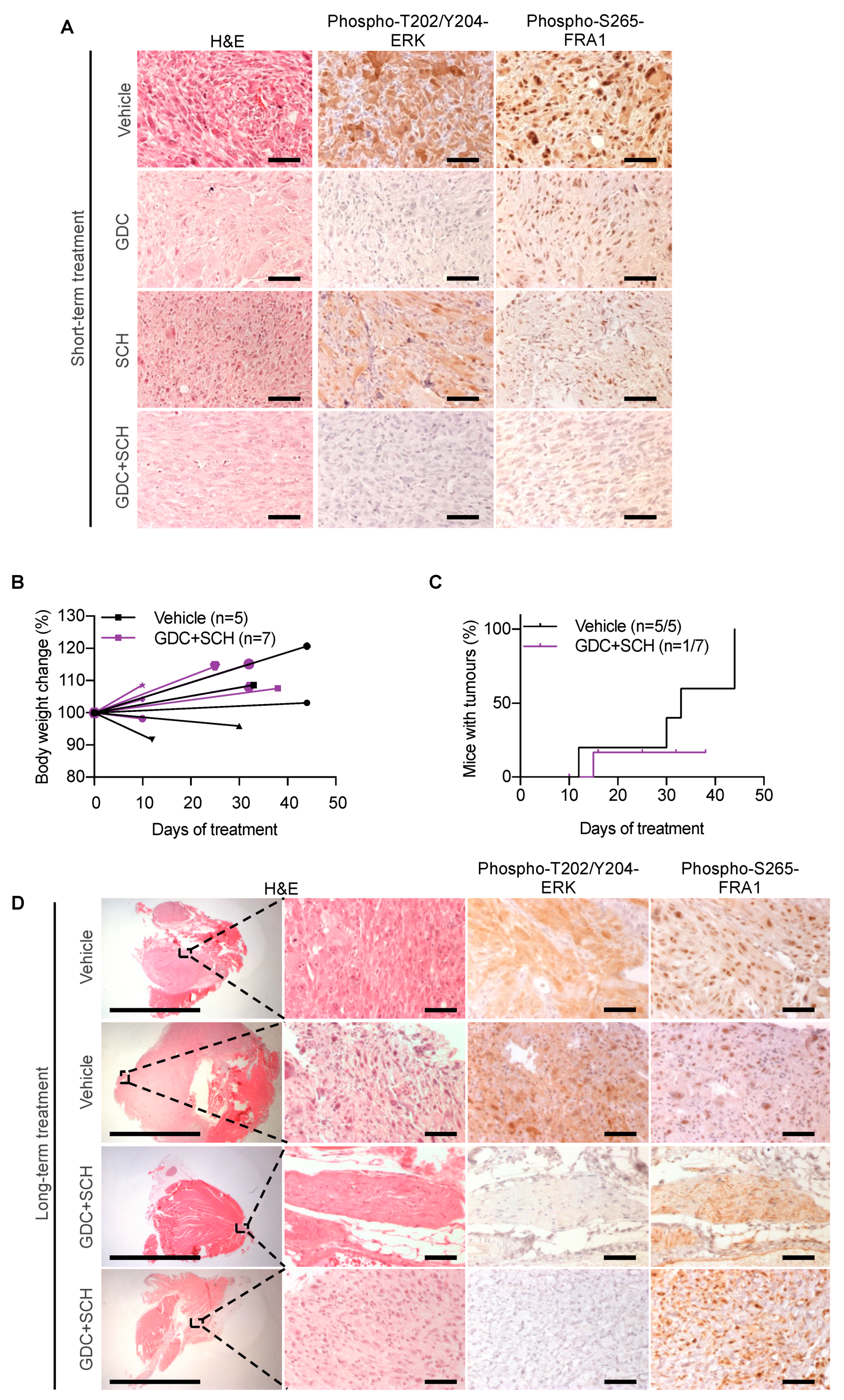
3.5. Testing Dual MEKi Plus ERKi Therapy In Vivo
3.6. Acquired Resistance to Single MEKi or ERKi Overcomes Susceptibility to Dual Inhibition
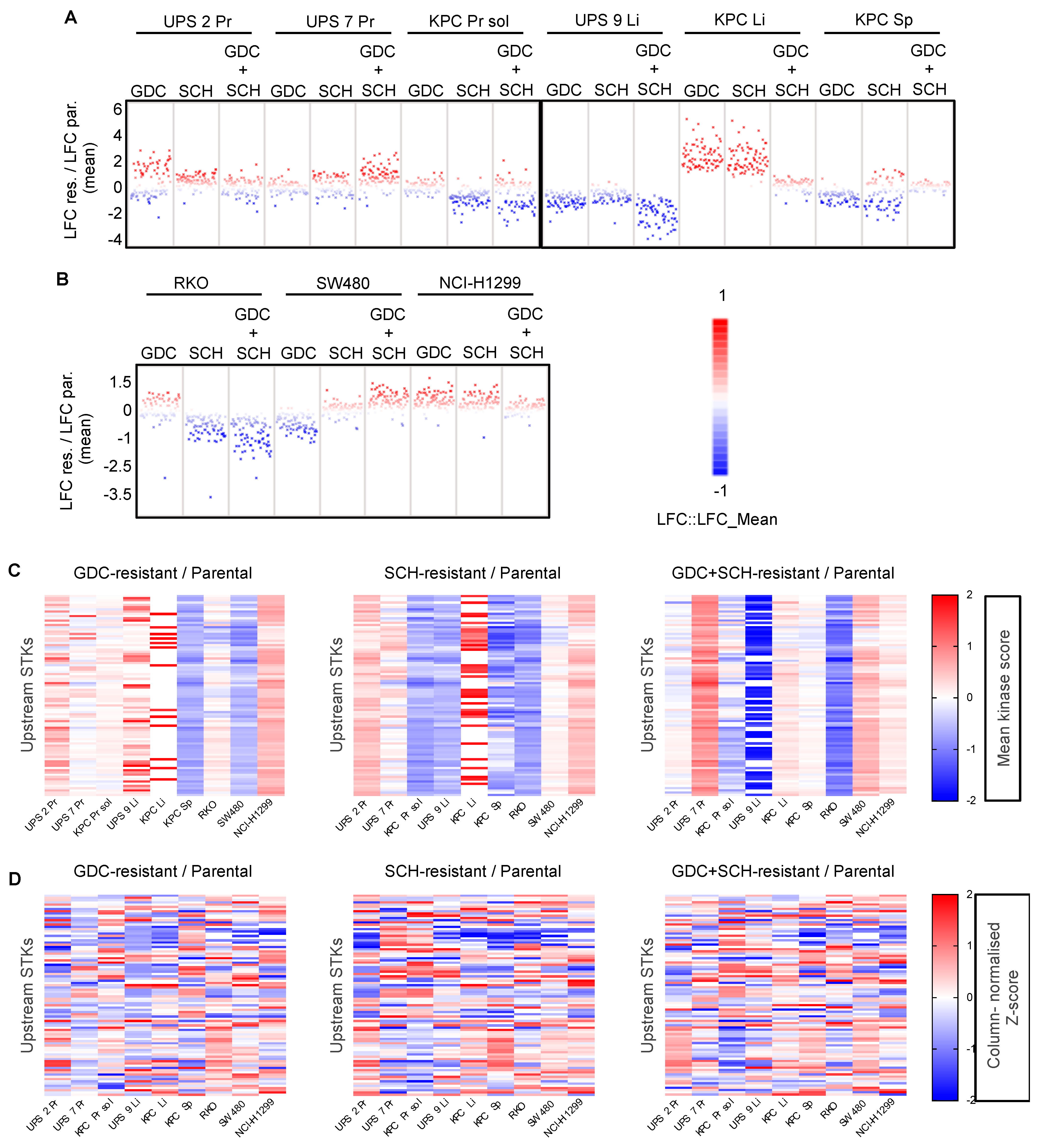
3.7. Kinome Profiling Shows Large Variation and Complex Signalling Rewiring in Resistant Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephen, A.G.; Esposito, D.; Bagni, R.K.; McCormick, F. Dragging ras back in the ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheffzek, K.; Ahmadian, M.R.; Kabsch, W.; Wiesmüller, L.; Lautwein, A.; Schmitz, F.; Wittinghofer, A. The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 1997, 277, 333–338. [Google Scholar] [CrossRef] [Green Version]
- John, J.; Sohmen, R.; Feuerstein, J.; Linke, R.; Wittinghofer, A.; Goody, R.S. Kinetics of interaction of nucleotides with nucleotide-free H-ras p21. Biochemistry 1990, 29, 6058–6065. [Google Scholar] [CrossRef]
- Nagasaka, M.; Li, Y.; Sukari, A.; Ou, S.-H.I.; Al-Hallak, M.N.; Azmi, A.S. KRAS G12C Game of Thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treat. Rev. 2020, 84, 101974. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Ryan, M.B.; Corcoran, R.B. Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol. 2018, 15, 709–720. [Google Scholar] [CrossRef]
- Ryan, M.B.; Der, C.J.; Wang-Gillam, A.; Cox, A.D. Targeting RAS-mutant cancers: Is ERK the key? Trends Cancer 2015, 1, 183–198. [Google Scholar] [CrossRef] [Green Version]
- Berndt, N.; Hamilton, A.D.; Sebti, S.M. Targeting protein prenylation for cancer therapy. Nat. Publ. Group 2011, 11, 775–791. [Google Scholar] [CrossRef] [Green Version]
- Basso, A.D.; Kirschmeier, P.; Bishop, W.R. Lipid posttranslational modifications. Farnesyl transferase inhibitors. J. Lipid Res. 2006, 47, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.K. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef] [Green Version]
- Rowell, C.A.; Kowalczyk, J.J.; Lewis, M.D.; Garcia, A.M. Direct demonstration of geranylgeranylation and farnesylation of Ki-Ras in vivo. J. Biol. Chem. 1997, 272, 14093–14097. [Google Scholar] [CrossRef] [Green Version]
- Downward, J. RAS Synthetic Lethal Screens Revisited: Still Seeking the Elusive Prize? Clin. Cancer Res. 2015, 21, 1802–1809. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [Green Version]
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF-MEK Pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Röring, M.; Brummer, T. Aberrant B-Raf signaling in human cancer—10 years from bench to bedside. Crit. Rev. Oncog. 2012, 17, 97–121. [Google Scholar] [CrossRef]
- Caunt, C.J.; Sale, M.J.; Smith, P.D.; Cook, S.J. MEK1 and MEK2 inhibitors and cancer therapy: The long and winding road. Nat. Rev. Cancer 2015, 15, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Moschos, S.J.; Sullivan, R.J.; Hwu, W.-J.; Ramanathan, R.K.; Adjei, A.A.; Fong, P.C.; Shapira-Frommer, R.; Tawbi, H.A.; Rubino, J.; Rush, T.S.; et al. Development of MK-8353, an orally administered ERK1/2 inhibitor, in patients with advanced solid tumors. JCI Insight 2018, 3, e92352. [Google Scholar] [CrossRef]
- Morris, E.J.; Jha, S.; Restaino, C.R.; Dayananth, P.; Zhu, H.; Cooper, A.; Carr, D.; Deng, Y.; Jin, W.; Black, S.; et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013, 3, 742–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazar-Rethinam, M.; Kleyman, M.; Han, G.C.; Liu, D.; Ahronian, L.G.; Shahzade, H.A.; Chen, L.; Parikh, A.R.; Allen, J.N.; Clark, J.W.; et al. Convergent Therapeutic Strategies to Overcome the Heterogeneity of Acquired Resistance in BRAFV600E Colorectal Cancer. Cancer Discov. 2018, 8, 417–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oddo, D.; Sennott, E.M.; Barault, L.; Valtorta, E.; Arena, S.; Cassingena, A.; Filiciotto, G.; Marzolla, G.; Elez, E.; van Geel, R.M.J.M.; et al. Molecular Landscape of Acquired Resistance to Targeted Therapy Combinations in BRAF-Mutant Colorectal Cancer. Cancer Res. 2016, 76, 4504–4515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahronian, L.G.; Sennott, E.M.; Van Allen, E.M.; Wagle, N.; Kwak, E.L.; Faris, J.E.; Godfrey, J.T.; Nishimura, K.; Lynch, K.D.; Mermel, C.H.; et al. Clinical Acquired Resistance to RAF Inhibitor Combinations in BRAF-Mutant Colorectal Cancer through MAPK Pathway Alterations. Cancer Discov. 2015, 5, 358–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagle, N.; Van Allen, E.M.; Treacy, D.J.; Frederick, D.T.; Cooper, Z.A.; Taylor-Weiner, A.; Rosenberg, M.; Goetz, E.M.; Sullivan, R.J.; Farlow, D.N.; et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014, 4, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, I.R.; Li, L.; Cabeceiras, P.K.; Mahdavi, M.; Gutschner, T.; Genovese, G.; Wang, G.; Fang, Z.; Tepper, J.M.; Stemke-Hale, K.; et al. The RAC1 P29S hotspot mutation in melanoma confers resistance to pharmacological inhibition of RAF. Cancer Res. 2014, 74, 4845–4852. [Google Scholar] [CrossRef] [Green Version]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. Dermatologic Cooperative Oncology Group of Germany (DeCOG) The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haarberg, H.E.; Smalley, K.S.M. Resistance to Raf inhibition in cancer. Drug Discov. Today Technol. 2014, 11, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Jha, S.; Morris, E.J.; Hruza, A.; Mansueto, M.S.; Schroeder, G.K.; Arbanas, J.; McMasters, D.; Restaino, C.R.; Dayananth, P.; Black, S.; et al. Dissecting Therapeutic Resistance to ERK Inhibition. Mol. Cancer Ther. 2016, 15, 548–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, B.S.; Durinck, S.; Stawiski, E.W.; Yin, J.; Wang, W.; Lin, E.; Moffat, J.; Martin, S.E.; Modrusan, Z.; Seshagiri, S. ERK Mutations and Amplification Confer Resistance to ERK-Inhibitor Therapy. Clin. Cancer Res. 2018, 24, 4044–4055. [Google Scholar] [CrossRef] [Green Version]
- Hatzivassiliou, G.; Liu, B.; O’Brien, C.; Spoerke, J.M.; Hoeflich, K.P.; Haverty, P.M.; Soriano, R.; Forrest, W.F.; Heldens, S.; Chen, H.; et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol. Cancer Ther. 2012, 11, 1143–1154. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Martelotto, L.; Baslan, T.; Vides, A.; Solomon, M.; Mai, T.T.; Chaudhary, N.; Riely, G.J.; Li, B.T.; Scott, K.; et al. An approach to suppress the evolution of resistance in BRAFV600E-mutant cancer. Nat. Med. 2017, 23, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Merchant, M.; Moffat, J.; Schaefer, G.; Chan, J.; Wang, X.; Orr, C.; Cheng, J.; Hunsaker, T.; Shao, L.; Wang, S.J.; et al. Combined MEK and ERK inhibition overcomes therapy-mediated pathway reactivation in RAS mutant tumors. PLoS ONE 2017, 12, e0185862. [Google Scholar] [CrossRef] [Green Version]
- Albers, J.; Danzer, C.; Rechsteiner, M.; Lehmann, H.; Brandt, L.P.; Hejhal, T.; Catalano, A.; Busenhart, P.; Gonçalves, A.F.; Brandt, S.; et al. A versatile modular vector system for rapid combinatorial mammalian genetics. J. Clin. Investig. 2015, 125, 1603–1619. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, L.A.; Tiriac, H.; Tuveson, D.A. Generation and Culture of Human Pancreatic Ductal Adenocarcinoma Organoids from Resected Tumor Specimens. Methods Mol. Biol. 2019, 1882, 97–115. [Google Scholar] [PubMed]
- Hussung, S.; Follo, M.; Klar, R.F.U.; Michalczyk, S.; Fritsch, K.; Nollmann, F.; Hipp, J.; Duyster, J.; Scherer, F.; von Bubnoff, N.; et al. Development and Clinical Validation of Discriminatory Multitarget Digital Droplet PCR Assays for the Detection of Hot Spot KRAS and NRAS Mutations in Cell-Free DNA. J. Mol. Diagn. 2020, 22, 943–956. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Tallarida, R.J. Quantitative methods for assessing drug synergism. Genes Cancer 2011, 2, 1003–1008. [Google Scholar] [CrossRef] [Green Version]
- Frew, I.J.; Thoma, C.R.; Georgiev, S.; Minola, A.; Hitz, M.; Montani, M.; Moch, H.; Krek, W. pVHL and PTEN tumour suppressor proteins cooperatively suppress kidney cyst formation. EMBO J. 2008, 27, 1747–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, E.L.; Willis, N.; Mercer, K.; Bronson, R.T.; Crowley, D.; Montoya, R.; Jacks, T.; Tuveson, D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001, 15, 3243–3248. [Google Scholar] [CrossRef] [Green Version]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lex, A.; Gehlenborg, N.; Strobelt, H.; Vuillemot, R.; Pfister, H. UpSet: Visualization of Intersecting Sets. IEEE Trans. Vis. Comput. Graph. 2014, 20, 1983–1992. [Google Scholar] [CrossRef]
- Thoma, C.R.; Frew, I.J.; Hoerner, C.R.; Montani, M.; Moch, H.; Krek, W. pVHL and GSK3beta are components of a primary cilium-maintenance signalling network. Nat. Cell Biol. 2007, 9, 588–595. [Google Scholar] [CrossRef]
- Fletcher, C.D.M.; Unni, K.K.; Mertens, F. Pathology and Genetics of Tumours of Soft Tissue and Bone; IARC: Lyon, France, 2002. [Google Scholar]
- Pappo, A.S.; Anderson, J.R.; Crist, W.M.; Wharam, M.D.; Breitfeld, P.P.; Hawkins, D.; Raney, R.B.; Womer, R.B.; Parham, D.M.; Qualman, S.J.; et al. Survival after relapse in children and adolescents with rhabdomyosarcoma: A report from the Intergroup Rhabdomyosarcoma Study Group. J. Clin. Oncol. 1999, 17, 3487–3493. [Google Scholar] [CrossRef]
- Le Doussal, V.; Coindre, J.M.; Leroux, A.; Hacene, K.; Terrier, P.; Bui, N.B.; Bonichon, F.; Collin, F.; Mandard, A.M.; Contesso, G. Prognostic factors for patients with localized primary malignant fibrous histiocytoma: A multicenter study of 216 patients with multivariate analysis. Cancer 1996, 77, 1823–1830. [Google Scholar] [CrossRef]
- Gibbs, J.F.; Huang, P.P.; Lee, R.J.; McGrath, B.; Brooks, J.; McKinley, B.; Driscoll, D.; Kraybill, W.G. Malignant fibrous histiocytoma: An institutional review. Cancer Investig. 2001, 19, 23–27. [Google Scholar] [CrossRef]
- Engellau, J. Prognostic factors in soft tissue sarcoma. Tissue microarray for immunostaining, the importance of whole-tumor sections and time-dependence. Acta Orthop. Scand. Suppl. 2004, 75, 2. [Google Scholar] [CrossRef] [Green Version]
- Kaseb, H.; Kuhn, J.; Babiker, H.M. Rhabdomyosarcoma. In Statpearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Lehnhardt, M.; Daigeler, A.; Homann, H.H.; Schwaiberger, V.; Goertz, O.; Kuhnen, C.; Steinau, H.U. MFH revisited: Outcome after surgical treatment of undifferentiated pleomorphic or not otherwise specified (NOS) sarcomas of the extremities—An analysis of 140 patients. Langenbecks Arch. Surg. 2009, 394, 313–320. [Google Scholar] [CrossRef]
- Mueller, S.; Engleitner, T.; Maresch, R.; Zukowska, M.; Lange, S.; Kaltenbacher, T.; Konukiewitz, B.; Öllinger, R.; Zwiebel, M.; Strong, A.; et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 2018, 554, 62–68. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Hatzivassiliou, G.; Haling, J.R.; Chen, H.; Song, K.; Price, S.; Heald, R.; Hewitt, J.F.M.; Zak, M.; Peck, A.; Orr, C.; et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature 2013, 501, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Xie, H.; Dou, Y.; Yuan, J.; Zeng, D.; Xiao, S. Expression and function of FRA1 protein in tumors. Mol. Biol. Rep. 2020, 47, 737–752. [Google Scholar] [CrossRef] [PubMed]
- Muzumdar, M.D.; Chen, P.-Y.; Dorans, K.J.; Chung, K.M.; Bhutkar, A.; Hong, E.; Noll, E.M.; Sprick, M.R.; Trumpp, A.; Jacks, T. Survival of pancreatic cancer cells lacking KRAS function. Nat. Commun. 2017, 8, 1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.-Y.; Muzumdar, M.D.; Dorans, K.J.; Robbins, R.; Bhutkar, A.; Del Rosario, A.; Mertins, P.; Qiao, J.; Schafer, A.C.; Gertler, F.; et al. Adaptive and Reversible Resistance to Kras Inhibition in Pancreatic Cancer Cells. Cancer Res. 2018, 78, 985–1002. [Google Scholar] [CrossRef] [Green Version]
- Fernandes Neto, J.M.; Nadal, E.; Bosdriesz, E.; Ooft, S.N.; Farre, L.; McLean, C.; Klarenbeek, S.; Jurgens, A.; Hagen, H.; Wang, L.; et al. Multiple low dose therapy as an effective strategy to treat EGFR inhibitor-resistant NSCLC tumours. Nat. Commun. 2020, 11, 3157–3159. [Google Scholar] [CrossRef]
- Algazi, A.P.; Othus, M.; Daud, A.I.; Lo, R.S.; Mehnert, J.M.; Truong, T.-G.; Conry, R.; Kendra, K.; Doolittle, G.C.; Clark, J.I.; et al. Continuous versus intermittent BRAF and MEK inhibition in patients with BRAF-mutated melanoma: A randomized phase 2 trial. Nat. Med. 2020, 26, 1564–1568. [Google Scholar] [CrossRef]
- Brummer, T.; McInnes, C. RAF kinase dimerization: Implications for drug discovery and clinical outcomes. Oncogene 2020, 39, 4155–4169. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Mori, M.; Pan, B.; Yau, C.; Wolf, D.M.; Ruiz-Saenz, A.; Brunen, D.; Prahallad, A.; Cornelissen-Steijger, P.; Kemper, K.; et al. Mapping phospho-catalytic dependencies of therapy-resistant tumours reveals actionable vulnerabilities. Nat. Cell Biol. 2019, 21, 778–790. [Google Scholar] [CrossRef]
- Gao, X.; Chen, G.; Gao, C.; Zhang, D.H.; Kuan, S.-F.; Stabile, L.P.; Liu, G.; Hu, J. MAP4K4 is a novel MAPK/ERK pathway regulator required for lung adenocarcinoma maintenance. Mol. Oncol. 2017, 11, 628–639. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catalano, A.; Adlesic, M.; Kaltenbacher, T.; Klar, R.F.U.; Albers, J.; Seidel, P.; Brandt, L.P.; Hejhal, T.; Busenhart, P.; Röhner, N.; et al. Sensitivity and Resistance of Oncogenic RAS-Driven Tumors to Dual MEK and ERK Inhibition. Cancers 2021, 13, 1852. https://doi.org/10.3390/cancers13081852
Catalano A, Adlesic M, Kaltenbacher T, Klar RFU, Albers J, Seidel P, Brandt LP, Hejhal T, Busenhart P, Röhner N, et al. Sensitivity and Resistance of Oncogenic RAS-Driven Tumors to Dual MEK and ERK Inhibition. Cancers. 2021; 13(8):1852. https://doi.org/10.3390/cancers13081852
Chicago/Turabian StyleCatalano, Antonella, Mojca Adlesic, Thorsten Kaltenbacher, Rhena F. U. Klar, Joachim Albers, Philipp Seidel, Laura P. Brandt, Tomas Hejhal, Philipp Busenhart, Niklas Röhner, and et al. 2021. "Sensitivity and Resistance of Oncogenic RAS-Driven Tumors to Dual MEK and ERK Inhibition" Cancers 13, no. 8: 1852. https://doi.org/10.3390/cancers13081852
APA StyleCatalano, A., Adlesic, M., Kaltenbacher, T., Klar, R. F. U., Albers, J., Seidel, P., Brandt, L. P., Hejhal, T., Busenhart, P., Röhner, N., Zodel, K., Fritsch, K., Wild, P. J., Duyster, J., Fritsch, R., Brummer, T., & Frew, I. J. (2021). Sensitivity and Resistance of Oncogenic RAS-Driven Tumors to Dual MEK and ERK Inhibition. Cancers, 13(8), 1852. https://doi.org/10.3390/cancers13081852