
Eye Tumors in Childhood as First Sign of Tumor Predisposition Syndromes: Insights from an Observational Study Conducted in Germany and Austria


 , , , , , , , , , add
Show full author list
, , , , , , , , , add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. Genetic Counselling and Molecular Genetic Analysis
2.3. Statistical Analysis
3. Results
3.1. Retinoblastoma Was the Most Common Malignant Eye Tumor in Childhood
3.2. The Age at Diagnosis Differed among the Different Eye Tumor Entities
3.3. Eye Tumors in Childhood Are often the First Sign of a Tumor Predisposition Syndrome
3.4. The Overall Survival of Children with Eye Tumors Is High
3.5. Trilateral Retinoblastoma Is a Rare Manifestation of Heritable Retinoblastoma
3.6. Clinical Presentation of Heritable and Non-Heritable Retinoblastoma Is Distinct
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kratz, C.P.; Jongmans, M.C.; Cave, H.; Wimmer, K.; Behjati, S.; Guerrini-Rousseau, L.; Milde, T.; Pajtler, K.W.; Golmard, L.; Gauthier-Villars, M.; et al. Predisposition to cancer in children and adolescents. Lancet Child. Adolesc. Health 2021, 5, 142–154. [Google Scholar] [CrossRef]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, N. Realizing the promise of cancer predisposition genes. Nature 2014, 505, 302–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saletta, F.; Dalla Pozza, L.; Byrne, J.A. Genetic causes of cancer predisposition in children and adolescents. Transl. Pediatr. 2015, 4, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.M.K.; Behnert, A.; Pietsch, T.; Vokuhl, C.; Kratz, C.P. Proportion of children with cancer that have an indication for genetic counseling and testing based on the cancer type irrespective of other features. Fam. Cancer 2021. [Google Scholar] [CrossRef] [PubMed]
- Postema, F.A.M.; Hopman, S.M.J.; Aalfs, C.M.; Berger, L.P.V.; Bleeker, F.E.; Dommering, C.J.; Jongmans, M.C.J.; Letteboer, T.G.W.; Olderode-Berends, M.J.W.; Wagner, A.; et al. Childhood tumours with a high probability of being part of a tumour predisposition syndrome; reason for referral for genetic consultation. Eur. J. Cancer 2017, 80, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Broaddus, E.; Topham, A.; Singh, A.D. Incidence of retinoblastoma in the USA: 1975–2004. Br. J. Ophthalmol. 2009, 93, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Seregard, S.; Lundell, G.; Svedberg, H.; Kivelä, T. Incidence of retinoblastoma from 1958 to 1998 in Northern Europe: Advantages of birth cohort analysis. Ophthalmology 2004, 111, 1228–1232. [Google Scholar] [CrossRef]
- Kivela, T. The epidemiological challenge of the most frequent eye cancer: Retinoblastoma, an issue of birth and death. Br. J. Ophthalmol. 2009, 93, 1129–1131. [Google Scholar] [CrossRef]
- Kleinerman, R.A.; Tucker, M.A.; Tarone, R.E.; Abramson, D.H.; Seddon, J.M.; Stovall, M.; Li, F.P.; Fraumeni, J.F., Jr. Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: An extended follow-up. J. Clin. Oncol. 2005, 23, 2272–2279. [Google Scholar] [CrossRef]
- Gregersen, P.A.; Olsen, M.H.; Urbak, S.F.; Funding, M.; Dalton, S.O.; Overgaard, J.; Alsner, J. Incidence and Mortality of Second Primary Cancers in Danish Patients With Retinoblastoma, 1943-2013. JAMA Netw. Open 2020, 3, e2022126. [Google Scholar] [CrossRef]
- De Jong, M.C.; Kors, W.A.; Moll, A.C.; de Graaf, P.; Castelijns, J.A.; Jansen, R.W.; Gallie, B.; Soliman, S.E.; Shaikh, F.; Dimaras, H.; et al. Screening for Pineal Trilateral Retinoblastoma Revisited: A Meta-analysis. Ophthalmology 2020, 127, 601–607. [Google Scholar] [CrossRef]
- De Jong, M.C.; Kors, W.A.; de Graaf, P.; Castelijns, J.A.; Kivelä, T.; Moll, A.C. Trilateral retinoblastoma: A systematic review and meta-analysis. Lancet Oncol. 2014, 15, 1157–1167. [Google Scholar] [CrossRef]
- Kaliki, S.; Shields, C.L.; Eagle, R.C., Jr.; Vemuganti, G.K.; Almeida, A.; Manjandavida, F.P.; Mulay, K.; Honavar, S.G.; Shields, J.A. Ciliary body medulloepithelioma: Analysis of 41 cases. Ophthalmology 2013, 120, 2552–2559. [Google Scholar] [CrossRef]
- Schultz, K.A.P.; Williams, G.M.; Kamihara, J.; Stewart, D.R.; Harris, A.K.; Bauer, A.J.; Turner, J.; Shah, R.; Schneider, K.; Schneider, K.W.; et al. DICER1 and Associated Conditions: Identification of at-risk Individuals and Recommended Surveillance Strategies. Clin. Cancer Res. 2018, 24, 2251–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, C.L.; Kaliki, S.; Arepalli, S.; Atalay, H.T.; Manjandavida, F.P.; Pieretti, G.; Shields, J.A. Uveal melanoma in children and teenagers. Saudi J. Ophthalmol. 2013, 27, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.E.; Karnell, L.H.; Menck, H.R. The National Cancer Data Base report on cutaneous and noncutaneous melanoma: A summary of 84,836 cases from the past decade. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer 1998, 83, 1664–1678. [Google Scholar] [CrossRef] [Green Version]
- Al-Jamal, R.T.; Cassoux, N.; Desjardins, L.; Damato, B.; Konstantinidis, L.; Coupland, S.E.; Heimann, H.; Petrovic, A.; Zografos, L.; Schalenbourg, A.; et al. The Pediatric Choroidal and Ciliary Body Melanoma Study: A Survey by the European Ophthalmic Oncology Group. Ophthalmology 2016, 123, 898–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walpole, S.; Pritchard, A.L.; Cebulla, C.M.; Pilarski, R.; Stautberg, M.; Davidorf, F.H.; de la Fouchardiere, A.; Cabaret, O.; Golmard, L.; Stoppa-Lyonnet, D.; et al. Comprehensive Study of the Clinical Phenotype of Germline BAP1 Variant-Carrying Families Worldwide. J. Natl. Cancer Inst. 2018, 110, 1328–1341. [Google Scholar] [CrossRef]
- Abdel-Rahman, M.H.; Pilarski, R.; Cebulla, C.M.; Massengill, J.B.; Christopher, B.N.; Boru, G.; Hovland, P.; Davidorf, F.H. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J. Med. Genet. 2011, 48, 856–859. [Google Scholar] [CrossRef]
- Navahi, R.A.; Babaheidarian, P.; Sedaghat, A.; Naseripour, M.; Aghai, G.; Mardani, A.; Ahangarani, M. Retinal anaplastic pleomorphic xanthoastrocytoma unassociated with phakomatosis. J. Curr. Ophthalmol. 2019, 31, 234–237. [Google Scholar] [CrossRef]
- Ulbright, T.M.; Fulling, K.H.; Helveston, E.M. Astrocytic tumors of the retina. Differentiation of sporadic tumors from phakomatosis-associated tumors. Arch. Pathol. Lab. Med. 1984, 108, 160–163. [Google Scholar]
- Shields, C.L.; Shields, J.A.; Eagle, R.C., Jr.; Cangemi, F. Progressive enlargement of acquired retinal astrocytoma in 2 cases. Ophthalmology 2004, 111, 363–368. [Google Scholar] [CrossRef]
- Rowley, S.A.; O’Callaghan, F.J.; Osborne, J.P. Ophthalmic manifestations of tuberous sclerosis: A population based study. Br. J. Ophthalmol. 2001, 85, 420–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binderup, M.L.M.; Stendell, A.S.; Galanakis, M.; Moller, H.U.; Kiilgaard, J.F.; Bisgaard, M.L. Retinal hemangioblastoma: Prevalence, incidence and frequency of underlying von Hippel-Lindau disease. Br. J. Ophthalmol. 2018, 102, 942–947. [Google Scholar] [CrossRef]
- Moll, A.C.; Kuik, D.J.; Bouter, L.M.; Den Otter, W.; Bezemer, P.D.; Koten, J.W.; Imhof, S.M.; Kuyt, B.P.; Tan, K.E. Incidence and survival of retinoblastoma in The Netherlands: A register based study 1862-1995. Br. J. Ophthalmol. 1997, 81, 559–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broaddus, E.; Topham, A.; Singh, A.D. Survival with retinoblastoma in the USA: 1975–2004. Br. J. Ophthalmol. 2009, 93, 24–27. [Google Scholar] [CrossRef]
- Bornfeld, N.; Schuler, A.; Boloni, R.; Jurklies, C.; Wieland, R.; Sauerwein, W.; Lohmann, D. [Retinoblastoma]. Ophthalmologe 2006, 103, 59–76; quiz 77–78. [Google Scholar] [CrossRef] [PubMed]
- Mourits, D.L.; Hartong, D.T.; Lissenberg-Witte, B.I.; Bosscha, M.I.; Tan, H.S.; Moll, A.C. Cosmetic results of enucleation and/or external beam radiation therapy in 195 retinoblastoma survivors. Acta Ophthalmol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Temming, P.; Viehmann, A.; Arendt, M.; Eisele, L.; Spix, C.; Bornfeld, N.; Sauerwein, W.; Jöckel, K.H.; Lohmann, D.R. Pediatric second primary malignancies after retinoblastoma treatment. Pediatr. Blood Cancer 2015, 62, 1799–1804. [Google Scholar] [CrossRef]
- Temming, P.; Arendt, M.; Viehmann, A.; Eisele, L.; Le Guin, C.H.; Schundeln, M.M.; Biewald, E.; Mausert, J.; Wieland, R.; Bornfeld, N.; et al. How Eye-Preserving Therapy Affects Long-Term Overall Survival in Heritable Retinoblastoma Survivors. J. Clin. Oncol. 2016, 34, 3183–3188. [Google Scholar] [CrossRef]
- Wong, J.R.; Morton, L.M.; Tucker, M.A.; Abramson, D.H.; Seddon, J.M.; Sampson, J.N.; Kleinerman, R.A. Risk of subsequent malignant neoplasms in long-term hereditary retinoblastoma survivors after chemotherapy and radiotherapy. J. Clin. Oncol. 2014, 32, 3284–3290. [Google Scholar] [CrossRef] [Green Version]
- Abramson, D.H.; Shields, C.L.; Munier, F.L.; Chantada, G.L. Treatment of Retinoblastoma in 2015: Agreement and Disagreement. JAMA Ophthalmol. 2015, 133, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, P.; Ansperger-Rescher, B.; Schuler, A.; Zeschnigk, M.; Gallie, B.; Lohmann, D.R. Spectrum of gross deletions and insertions in the RB1 gene in patients with retinoblastoma and association with phenotypic expression. Hum. Mutat. 2005, 26, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, D.R.; Gerick, M.; Brandt, B.; Oelschlager, U.; Lorenz, B.; Passarge, E.; Horsthemke, B. Constitutional RB1-gene mutations in patients with isolated unilateral retinoblastoma. Am. J. Hum. Genet. 1997, 61, 282–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeschnigk, M.; Bohringer, S.; Price, E.A.; Onadim, Z.; Masshofer, L.; Lohmann, D.R. A novel real-time PCR assay for quantitative analysis of methylated alleles (QAMA): Analysis of the retinoblastoma locus. Nucleic Acids Res. 2004, 32, e125. [Google Scholar] [CrossRef]
- Zhang, K.; Nowak, I.; Rushlow, D.; Gallie, B.L.; Lohmann, D.R. Patterns of missplicing caused by RB1 gene mutations in patients with retinoblastoma and association with phenotypic expression. Hum. Mutat. 2008, 29, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Tschentscher, F.; Husing, J.; Holter, T.; Kruse, E.; Dresen, I.G.; Jockel, K.H.; Anastassiou, G.; Schilling, H.; Bornfeld, N.; Horsthemke, B.; et al. Tumor classification based on gene expression profiling shows that uveal melanomas with and without monosomy 3 represent two distinct entities. Cancer Res. 2003, 63, 2578–2584. [Google Scholar]
- Le Guin, C.H.D.; Metz, K.A.; Kreis, S.H.; Bechrakis, N.E.; Bornfeld, N.; Zeschnigk, M.; Lohmann, D.R. GNAQ Q209R Mutations Are Highly Specific for Circumscribed Choroidal Hemangioma. Cancers 2019, 11, 31. [Google Scholar] [CrossRef] [Green Version]
- GENESIS-Online Nutzerservice, DESTATIS Statistisches Bundesamt. Available online: https://www-genesis.destatis.de/genesis/online (accessed on 26 January 2021).
- Statistik Austria, Die Informationsmanager. Available online: https://statistik.at (accessed on 26 January 2021).
- Stacey, A.W.; Bowman, R.; Foster, A.; Kivela, T.T.; Munier, F.L.; Cassoux, N.; Fabian, I.D.; Global Retinoblastoma Study Group. Incidence of Retinoblastoma Has Increased: Results from 40 European Countries. Ophthalmology 2021. [Google Scholar] [CrossRef]
- Temming, P.; Viehmann, A.; Biewald, E.; Lohmann, D.R. Sporadic unilateral retinoblastoma or first sign of bilateral disease? Br. J. Ophthalmol. 2013, 97, 475–480. [Google Scholar] [CrossRef]
- Mastrangelo, D.; Hadjistilianou, T.; Di Pisa, F.; Capretti, M.C.; Frezzotti, R. Metachronous tumor development in unilateral retinoblastoma. Eur. J. Ophthalmol. 2000, 10, 149–152. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [Green Version]
- Soliman, S.E.; Racher, H.; Zhang, C.; MacDonald, H.; Gallie, B.L. Genetics and Molecular Diagnostics in Retinoblastoma—An Update. Asia Pac. J. Ophthalmol. 2017, 6, 197–207. [Google Scholar] [CrossRef]
- Robertson, J.C.; Jorcyk, C.L.; Oxford, J.T. DICER1 Syndrome: DICER1 Mutations in Rare Cancers. Cancers 2018, 10, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theurer, S.; Biewald, E.; Kuchelmeister, K.; Temming, P.; Kuechler, A.; Oeffner, F.; Bornfeld, N.; Sirin, S.; Schmid, K.W.; Metz, K. Primary meningioma of the optical nerve sheet in infancy as initial presentation of neurofibromatosis type 2. Pathologe 2019, 40, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Kivela, T. Trilateral retinoblastoma: A meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J. Clin. Oncol. 1999, 17, 1829–1837. [Google Scholar] [CrossRef] [PubMed]
- Ripperger, T.; Bielack, S.S.; Borkhardt, A.; Brecht, I.B.; Burkhardt, B.; Calaminus, G.; Debatin, K.M.; Deubzer, H.; Dirksen, U.; Eckert, C.; et al. Childhood cancer predisposition syndromes-A concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. Am. J. Med. Genet. A 2017, 173, 1017–1037. [Google Scholar] [CrossRef] [PubMed]
- Jongmans, M.C.; Loeffen, J.L.; Waanders, E.; Hoogerbrugge, P.M.; Ligtenberg, M.J.; Kuiper, R.P.; Hoogerbrugge, N. Recognition of genetic predisposition in pediatric cancer patients: An easy-to-use selection tool. Eur. J. Med. Genet. 2016, 59, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerrish, A.; Stone, E.; Clokie, S.; Ainsworth, J.R.; Jenkinson, H.; McCalla, M.; Hitchcott, C.; Colmenero, I.; Allen, S.; Parulekar, M.; et al. Non-invasive diagnosis of retinoblastoma using cell-free DNA from aqueous humour. Br. J. Ophthalmol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Villani, A.; Tabori, U.; Schiffman, J.; Shlien, A.; Beyene, J.; Druker, H.; Novokmet, A.; Finlay, J.; Malkin, D. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: A prospective observational study. Lancet Oncol. 2011, 12, 559–567. [Google Scholar] [CrossRef]
- Tonorezos, E.S.; Friedman, D.N.; Barnea, D.; Bosscha, M.I.; Chantada, G.; Dommering, C.J.; de Graaf, P.; Dunkel, I.J.; Fabius, A.W.M.; Francis, J.H.; et al. Recommendations for Long-Term Follow-up of Adults with Heritable Retinoblastoma. Ophthalmology 2020, 127, 1549–1557. [Google Scholar] [CrossRef]
- Kamihara, J.; Bourdeaut, F.; Foulkes, W.D.; Molenaar, J.J.; Mosse, Y.P.; Nakagawara, A.; Parareda, A.; Scollon, S.R.; Schneider, K.W.; Skalet, A.H.; et al. Retinoblastoma and Neuroblastoma Predisposition and Surveillance. Clin. Cancer Res. 2017, 23, e98–e106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, D.G.R.; Salvador, H.; Chang, V.Y.; Erez, A.; Voss, S.D.; Schneider, K.W.; Scott, H.S.; Plon, S.E.; Tabori, U. Cancer and Central Nervous System Tumor Surveillance in Pediatric Neurofibromatosis 1. Clin. Cancer Res. 2017, 23, e46–e53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, D.G.R.; Salvador, H.; Chang, V.Y.; Erez, A.; Voss, S.D.; Druker, H.; Scott, H.S.; Tabori, U. Cancer and Central Nervous System Tumor Surveillance in Pediatric Neurofibromatosis 2 and Related Disorders. Clin. Cancer Res. 2017, 23, e54–e61. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
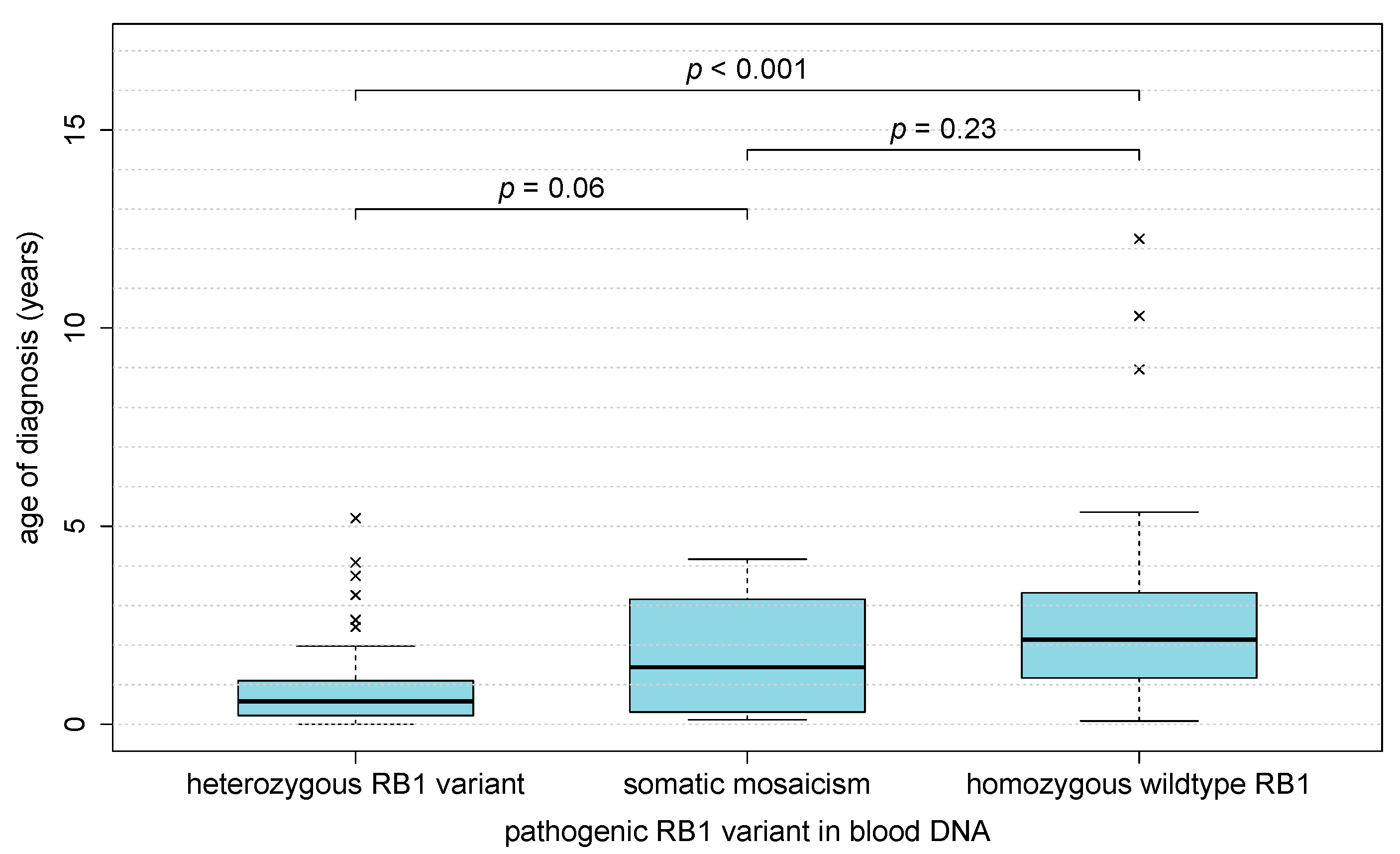{kind=link}
| Clinical Characteristics | Retinoblastoma | Uveal Melanoma | Ciliary Body Medulloepithelioma | Retinal Astrocytoma | Meningioma Optic Nerve with Intraocular Extension | |
|---|---|---|---|---|---|---|
| all | 287 | 7 | 3 | 2 | 1 | |
| gender | female | 138 | 4 | 1 | 1 | 0 |
| male | 149 | 3 | 2 | 1 | 1 | |
| age at diagnosis of eye tumor (years) | median | 1.3 | 11.9 | 5.5 | 9.0 | 0.28 |
| range | 0.0–12.3 | 9.9–16.3 | 2.1–11.8 | 3.6–14.4 | ||
| status | alive | 284 | 6 | 3 | 2 | 1 |
| deceased | 3 | 1 | 0 | 0 | 0 | |
| laterality | unilateral | 184 | 7 | 3 | 2 | 1 |
| bilateral | 99 | |||||
| no data | 4 | |||||
| Known associated cancer predisposition genes | RB1 | BAP1 | DICER1 | TSC1 TSC2 NF1 | NF2 | |
| Pathogenetic variant in cancer predisposition gene | yes | 99 | 0 | 0 | 0 | 1 |
| no evidence | 136 | 3 | 1 | 0 | 0 | |
| not tested or inconclusive | 52 | 4 | 0 | 0 | 0 | |
| Number of affected eyes | 386 | 7 | 3 | 2 | 1 | |
| Genetic and Clinical Criteria | Number of Patients (in %) | |
|---|---|---|
| All | 287 | |
| heritability of retinoblastoma | heritable RB | 129 (44.9) |
| (clinical and/or genetic criteria) | non-heritable RB with genetic analysis | 130 (45.3) |
| unilateral without genetic analysis | 28 (9.8) | |
| genetic analysis of RB1 | constitutional RB1 variant | 87 (30.3) |
| somatic mosaic RB1 variant in blood | 12 (4.2) | |
| no RB1 variant detected in blood | 136 (47.4) | |
| no data | 52 (18.1) | |
| type of heterozygous constitutional variant | nonsense or frameshift variant | 36 (41.4) |
| splice-site variant | 29 (33.3) | |
| missense or in-frame | 8 (9.2) | |
| large deletion | 14 (16.1) | |
| family history | isolated | 246 (85.7) |
| familial | 33 (11.5) | |
| data missing | 8 (2.8) | |
| number of affected eyes | 386 | |
| ICRB Group | ICRB A | 37 (9.6) |
| ICRB B | 87 (22.5) | |
| ICRB C | 21 (5.4) | |
| ICRB D | 70 (18.1) | |
| ICRB E | 147 (38.1) | |
| no data | 24 (6.2) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reschke, M.; Biewald, E.; Bronstein, L.; Brecht, I.B.; Dittner-Moormann, S.; Driever, F.; Ebinger, M.; Fleischhack, G.; Grabow, D.; Geismar, D.; et al. Eye Tumors in Childhood as First Sign of Tumor Predisposition Syndromes: Insights from an Observational Study Conducted in Germany and Austria. Cancers 2021, 13, 1876. https://doi.org/10.3390/cancers13081876
Reschke M, Biewald E, Bronstein L, Brecht IB, Dittner-Moormann S, Driever F, Ebinger M, Fleischhack G, Grabow D, Geismar D, et al. Eye Tumors in Childhood as First Sign of Tumor Predisposition Syndromes: Insights from an Observational Study Conducted in Germany and Austria. Cancers. 2021; 13(8):1876. https://doi.org/10.3390/cancers13081876
Chicago/Turabian StyleReschke, Madlen, Eva Biewald, Leo Bronstein, Ines B. Brecht, Sabine Dittner-Moormann, Frank Driever, Martin Ebinger, Gudrun Fleischhack, Desiree Grabow, Dirk Geismar, and et al. 2021. "Eye Tumors in Childhood as First Sign of Tumor Predisposition Syndromes: Insights from an Observational Study Conducted in Germany and Austria" Cancers 13, no. 8: 1876. https://doi.org/10.3390/cancers13081876
APA StyleReschke, M., Biewald, E., Bronstein, L., Brecht, I. B., Dittner-Moormann, S., Driever, F., Ebinger, M., Fleischhack, G., Grabow, D., Geismar, D., Göricke, S., Guberina, M., Le Guin, C. H. D., Kiefer, T., Kratz, C. P., Metz, K., Müller, B., Ryl, T., Schlamann, M., ... Ketteler, P. (2021). Eye Tumors in Childhood as First Sign of Tumor Predisposition Syndromes: Insights from an Observational Study Conducted in Germany and Austria. Cancers, 13(8), 1876. https://doi.org/10.3390/cancers13081876